

Abstract
Chronic pancreatitis (CP) is an inflammatory disease of the pancreas characterized by progressive fibrotic destruction of the pancreatic secretory parenchyma. Despite the heterogeneity in pathogenesis and involved risk factors, processes such as necrosis/apoptosis, inflammation or duct obstruction are involved. This fibrosing process ultimately leads to progressive loss of the lobular morphology and structure of the pancreas, deformation of the large ducts and severe changes in the arrangement and composition of the islets. These conditions lead to irreversible morphological and structural changes resulting in impairment of both exocrine and endocrine functions. The prevalence of the disease is largely dependent on culture and geography. The etiological risk-factors associated with CP are multiple and involve both genetic and environmental factors. Throughout this review the M-ANNHEIM classification system will be used, comprising a detailed description of risk factors such as: alcohol-consumption, nicotine-consumption, nutritional factors, hereditary factors, efferent duct factors, immunological factors and miscellaneous and rare metabolic factors. Increased knowledge of the different etiological factors may encourage the use of further advanced diagnostic tools, which potentially will help clinicians to diagnose CP at an earlier stage. However, in view of the multi factorial disease and the complex clinical picture, it is not surprising that treatment of patients with CP is challenging and often unsuccessful.
Keywords: Chronic pancreatitis, Pathogenesis, Risk factors, Etiology
Core tip: The reported prevalence of chronic pancreatitis (CP) is approximately 0.5%. Etiological risk-factors associated with CP are multiple and throughout the review the M-ANNHEIM classification is used comprising environmental factors (alcohol consumption, nicotine habits and nutrition), hereditary, well characterized mutations, ductal obstruction and autoimmune factors. CP is characterized by progressive fibrotic destruction of glandular tissue, inflammation or duct obstruction, leading to irreversible functional impairment of both exocrine and endocrine functions. In view of the multi-factorial disease and the complex clinical picture, it is not surprising that treatment of patients with CP is challenging and often unsuccessful.
INTRODUCTION
The reported prevalence of chronic pancreatitis (CP) varies due to differences in study design, diagnostic criteria, culture and geography; however in Europe and United States it is relatively rare varying between 0.2% and 0.6%[1]. The annual incidence is estimated to be approximately 7-10 per 100000[2]. The etiological risk-factors associated with CP are multiple and involve environmental factors (alcohol consumption, nicotine habits and nutrition), hereditary well characterized mutations, ductal obstruction and autoimmune factors[3]. CP is characterized by progressive fibrotic destruction of the glandular tissue. The secretory parenchyma is destroyed by processes such as necrosis/apoptosis, inflammation or duct obstruction. Increasing evidence indicates that pancreatic stellate cells (PSC) are the major mediators of fibrosis, resulting in the formation of extracellular matrix (ECM) in the interstitial spaces and in the areas where acinar cells disappear or duct cells are injured. This process ultimately leads to progressive loss of the lobular morphology and structure of the pancreas, bizarre deformation of the large ducts and severe changes in the arrangement and composition of the islets. The fibrotic destruction of the pancreatic gland is irreversible and the morphological and structural changes lead to functional impairment of both exocrine and endocrine functions, eventually leading to malnutrition and/or diabetes[3-6]. In many aspects the PSCs share many similar features to hepatic stellate cells and glomerular mesangial cells. The onset of pancreatic fibrogenesis is caused by injury which may involve interstitial mesenchymal cells, the duct cells and/or the acinar cells. Which of these elements is affected depends on the etiological risk factor. However, destruction to any one of these pancreatic tissue compartments is associated with transformation of resident fibroblasts/pancreatic stellate cells into myofibroblast-like phenotypes; a process called activation. In the activated state PSCs express α-smooth muscle actin (α-SMA), proliferate, and secrete fibrillar collagens, including collagen I and III and fibronectin. All together this production and deposition of ECM is characteristic in chronic pancreatic fibrosis, and the PSCs likely represent the wound-healing myofibroblasts of the pancreas[7,8]. The exact pathophysiological mechanisms initiating and maintaining the development of fibrosis in the pancreas are poorly understood, but may be viewed as a progression similar to, e.g., liver fibrosis[9]. Hence, the initial injury to one or all of the various tissue compartments or cell types of the pancreas, leads to cell necrosis and/or apoptosis and consequently release of cytokines/growth factors (e.g., tumor growth factor b1, interleukin-8, platelet-derived growth factor and CC-chemokines), either from immigrating inflammatory cells, especially macrophages, and/or nearby preexistent epithelial or mesenchymal cells[10-13]. Thereafter damaged cells are phagocytosed by macrophages, causing release of cytokines, which in turn causes activation and proliferation of resident fibroblasts/PCS situated in the immediate surroundings of the original site of injury, which accordingly induces transformation into myofibroblast cells[7,14]. However, it has also been suggested as an alternative hypothesis, that the abovementioned progression is bypassed, and the initiating etiological factor (for instance, ethanol consumption) activates resident fibroblasts directly. In the final stage, myofibroblasts produce and extracellular matrix deposits replace the inflammatory infiltrate and affect the architecture and function of the remaining pancreatic tissues[4]. Subsequently, a vicious circle has started, because in order to facilitate the deposition of the newly formed ECM, myofibroblasts enhance production of specialized enzymes, such as metalloproteinase matrix metalloproteinase (MMP)-3 and MMP-9, which are able to demolish the normal pericellular ECM. These metalloproteinases are in return regulated by cytokine tumor growth factor (TGF)-β1s, which through autocrine inhibition enhances pancreatic fibrogenesis by reducing collagen degradation[8]. As a consequence of declined ECM production (due to withdrawal of the initiating factor), myofibroblasts may disappear either through apoptosis or retransformation into fibroblasts. Furthermore, pancreatic stellate cells express both mediators of matrix remodeling and the regulatory cytokine TGF-β1 that, by autocrine inhibition of MMP-3 and MMP-9, may enhance fibrogenesis by reducing collagen degradation[8]. Pancreatic stellate cells express both mediators of matrix remodeling and the regulatory cytokine TGF-β1 that, by autocrine inhibition of MMP-3 and MMP-9, may enhance fibrogenesis by reducing collagen degradation[8].
To describe the heterogeneity of the underlying pathophysiology we have throughout the review used the M-ANNHEIM classification[3], comprising detailed description of Multiple risk factors such as: alcohol-consumption, nicotine-consumption, nutritional factors, hereditary factors, efferent duct factors, immunological factors and miscellaneous including rare metabolic factors (Figure 1). Increased knowledge of the different etiological factors may encourage the use of further advanced diagnostic tools, which potentially will help clinicians to diagnose CP at an earlier stage.
Figure 1.
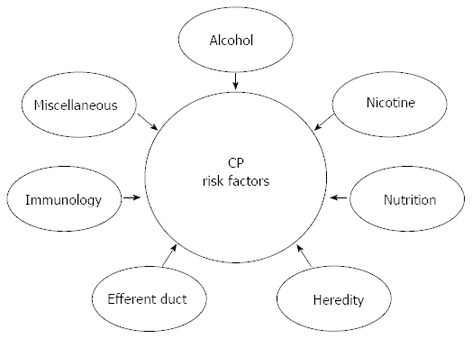
The etiological risk-factors associated with chronic pancreatitis are multiple and involve both genetic and environmental factors. According to the M-ANNHEIM classification system, different synergistic risk factors are known such as: alcohol-consumption, nicotine-consumption, nutritional factors, hereditary factors, efferent duct factors, immunological factors and miscellaneous and rare metabolic factors. CP: Chronic pancreatitis.
However, in view of the multi-factorial disease and the complex clinical picture, it is not surprising that treatment of patients with CP is challenging and often unsuccessful.
According to the M-ANNHEIM classification, the following etiological risk factors are involved in the pathogenesis of chronic pancreatitis.
ALCOHOL CONSUMPTION
A relationship between alcohol consumption and pancreatic impairment has been reported as early as 1878[15] and hence, alcohol intake has long been regarded as the primary cause of chronic pancreatitis. Nowadays, there is satisfactory indication that the pancreas has the ability to metabolize ethanol via both oxidative and the non-oxidative pathways[16]. The metabolites and their byproducts injure acinal cells and activate stellate cells to produce and deposit ECM (Figure 2). A study by Dufour et al[17] suggested that CP development is associated to the dose and duration of alcohol consumption. It was estimated that approximately 80 g of alcohol per day for a minimum of 6-12 years is required to produce symptomatic pancreatitis. However, the consumption of lesser quantities may also lead to pancreatic injury and may have an impact on the progression of the disease[18]. In order to consider the risks associated with lower consumption of alcohol, the M-ANNHEIM classification system of CP grouped alcohol consumption into patterns of moderate (< 20 g pure ethanol per day), increased (20-80 g pure ethanol per day), or excessive (> 80 g pure ethanol per day)[3]. While alcohol consumption is doubtlessly a contributing factor in CP, it must be noted, that considerable amount of recent epidemiological studies and animal experiments suggest that alcohol alone is not sufficient to induce CP. An overview of the associations between alcohol consumption and other risk factors are listed in Table 1.
Figure 2.
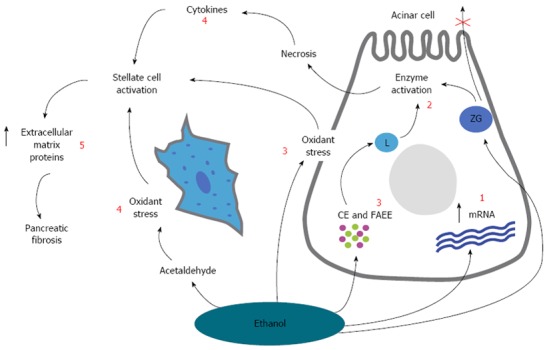
A schematic overview showing the overall hypothesis for the pathogenesis of alcoholic chronic pancreatitis. The effect of ethanol and its metabolites on the subcellular organelles include increased digestive and lysosomal enzyme content [due to increased synthesis (increased mRNA) and impaired secretion (1)] and destabilization of lysosomes (L) (2) and zymogen granules (ZG) [mediated by oxidant stress, cholesteryl esters (CE) and fatty acid ethyl esters (FAEE) (3)]. These changes will make the cell more sensitive to trigger factors and in the presence of appropriate trigger factors, overt acinar cell injury is initiated (alcoholic acute pancreatitis). Pancreatic stellate cells are activated by cytokines during alcohol-induced necroinflammation, or directly by ethanol via its metabolism to acetaldehyde and the subsequent generation of oxidant stress (4). Activated pancreatic stellate cell then increases the synthesis of extracellular matrix proteins leading to pancreatic fibrosis. Modified from Vonlaufen et al[84] (5).
Table 1.
Association with alcoholic chronic pancreatitis
| Factor | Association |
| Drink type | Yes: Vonlaufen et al[83] |
| Nakamura et al[84] | |
| No: Levy et al[29] | |
| Wilson et al[85] | |
| Drinking pattern | Yes: Lankisch et al[18] |
| No: Levy et al[29] | |
| Wilson et al[85] | |
| Diet | Yes: Levy et al[29] |
| No: Wilson et al[85] | |
| Tobacco | Yes: Rebours et al[28] |
| Maisonneuve[27] | |
| Imoto[26] | |
| Lowenfels[86] | |
| No: Levy[29] | |
| Haber et al[87] | |
| Genetics | Yes: Whitcomb et al[24] (PRSS1-PRSS2 and X- linked CLDN 2) |
| Rosendahl[88] (Chymotrypsin C gene mutation) | |
| Miyasaka et al[89] (Cholesteryl esterlipase polymorphism) | |
| Ockenga et al[90] (UDP-glucuronosyl transferase) | |
| Witt et al[91] (SPINK1 mutations) | |
| No: Schneider et al[92] | |
| Perri et al[93] | |
| Frenzer et al[94] | |
| Schneider et al[82] | |
| Norton et al[95] | |
| Haber et al[96] | |
| Wilson et al[85] |
PRSS1: Cationic trypsinogen gene; PRSS2: Anionic trypsinogen gene; CLDN 2: Claudin 2 gene; UDP-glucuronosyltransferase: Uridine 5´-diphospho-glucuronosyltransferase; SPINK1: Serine protease inhibitor, kazal type 1, gene (encodes for pancreatic secretory trypsin inhibitor).
A multi-centre study from Italy reported that excessive alcohol consumption was the principal factor in only 34% of cases of CP[19] and an assessment from the United States concluded it was the main factor in 44% of CP cases[20]. Moreover, African-Americans are at particular risk of developing alcoholic CP[21]. Overall, less than 10% of heavy alcohol consumers develop alcoholic induced CP[17]. Hence, several theories have been proposed as to how alcohol might lead to CP, but no clear-cut answer exists as prolonged feeding of ethanol does not trigger the onset of CP in itself. Therefore, there are indications that alcohol sensitizes the pancreas to other external factors which interact to increase ethanol toxicity in vivo, such as cigarette smoking and diet[22] or genetic predisposition). Interestingly, alteration of pancreatic secretory trypsin inhibitor (SPINK1, Figure 3) and CFTR genes was present in patients with alcoholic CP and the increase of those genes was moreover associated with higher levels of alcohol consumption[23]. In this line, a recent study[24] found a genetic variant on chromosome X near the CLDN2 gene that predicts which heavy drinking males were in higher risk of developing CP.
Figure 3.
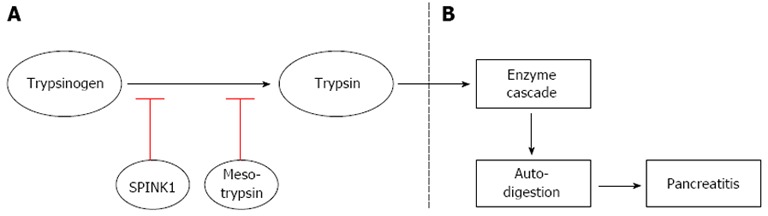
A scheme representing the role of digestive enzymes in normal pancreatic tissue and in the case of pancreatitis. A: In a normal pancreas, SPINK1 (first line of defense) and mesotrypsin (second line of defense) inhibit the generation of trypsin resulting from auto-activation of trypsinogen. These defense mechanisms prevent the pancreas from activating the pancreatic enzyme cascade and auto-digestion; B: If mutations are present in the SPINK1 and/or in the mesotrypsin gene, they losses their ability to inhibit the generation of trypsin resulting in activation of enzyme cascade and subsequent pancreatic auto-digestion leading to pancreatitis. SPINK1: Serine protease inhibitor, kazal type 1, gene (encodes for pancreatic secretory trypsin inhibitor).
NICOTINE CONSUMPTION
Smoking has been identified as an important risk factor for development of chronic pancreatitis. The effect of tobacco in CP was first described in 1994[25], where heavy smokers had a significantly increased risk of developing pancreatic calcifications but no effects of alcohol were found. Another study reported that cigarette smoking increased the risk of pancreatic calcifications in late onset idiopathic CP in a population that never drank alcohol[26]. Moreover, a study by Maisonneuve et al[27] showed that smoking accelerated the degenerative alcohol induced pancreatitis quantified by the presence of calcifications and diabetes. Recently, a study reported that tobacco increased the occurrence of all major complications of alcoholic CP depending on the amount of smoking (1 pack-year is calculated as the number of cigarettes per day, multiplied by the number of years of smoking divided by 20 cigarettes/pack)[28]. No differences in CP outcome were seen at nicotine consumption corresponding to 10 pack-years. At a threshold of 15 pack-years CP was diagnosed earlier (36 years vs 46 years) and at nicotine consumption threshold of 20 pack-years up to 76% of patients were presented with pancreatic calcifications and ductal changes[28]. Tobacco effect on CP has long been considered to merely potentiate the main pancreatic toxic role of alcohol. However, although heavy smokers tended to be heavy drinkers, these recent findings indicate that tobacco intake is an independent risk factor in CP and accelerates the course of the disease.
NUTRITIONAL FACTORS
Only a few studies were conducted to investigate nutritional effect on CP[29-31], finding that diets rich in fat and protein possibly have an impact on the development of CP. However, due to subjective descriptions of daily nutritional customs and determination of the retrospective body mass index make it nearly impossible to deliver a simple report of past daily nutrition in majority of patients with CP. There is a type of CP named tropical CP which has been first described in Indonesia in 1959[32]. Tropical CP is a form of non-alcoholic CP existing in tropical developing countries. There is some confusion regarding the nomenclature sine tropical CP previously was hypothesized to be linked to diets high in consumption of cassava[33], which is a root high in carbohydrates but low in proteins and is cultivated in tropical and subtropical regions of the world. However, a study in rats which were fed cassava for up to one year failed to produce CP[34]. Moreover, a study in humans comparing the amount of consumption of cassava did not observe a difference between healthy controls and tropical CP patients[35], and today tropical pancreatitis is - at least partly - linked to genetic mutations[36]. Hence, tropical CP is widespread in parts of India and Africa where cassava is not a part of nutrition and tropical CP is not observed in rural West Africa where cassava intake is high[37]. Therefore, the cassava theory lacks evidence.
HEREDITARY FACTORS
Over the last decades an association between different types of chronic pancreatitis and hereditary factors has been reported. Hereditary chronic pancreatitis is a rare form which is characterized by an early onset of recurrent attacks of severe epigastric pain usually before ten years of age[38]. Back in 1952, Comfort and Steinberg[39] first described hereditary chronic pancreatitis as an autosomal dominant disease. In 1996, the hereditary chronic pancreatitis disease gene was mapped to chromosome 7q35, which encodes the cationic trypsinogen gene (PRSS1), and the first mutation, R122H, was detected[40]. Ever since then, many others mutations have been identified (A16V, D22G, K23R, N29I, N29T, R122C and R122H)[41-46] and nowadays it is known that approximately 80% of hereditary chronic pancreatitis patients have a PRSS1 mutation[47]. R122H is the most frequent mutation causing hereditary chronic pancreatitis, where Arginine (Arg) is substituted with Histidine (His) at residue 117 on codon 122[5].
Trypsinogen is the inactive pro-enzyme for trypsin and cationic trypsinogen is the most abundant isoform in the human pancreatic juice (Figure 3). Trypsin becomes active when an eight-amino acid amino-terminal peptide is removed by an enteropeptidase, and the active trypsin plays a central role in pancreatic exocrine physiology as it initiates the cascade activation of other pancreatic digestive enzymes. Normally minor amounts of trypsin are activated within pancreatic acinar cells. Despite the small amount of active trypsin, an ongoing rapid inactivation takes place in order to prevent the digestion enzymes activation cascade and pancreatic auto digestion. To avoid auto digestion, two inhibitory mechanisms are present. First line of defense is the pancreatic secretory trypsin inhibitor (PSTI, gene name: serine protease inhibitor, kazal type 1, SPINK1), which inhibits up to 20 % of the trypsin activity[38,48]. If SPINK1 fails to inhibit the trypsin activity, the trypsin-like enzymes (e.g., mesotrypsin) are activated which hydrolyses trypsin and other zymogens (second line of defense)[49,50]. Hence, any mutation in SPINK1 or the mesotrypsin gene will delay the protection, and therefore hereditary chronic pancreatitis patients are only protected against pancreatic auto digestion and progressive pancreatitis as long as the level of trypsin activity is less than SPINK1 level.
In addition, a diagnosis of familial pancreatitis refers to pancreatitis from any cause that occurs in family with an incidence greater than expected by chance alone, given the size of the family and incidence of pancreatitis within a defined population[51]. Familial pancreatitis may as well be caused by genetic mutations.
As mentioned above SPINK1 encodes PSTI (one of the defensive mechanisms against prematurely trypsin activity within the acinar cells) and SPINK1 mutations are thought to be a disease modifying factor rather than being disease causing factor in idiopathic chronic pancreatitis[52,53].
Idiopathic chronic pancreatitis is defined as cases of pancreatitis within a family where no associated factor can be identified and represents 20%-30% of cases of chronic pancreatitis[54,55]. Most of the idiopathic chronic pancreatitis cases may be due to a variety of processes such as mutations in SPINK1 and cystic fibrosis transmembrane conductance regulator (CFTR) gene[54-56], or Sjögren’s syndrome[57,58].
In 1998 a relationship between the CFTR gene and idiopathic chronic pancreatitis was described[59,60], but the underlying mechanisms leading to the development of chronic pancreatitis are still poorly understood. CFTR is identified as the cystic fibrosis gene and the link between cystic fibrosis and chronic pancreatitis is that both conditions may show abnormal sweat chloride concentrations together with pancreatic ductal obstruction caused by inspissated secretions. Furthermore, 1%-2% of patients with cystic fibrosis may suffer from recurrent pancreatitis[60,61], however up to 85% suffer from exocrine insufficiency and even up to 93% take exogenous pancreatic supplements[62].
There are two forms of idiopathic chronic pancreatitis: early- and late-onset pancreatitis, which both differ from alcoholic pancreatitis. Patients with early-onset develop calcification and exocrine and endocrine insufficiency more slowly than patients with late-onset disease, but they experience more severe pain[63].
EFFERENT DUCT FACTORS
Anatomically, the main pancreatic duct joins the common bile duct, after which both ducts perforate the medial side of the second portion of the duodenum at the major duodenal papilla. Hence any obstruction (partial or complete), compression or inflammation of the pancreatic tissue will increase the pressure within the pancreatic efferent ducts leading to ductal dilation proximal (upstream) of the stenosis and to atrophy of the acinar cells and replacement by fibrous tissue[4]. During embryogenesis, two efferent pancreatic ducts, a ventral and a dorsal duct fuse together to form one main pancreatic duct. When this fusion fails to occur, a pancreas divisum is formed, which is one of the most frequent congenital ductal anomalies. Theoretically this may cause flow problems within the pancreatic duct. However, pancreas divisum is present in up to 9% of autopsy studies and controversies exist whether presence of this abnormality is overrepresented in chronic pancreatitis in comparison to the normal population. Despite the rarity (5-15 cases per 100000), annular pancreas is another congenital disease which may be linked to linked to chronic pancreatitis, as the pancreatic tissue forms a complete or partial ring around the duodenum[64].
There are various other possible causes for a duct obstruction, but the most important and common is the narrowing and eventual occlusion of the main pancreatic duct in the head of the pancreas due to a ductal adenocarcinoma. Other causes for obstruction of the main pancreatic duct include intraductal papillary-mucinous neoplasms, cystic and endocrine neoplasms and acquired fibrous strictures. In the smaller pancreatic ducts, ductal papillary hyperplasia is a common cause of narrowing of the duct lumen. Finally, obstruction caused by a gallstones in the common bile duct (choledocholithiasis) or sphincter of Oddi dysmotility can obstruct flow and therefore cause retention of bile in the biliary tree and pancreatic juice in the pancreatic duct. The former is a frequent and well-known cause for acute pancreatitis.
IMMUNOLOGICAL RISK FACTORS
Immunological risk factors involved in chronic pancreatitis leading to autoimmune pancreatitis (AIP) deserve increased awareness as the underlying pathogenesis is not fully elucidated. The effort to diagnose autoimmune pancreatitis is mainly focused on the treatment possibilities with e.g., glucocorticosteroids and differentiating the disease from pancreatic cancer. There are several lines of evidence that immunological factors play a key role in CP. Even though lymphoplasmacytic infiltration in AIP is most pronounced in the pancreatic ducts, advanced cases also show intralobular lymphoplasmacytic infiltration, and the inflammation may also specifically attack the acinar cells. IgG antibodies to a plasminogen-binding protein homologous to the human ubiquitin-protein ligase E3 component n-recognin 2, is expressed in pancreatic acinar cells, and have been found in the sera of AIP patients[65]. In AIP patients auto-antibodies to trypsinogens are also upregulated, which is not the case in alcohol induced chronic pancreatitis patients, corresponding to loss of trypsinogen-positive acinar cells in AIP tissues[66]. Furthermore, basement membrane deposits of complement C3c, IgG4 and IgG not only around pancreatic ducts but also around acini have been demonstrated in AIP[67].
Even though it has not yet been fully proved that there are differences in the pathogenesis, AIP is separated into two distinct types: Type 1 and type 2 AIP.
Type 1 AIP patients (most common type in East Asia) are typically older than type 2 patients, with a mean age at disease onset of 62 years[68]. Plasma levels of IgG4 are increased and in approximately 50% of the cases other organs are affected[68,69]. The key histological feature that distinguishes type 1 AIP from type 2 AIP is strong infiltration of IgG4-immunopositive plasma cells and lymphoplasmacytic infiltration[70,71] IgG4-positive sclerosing cholangitis represents one of the main extrapancreatic manifestations of type 1 AIP, and hence jaundice is a common symptom[72]. Due to the multiple extrapancreatic manifestations of type 1 AIP, Kamisawa et al[73] promoted the concept of AIP as part of a clinicopathological entity of IgG4-associated systemic disease, together with retroperitoneal fibrosis, sclerosing sialadenitis and sclerosing cholangitis. This systemic fibroinflammatory disease probably also includes lesions in the aorta, breast and prostate[73-76].
Type 2 AIP patients (45% of the cases in United States and Europe) are typically younger than type 1 AIP, with a mean age of 40-48 years[68,77]. AIP is often associated with Crohn’s disease and ulcerative colitis[78]. The most characteristic histological feature which distinguishes type 2 from type 1 AIP is the so-called granulocytic epithelial lesions which are a hallmark of type 2 AIP[77,79]. Besides, type 2 AIP is usually not associated with the extrapancreatic manifestations observed in type 1 AIP.
MISCELLANEOUS
Tropical chronic pancreatitis
Tropical chronic pancreatitis may be referred to an idiopathic, juvenile, non-alcoholic form of chronic pancreatitis widely prevalent in the developing countries of the tropical world. Tropical chronic pancreatitis can be sub-grouped in two entities: Tropical calcific pancreatitis and fibrocalculous pancreatic diabetes. Tropical calcific pancreatitis describes the early pre-diabetic stage of the disease and affects younger people. Tropical calcific pancreatitis is characterized by severe abdominal pain, pancreatic calcification, and signs of pancreatic dysfunction (no diabetes mellitus at the time of diagnosis). Fibrocalculous pancreatic diabetes describes the late diabetic stage of the disease where diabetes mellitus is the first major clinical sign to determine the diagnosis of fibrocalculous pancreatic diabetes. The etiology of tropical calcific pancreatitis and fibrocalculous pancreatic diabetes has shown to be related to genetic mutations in the SPINK1 gene, but it is still unknown if environmental factors have an influence[36,80-82].
Primary hypercalcemia
It has been suggested that higher serum calcium levels may contribute to pancreatitis, as hypercalcemia may predispose the pancreatic acinar cell to abnormal, sustained calcium levels, which leads to premature pancreatic protease activation, and consequently pancreatitis.
Hyperparathyreoidisme
Controversies exist regarding associations between primary hyperparathyroidism and acute or chronic pancreatitis. However, most studies show an increased rate of pancreatitis among patients with primary hyperparathyroidism in comparison to general hospitalized patients without the disease.
Hyperlipidemia
Hyperlipidemia covers abnormally elevated levels of any or all lipids, lipoproteins or both in the blood. Hyperlipidemia can be divided in primary and secondary subtypes. Lipid and lipoprotein abnormalities are relatively common in the general population, and are regarded as a modifiable risk factor associated with acute pancreatitis.
CONCLUSION
Increased knowledge of different etiological factors and how they interact may encourage the use of further advanced diagnostic tools, which potentially will help clinicians to diagnose and treat CP at an earlier stage. Recent research has increased our understanding of the disease and has changed the approach to CP. However, in view of the multi-factorial disease and the complex clinical picture, it is not surprising that treatment of patients with CP is challenging and often unsuccessful.
Footnotes
Supported by The Danish Council for Strategic Research
P- Reviewers: Bramhall SR, Dambrauskas Z, Falconi M, Sakata N S- Editor: Wen LL L- Editor: A E- Editor: Zhang DN
References
- 1.Lévy P, Barthet M, Mollard BR, Amouretti M, Marion-Audibert AM, Dyard F. Estimation of the prevalence and incidence of chronic pancreatitis and its complications. Gastroenterol Clin Biol. 2006;30:838–844. doi: 10.1016/s0399-8320(06)73330-9. [DOI] [PubMed] [Google Scholar]
- 2.Andersen BN, Pedersen NT, Scheel J, Worning H. Incidence of alcoholic chronic pancreatitis in Copenhagen. Scand J Gastroenterol. 1982;17:247–252. doi: 10.3109/00365528209182047. [DOI] [PubMed] [Google Scholar]
- 3.Schneider A, Löhr JM, Singer MV. The M-ANNHEIM classification of chronic pancreatitis: introduction of a unifying classification system based on a review of previous classifications of the disease. J Gastroenterol. 2007;42:101–119. doi: 10.1007/s00535-006-1945-4. [DOI] [PubMed] [Google Scholar]
- 4.Klöppel G, Detlefsen S, Feyerabend B. Fibrosis of the pancreas: the initial tissue damage and the resulting pattern. Virchows Arch. 2004;445:1–8. doi: 10.1007/s00428-004-1021-5. [DOI] [PubMed] [Google Scholar]
- 5.Etemad B, Whitcomb DC. Chronic pancreatitis: diagnosis, classification, and new genetic developments. Gastroenterology. 2001;120:682–707. doi: 10.1053/gast.2001.22586. [DOI] [PubMed] [Google Scholar]
- 6.Andrén-Sandberg A, Hoem D, Gislason H. Pain management in chronic pancreatitis. Eur J Gastroenterol Hepatol. 2002;14:957–970. doi: 10.1097/00042737-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44:534–541. doi: 10.1136/gut.44.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shek FW, Benyon RC, Walker FM, McCrudden PR, Pender SL, Williams EJ, Johnson PA, Johnson CD, Bateman AC, Fine DR, et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am J Pathol. 2002;160:1787–1798. doi: 10.1016/s0002-9440(10)61125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bedossa P, Paradis V. Liver extracellular matrix in health and disease. J Pathol. 2003;200:504–515. doi: 10.1002/path.1397. [DOI] [PubMed] [Google Scholar]
- 10.Andoh A, Takaya H, Saotome T, Shimada M, Hata K, Araki Y, Nakamura F, Shintani Y, Fujiyama Y, Bamba T. Cytokine regulation of chemokine (IL-8, MCP-1, and RANTES) gene expression in human pancreatic periacinar myofibroblasts. Gastroenterology. 2000;119:211–219. doi: 10.1053/gast.2000.8538. [DOI] [PubMed] [Google Scholar]
- 11.Ebert M, Kasper HU, Hernberg S, Friess H, Büchler MW, Roessner A, Korc M, Malfertheiner P. Overexpression of platelet-derived growth factor (PDGF) B chain and type beta PDGF receptor in human chronic pancreatitis. Dig Dis Sci. 1998;43:567–574. doi: 10.1023/a:1018867209170. [DOI] [PubMed] [Google Scholar]
- 12.Luttenberger T, Schmid-Kotsas A, Menke A, Siech M, Beger H, Adler G, Grünert A, Bachem MG. Platelet-derived growth factors stimulate proliferation and extracellular matrix synthesis of pancreatic stellate cells: implications in pathogenesis of pancreas fibrosis. Lab Invest. 2000;80:47–55. doi: 10.1038/labinvest.3780007. [DOI] [PubMed] [Google Scholar]
- 13.Saurer L, Reber P, Schaffner T, Büchler MW, Buri C, Kappeler A, Walz A, Friess H, Mueller C. Differential expression of chemokines in normal pancreas and in chronic pancreatitis. Gastroenterology. 2000;118:356–367. doi: 10.1016/s0016-5085(00)70218-6. [DOI] [PubMed] [Google Scholar]
- 14.Haber PS, Keogh GW, Apte MV, Moran CS, Stewart NL, Crawford DH, Pirola RC, McCaughan GW, Ramm GA, Wilson JS. Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am J Pathol. 1999;155:1087–1095. doi: 10.1016/S0002-9440(10)65211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedreich N. Cyclopoedia of the Practice of Medicine. New York: William Wood; 1878. Disease of the pancreas. [Google Scholar]
- 16.Vonlaufen A, Wilson JS, Pirola RC, Apte MV. Role of alcohol metabolism in chronic pancreatitis. Alcohol Res Health. 2007;30:48–54. [PMC free article] [PubMed] [Google Scholar]
- 17.Dufour MC, Adamson MD. The epidemiology of alcohol-induced pancreatitis. Pancreas. 2003;27:286–290. doi: 10.1097/00006676-200311000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Lankisch MR, Imoto M, Layer P, DiMagno EP. The effect of small amounts of alcohol on the clinical course of chronic pancreatitis. Mayo Clin Proc. 2001;76:242–251. doi: 10.4065/76.3.242. [DOI] [PubMed] [Google Scholar]
- 19.Frulloni L, Gabbrielli A, Pezzilli R, Zerbi A, Cavestro GM, Marotta F, Falconi M, Gaia E, Uomo G, Maringhini A, et al. Chronic pancreatitis: report from a multicenter Italian survey (PanCroInfAISP) on 893 patients. Dig Liver Dis. 2009;41:311–317. doi: 10.1016/j.dld.2008.07.316. [DOI] [PubMed] [Google Scholar]
- 20.Yadav D, Whitcomb DC. The role of alcohol and smoking in pancreatitis. Nat Rev Gastroenterol Hepatol. 2010;7:131–145. doi: 10.1038/nrgastro.2010.6. [DOI] [PubMed] [Google Scholar]
- 21.Herreros-Villanueva M, Hijona E, Bañales JM, Cosme A, Bujanda L. Alcohol consumption on pancreatic diseases. World J Gastroenterol. 2013;19:638–647. doi: 10.3748/wjg.v19.i5.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandol SJ, Gukovsky I, Satoh A, Lugea A, Gukovskaya AS. Emerging concepts for the mechanism of alcoholic pancreatitis from experimental models. J Gastroenterol. 2003;38:623–628. doi: 10.1007/s00535-003-1134-7. [DOI] [PubMed] [Google Scholar]
- 23.Keim V. [Chronic pancreatitis--pancreas cancer: influence of genetic factors] Praxis (Bern 1994) 2005;94:811–817. doi: 10.1024/0369-8394.94.20.811. [DOI] [PubMed] [Google Scholar]
- 24.Whitcomb DC, LaRusch J, Krasinskas AM, Klei L, Smith JP, Brand RE, Neoptolemos JP, Lerch MM, Tector M, Sandhu BS, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet. 2012;44:1349–1354. doi: 10.1038/ng.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavallini G, Talamini G, Vaona B, Bovo P, Filippini M, Rigo L, Angelini G, Vantini I, Riela A, Frulloni L. Effect of alcohol and smoking on pancreatic lithogenesis in the course of chronic pancreatitis. Pancreas. 1994;9:42–46. doi: 10.1097/00006676-199401000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Imoto M, DiMagno EP. Cigarette smoking increases the risk of pancreatic calcification in late-onset but not early-onset idiopathic chronic pancreatitis. Pancreas. 2000;21:115–119. doi: 10.1097/00006676-200008000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Maisonneuve P, Lowenfels AB, Müllhaupt B, Cavallini G, Lankisch PG, Andersen JR, Dimagno EP, Andrén-Sandberg A, Domellöf L, Frulloni L, et al. Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut. 2005;54:510–514. doi: 10.1136/gut.2004.039263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rebours V, Vullierme MP, Hentic O, Maire F, Hammel P, Ruszniewski P, Lévy P. Smoking and the course of recurrent acute and chronic alcoholic pancreatitis: a dose-dependent relationship. Pancreas. 2012;41:1219–1224. doi: 10.1097/MPA.0b013e31825de97d. [DOI] [PubMed] [Google Scholar]
- 29.Lévy P, Mathurin P, Roqueplo A, Rueff B, Bernades P. A multidimensional case-control study of dietary, alcohol, and tobacco habits in alcoholic men with chronic pancreatitis. Pancreas. 1995;10:231–238. doi: 10.1097/00006676-199504000-00003. [DOI] [PubMed] [Google Scholar]
- 30.Grendell JH. Nutrition and absorption in diseases of the pancreas. Clin Gastroenterol. 1983;12:551–562. [PubMed] [Google Scholar]
- 31.Uscanga L, Robles-Díaz G, Sarles H. Nutritional data and etiology of chronic pancreatitis in Mexico. Dig Dis Sci. 1985;30:110–113. doi: 10.1007/BF01308194. [DOI] [PubMed] [Google Scholar]
- 32.Zuidema PJ. Cirrhosis and disseminated calcification of the pancreas in patients with malnutrition. Trop Geogr Med. 1959;11:70–74. [PubMed] [Google Scholar]
- 33.Barman KK, Premalatha G, Mohan V. Tropical chronic pancreatitis. Postgrad Med J. 2003;79:606–615. doi: 10.1136/pmj.79.937.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathangi DC, Deepa R, Mohan V, Govindarajan M, Namasivayam A. Long-term ingestion of cassava (tapioca) does not produce diabetes or pancreatitis in the rat model. Int J Pancreatol. 2000;27:203–208. doi: 10.1385/IJGC:27:3:203. [DOI] [PubMed] [Google Scholar]
- 35.Balakrishnan V, Hariharan M, Sindhu S. Pathogenesis of pancreatitis in India leading on to diabetes mellitus. Proc Nutr Soc India. 1990;36:1–7. [Google Scholar]
- 36.Chandak GR, Idris MM, Reddy DN, Bhaskar S, Sriram PV, Singh L. Mutations in the pancreatic secretory trypsin inhibitor gene (PSTI/SPINK1) rather than the cationic trypsinogen gene (PRSS1) are significantly associated with tropical calcific pancreatitis. J Med Genet. 2002;39:347–351. doi: 10.1136/jmg.39.5.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teuscher T, Baillod P, Rosman JB, Teuscher A. Absence of diabetes in a rural West African population with a high carbohydrate/cassava diet. Lancet. 1987;1:765–768. doi: 10.1016/s0140-6736(87)92797-8. [DOI] [PubMed] [Google Scholar]
- 38.Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, Martin SP, Gates LK, Amann ST, Toskes PP, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141–145. doi: 10.1038/ng1096-141. [DOI] [PubMed] [Google Scholar]
- 39.Comfort MW, Steinberg AG. Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology. 1952;21:54–63. [PubMed] [Google Scholar]
- 40.Whitcomb DC, Preston RA, Aston CE, Sossenheimer MJ, Barua PS, Zhang Y, Wong-Chong A, White GJ, Wood PG, Gates LK, et al. A gene for hereditary pancreatitis maps to chromosome 7q35. Gastroenterology. 1996;110:1975–1980. doi: 10.1053/gast.1996.v110.pm8964426. [DOI] [PubMed] [Google Scholar]
- 41.Gorry MC, Gabbaizedeh D, Furey W, Gates LK, Preston RA, Aston CE, Zhang Y, Ulrich C, Ehrlich GD, Whitcomb DC. Mutations in the cationic trypsinogen gene are associated with recurrent acute and chronic pancreatitis. Gastroenterology. 1997;113:1063–1068. doi: 10.1053/gast.1997.v113.pm9322498. [DOI] [PubMed] [Google Scholar]
- 42.Férec C, Raguénès O, Salomon R, Roche C, Bernard JP, Guillot M, Quéré I, Faure C, Mercier B, Audrézet MP, et al. Mutations in the cationic trypsinogen gene and evidence for genetic heterogeneity in hereditary pancreatitis. J Med Genet. 1999;36:228–232. [PMC free article] [PubMed] [Google Scholar]
- 43.Witt H, Luck W, Becker M. A signal peptide cleavage site mutation in the cationic trypsinogen gene is strongly associated with chronic pancreatitis. Gastroenterology. 1999;117:7–10. doi: 10.1016/s0016-5085(99)70543-3. [DOI] [PubMed] [Google Scholar]
- 44.Pfützer R, Myers E, Applebaum-Shapiro S, Finch R, Ellis I, Neoptolemos J, Kant JA, Whitcomb DC. Novel cationic trypsinogen (PRSS1) N29T and R122C mutations cause autosomal dominant hereditary pancreatitis. Gut. 2002;50:271–272. doi: 10.1136/gut.50.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simon P, Weiss FU, Sahin-Toth M, Parry M, Nayler O, Lenfers B, Schnekenburger J, Mayerle J, Domschke W, Lerch MM. Hereditary pancreatitis caused by a novel PRSS1 mutation (Arg-122 --& gt; Cys) that alters autoactivation and autodegradation of cationic trypsinogen. J Biol Chem. 2002;277:5404–5410. doi: 10.1074/jbc.M108073200. [DOI] [PubMed] [Google Scholar]
- 46.Rebours V, Lévy P, Ruszniewski P. An overview of hereditary pancreatitis. Dig Liver Dis. 2012;44:8–15. doi: 10.1016/j.dld.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 47.Joergensen MT, Brusgaard K, Crüger DG, Gerdes AM, Schaffalitzky de Muckadell OB. Genetic, epidemiological, and clinical aspects of hereditary pancreatitis: a population-based cohort study in Denmark. Am J Gastroenterol. 2010;105:1876–1883. doi: 10.1038/ajg.2010.193. [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto T, Nakamura Y, Nishide J, Emi M, Ogawa M, Mori T, Matsubara K. Molecular cloning and nucleotide sequence of human pancreatic secretory trypsin inhibitor (PSTI) cDNA. Biochem Biophys Res Commun. 1985;132:605–612. doi: 10.1016/0006-291x(85)91176-3. [DOI] [PubMed] [Google Scholar]
- 49.Rinderknecht H, Renner IG, Abramson SB, Carmack C. Mesotrypsin: a new inhibitor-resistant protease from a zymogen in human pancreatic tissue and fluid. Gastroenterology. 1984;86:681–692. [PubMed] [Google Scholar]
- 50.Rinderknecht H, Adham NF, Renner IG, Carmack C. A possible zymogen self-destruct mechanism preventing pancreatic autodigestion. Int J Pancreatol. 1988;3:33–44. doi: 10.1007/BF02788221. [DOI] [PubMed] [Google Scholar]
- 51.Whitcomb DC. Hereditary diseases of the pancreas. In: Yamada T, Alpers DH, Kaplowitz N, Laine L, Owyang C, et al., editors. Textbook of Gastroenterology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2003. pp. 2147–2165. [Google Scholar]
- 52.Pfützer RH, Barmada MM, Brunskill AP, Finch R, Hart PS, Neoptolemos J, Furey WF, Whitcomb DC. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology. 2000;119:615–623. doi: 10.1053/gast.2000.18017. [DOI] [PubMed] [Google Scholar]
- 53.Chen JM, Férec C. Chronic pancreatitis: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2009;10:63–87. doi: 10.1146/annurev-genom-082908-150009. [DOI] [PubMed] [Google Scholar]
- 54.Naruse S, Kitagawa M, Ishiguro H. Molecular understanding of chronic pancreatitis: a perspective on the future. Mol Med Today. 1999;5:493–499. doi: 10.1016/s1357-4310(99)01595-6. [DOI] [PubMed] [Google Scholar]
- 55.Cohn JA. Reduced CFTR function and the pathobiology of idiopathic pancreatitis. J Clin Gastroenterol. 2005;39:S70–S77. doi: 10.1097/01.mcg.0000155522.89005.bf. [DOI] [PubMed] [Google Scholar]
- 56.Midha S, Khajuria R, Shastri S, Kabra M, Garg PK. Idiopathic chronic pancreatitis in India: phenotypic characterisation and strong genetic susceptibility due to SPINK1 and CFTR gene mutations. Gut. 2010;59:800–807. doi: 10.1136/gut.2009.191239. [DOI] [PubMed] [Google Scholar]
- 57.Onodera M, Okazaki K, Morita M, Nishimori I, Yamamoto Y. Immune complex specific for the pancreatic duct antigen in patients with idiopathic chronic pancreatitis and Sjögren syndrome. Autoimmunity. 1994;19:23–29. doi: 10.3109/08916939409008005. [DOI] [PubMed] [Google Scholar]
- 58.Nishimori I, Yamamoto Y, Okazaki K, Morita M, Onodera M, Kino J, Tamura S, Yamamoto Y. Identification of autoantibodies to a pancreatic antigen in patients with idiopathic chronic pancreatitis and Sjögren’s syndrome. Pancreas. 1994;9:374–381. doi: 10.1097/00006676-199405000-00015. [DOI] [PubMed] [Google Scholar]
- 59.Sharer N, Schwarz M, Malone G, Howarth A, Painter J, Super M, Braganza J. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339:645–652. doi: 10.1056/NEJM199809033391001. [DOI] [PubMed] [Google Scholar]
- 60.Atlas AB, Orenstein SR, Orenstein DM. Pancreatitis in young children with cystic fibrosis. J Pediatr. 1992;120:756–759. doi: 10.1016/s0022-3476(05)80242-2. [DOI] [PubMed] [Google Scholar]
- 61.Shwachman H, Lebenthal E, Khaw KT. Recurrent acute pancreatitis in patients with cystic fibrosis with normal pancreatic enzymes. Pediatrics. 1975;55:86–95. [PubMed] [Google Scholar]
- 62.Borowitz D, Baker SS, Duffy L, Baker RD, Fitzpatrick L, Gyamfi J, Jarembek K. Use of fecal elastase-1 to classify pancreatic status in patients with cystic fibrosis. J Pediatr. 2004;145:322–326. doi: 10.1016/j.jpeds.2004.04.049. [DOI] [PubMed] [Google Scholar]
- 63.Layer P, Yamamoto H, Kalthoff L, Clain JE, Bakken LJ, DiMagno EP. The different courses of early- and late-onset idiopathic and alcoholic chronic pancreatitis. Gastroenterology. 1994;107:1481–1487. doi: 10.1016/0016-5085(94)90553-3. [DOI] [PubMed] [Google Scholar]
- 64.Tas A, Köklü S, Kocak E, Akbal E, Ergul B. An unusual cause of acute pancreatitis: annular pancreas and papillary opening of the cystic duct. Gut Liver. 2012;6:403–404. doi: 10.5009/gnl.2012.6.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frulloni L, Lunardi C, Simone R, Dolcino M, Scattolini C, Falconi M, Benini L, Vantini I, Corrocher R, Puccetti A. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med. 2009;361:2135–2142. doi: 10.1056/NEJMoa0903068. [DOI] [PubMed] [Google Scholar]
- 66.Löhr JM, Faissner R, Koczan D, Bewerunge P, Bassi C, Brors B, Eils R, Frulloni L, Funk A, Halangk W, et al. Autoantibodies against the exocrine pancreas in autoimmune pancreatitis: gene and protein expression profiling and immunoassays identify pancreatic enzymes as a major target of the inflammatory process. Am J Gastroenterol. 2010;105:2060–2071. doi: 10.1038/ajg.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Detlefsen S, Bräsen JH, Zamboni G, Capelli P, Klöppel G. Deposition of complement C3c, immunoglobulin (Ig)G4 and IgG at the basement membrane of pancreatic ducts and acini in autoimmune pancreatitis. Histopathology. 2010;57:825–835. doi: 10.1111/j.1365-2559.2010.03717.x. [DOI] [PubMed] [Google Scholar]
- 68.Sah RP, Chari ST, Pannala R, Sugumar A, Clain JE, Levy MJ, Pearson RK, Smyrk TC, Petersen BT, Topazian MD, et al. Differences in clinical profile and relapse rate of type 1 versus type 2 autoimmune pancreatitis. Gastroenterology. 2010;139:140–18; quiz 140-18;. doi: 10.1053/j.gastro.2010.03.054. [DOI] [PubMed] [Google Scholar]
- 69.Chari ST, Longnecker DS, Klöppel G. The diagnosis of autoimmune pancreatitis: a Western perspective. Pancreas. 2009;38:846–848. doi: 10.1097/MPA.0b013e3181bba281. [DOI] [PubMed] [Google Scholar]
- 70.Kamisawa T, Okamoto A. Autoimmune pancreatitis: proposal of IgG4-related sclerosing disease. J Gastroenterol. 2006;41:613–625. doi: 10.1007/s00535-006-1862-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gardner TB, Chari ST. Autoimmune pancreatitis. Gastroenterol Clin North Am. 2008;37:439–60, vii. doi: 10.1016/j.gtc.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 72.Kamisawa T, Anjiki H, Egawa N. Rapid changes in sclerosing cholangitis associated with autoimmune pancreatitis. Pancreas. 2009;38:601–602. doi: 10.1097/MPA.0b013e31819e0cdb. [DOI] [PubMed] [Google Scholar]
- 73.Cheuk W, Chan AC, Lam WL, Chow SM, Crowley P, Lloydd R, Campbell I, Thorburn M, Chan JK. IgG4-related sclerosing mastitis: description of a new member of the IgG4-related sclerosing diseases. Am J Surg Pathol. 2009;33:1058–1064. doi: 10.1097/PAS.0b013e3181998cbe. [DOI] [PubMed] [Google Scholar]
- 74.Kasashima S, Zen Y, Kawashima A, Endo M, Matsumoto Y, Kasashima F. A new clinicopathological entity of IgG4-related inflammatory abdominal aortic aneurysm. J Vasc Surg. 2009;49:1264–171; discussion 1271. doi: 10.1016/j.jvs.2008.11.072. [DOI] [PubMed] [Google Scholar]
- 75.Sakata N, Tashiro T, Uesugi N, Kawara T, Furuya K, Hirata Y, Iwasaki H, Kojima M. IgG4-positive plasma cells in inflammatory abdominal aortic aneurysm: the possibility of an aortic manifestation of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32:553–559. doi: 10.1097/PAS.0b013e31815a04db. [DOI] [PubMed] [Google Scholar]
- 76.Uehara T, Hamano H, Kawakami M, Koyama M, Kawa S, Sano K, Honda T, Oki K, Ota H. Autoimmune pancreatitis-associated prostatitis: distinct clinicopathological entity. Pathol Int. 2008;58:118–125. doi: 10.1111/j.1440-1827.2007.02199.x. [DOI] [PubMed] [Google Scholar]
- 77.Zamboni G, Lüttges J, Capelli P, Frulloni L, Cavallini G, Pederzoli P, Leins A, Longnecker D, Klöppel G. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch. 2004;445:552–563. doi: 10.1007/s00428-004-1140-z. [DOI] [PubMed] [Google Scholar]
- 78.Klöppel G, Detlefsen S, Chari ST, Longnecker DS, Zamboni G. Autoimmune pancreatitis: the clinicopathological characteristics of the subtype with granulocytic epithelial lesions. J Gastroenterol. 2010;45:787–793. doi: 10.1007/s00535-010-0265-x. [DOI] [PubMed] [Google Scholar]
- 79.Detlefsen S, Sipos B, Zhao J, Drewes AM, Klöppel G. Autoimmune pancreatitis: expression and cellular source of profibrotic cytokines and their receptors. Am J Surg Pathol. 2008;32:986–995. doi: 10.1097/PAS.0b013e31815d2583. [DOI] [PubMed] [Google Scholar]
- 80.Hassan Z, Mohan V, Ali L, Allotey R, Barakat K, Faruque MO, Deepa R, McDermott MF, Jackson AE, Cassell P, et al. SPINK1 is a susceptibility gene for fibrocalculous pancreatic diabetes in subjects from the Indian subcontinent. Am J Hum Genet. 2002;71:964–968. doi: 10.1086/342731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bhatia E, Choudhuri G, Sikora SS, Landt O, Kage A, Becker M, Witt H. Tropical calcific pancreatitis: strong association with SPINK1 trypsin inhibitor mutations. Gastroenterology. 2002;123:1020–1025. doi: 10.1053/gast.2002.36028. [DOI] [PubMed] [Google Scholar]
- 82.Schneider A, Suman A, Rossi L, Barmada MM, Beglinger C, Parvin S, Sattar S, Ali L, Khan AK, Gyr N, et al. SPINK1/PSTI mutations are associated with tropical pancreatitis and type II diabetes mellitus in Bangladesh. Gastroenterology. 2002;123:1026–1030. doi: 10.1053/gast.2002.36059. [DOI] [PubMed] [Google Scholar]
- 83.Vonlaufen A, Wilson JS, Apte MV. Molecular mechanisms of pancreatitis: current opinion. J Gastroenterol Hepatol. 2008;23:1339–1348. doi: 10.1111/j.1440-1746.2008.05520.x. [DOI] [PubMed] [Google Scholar]
- 84.Nakamura Y, Kobayashi Y, Ishikawa A, Maruyama K, Higuchi S. Severe chronic pancreatitis and severe liver cirrhosis have different frequencies and are independent risk factors in male Japanese alcoholics. J Gastroenterol. 2004;39:879–887. doi: 10.1007/s00535-004-1405-y. [DOI] [PubMed] [Google Scholar]
- 85.Wilson JS, Bernstein L, McDonald C, Tait A, McNeil D, Pirola RC. Diet and drinking habits in relation to the development of alcoholic pancreatitis. Gut. 1985;26:882–887. doi: 10.1136/gut.26.9.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lowenfels AB, Zwemer FL, Jhangiani S, Pitchumoni CS. Pancreatitis in a native American Indian population. Pancreas. 1987;2:694–697. doi: 10.1097/00006676-198711000-00012. [DOI] [PubMed] [Google Scholar]
- 87.Haber PS, Wilson JS, Pirola RC. Smoking and alcoholic pancreatitis. Pancreas. 1993;8:568–572. doi: 10.1097/00006676-199309000-00007. [DOI] [PubMed] [Google Scholar]
- 88.Rosendahl J, Witt H, Szmola R, Bhatia E, Ozsvári B, Landt O, Schulz HU, Gress TM, Pfützer R, Löhr M, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40:78–82. doi: 10.1038/ng.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miyasaka K, Ohta M, Takano S, Hayashi H, Higuchi S, Maruyama K, Tando Y, Nakamura T, Takata Y, Funakoshi A. Carboxylester lipase gene polymorphism as a risk of alcohol-induced pancreatitis. Pancreas. 2005;30:e87–e91. doi: 10.1097/01.mpa.0000160960.21580.ml. [DOI] [PubMed] [Google Scholar]
- 90.Ockenga J, Vogel A, Teich N, Keim V, Manns MP, Strassburg CP. UDP glucuronosyltransferase (UGT1A7) gene polymorphisms increase the risk of chronic pancreatitis and pancreatic cancer. Gastroenterology. 2003;124:1802–1808. doi: 10.1016/s0016-5085(03)00294-4. [DOI] [PubMed] [Google Scholar]
- 91.Witt H, Luck W, Becker M, Böhmig M, Kage A, Truninger K, Ammann RW, O’Reilly D, Kingsnorth A, Schulz HU, et al. Mutation in the SPINK1 trypsin inhibitor gene, alcohol use, and chronic pancreatitis. JAMA. 2001;285:2716–2717. doi: 10.1001/jama.285.21.2716-a. [DOI] [PubMed] [Google Scholar]
- 92.Schneider A, Barmada MM, Slivka A, Martin JA, Whitcomb DC. Transforming growth factor-beta1, interleukin-10 and interferon-gamma cytokine polymorphisms in patients with hereditary, familial and sporadic chronic pancreatitis. Pancreatology. 2004;4:490–494. doi: 10.1159/000080245. [DOI] [PubMed] [Google Scholar]
- 93.Perri F, Piepoli A, Stanziale P, Merla A, Zelante L, Andriulli A. Mutation analysis of the cystic fibrosis transmembrane conductance regulator (CFTR) gene, the cationic trypsinogen (PRSS1) gene, and the serine protease inhibitor, Kazal type 1 (SPINK1) gene in patients with alcoholic chronic pancreatitis. Eur J Hum Genet. 2003;11:687–692. doi: 10.1038/sj.ejhg.5201035. [DOI] [PubMed] [Google Scholar]
- 94.Frenzer A, Butler WJ, Norton ID, Wilson JS, Apte MV, Pirola RC, Ryan P, Roberts-Thomson IC. Polymorphism in alcohol-metabolizing enzymes, glutathione S-transferases and apolipoprotein E and susceptibility to alcohol-induced cirrhosis and chronic pancreatitis. J Gastroenterol Hepatol. 2002;17:177–182. doi: 10.1046/j.1440-1746.2002.02670.x. [DOI] [PubMed] [Google Scholar]
- 95.Norton ID, Apte MV, Dixson H, Trent RJ, Haber PS, Pirola RC, Wilson JS. Cystic fibrosis genotypes and alcoholic pancreatitis. J Gastroenterol Hepatol. 1998;13:496–499. doi: 10.1111/j.1440-1746.1998.tb00675.x. [DOI] [PubMed] [Google Scholar]
- 96.Haber PS, Wilson JS, McGarity BH, Hall W, Thomas MC, Pirola RC. Alpha 1 antitrypsin phenotypes and alcoholic pancreatitis. Gut. 1991;32:945–948. doi: 10.1136/gut.32.8.945. [DOI] [PMC free article] [PubMed] [Google Scholar]