

Abstract
Despite multiple theories on the pathogenesis of pain in chronic pancreatitis, no uniform and consistently successful treatment strategy exists and abdominal pain still remains the dominating symptom for most patients and a major challenge for clinicians. Traditional theories focussed on a mechanical cause of pain related to anatomical changes and evidence of increased ductal and interstitial pressures. These observations form the basis for surgical and endoscopic drainage procedures, but the outcome is variable and often unsatisfactory. This underscores the fact that other factors must contribute to pathogenesis of pain, and has shifted the focus towards a more complex neurobiological understanding of pain generation. Amongst other explanations for pain, experimental and human studies have provided evidence that pain perception at the peripheral level and central pain processing of the nociceptive information is altered in patients with chronic pancreatitis, and resembles that seen in neuropathic and chronic pain disorders. However, pain due to e.g., complications to the disease and adverse effects to treatment must not be overlooked as an additional source of pain. This review outlines the current theories on pain generation in chronic pancreatitis which is crucial in order to understand the complexity and limitations of current therapeutic approaches. Furthermore, it may also serve as an inspiration for further research and development of methods that can evaluate the relative contribution and interplay of different pain mechanisms in the individual patients, before they are subjected to more or less empirical treatment.
Keywords: Chronic pancreatitis, Abdominal pain, Pain mechanisms, Neuropathy, Sensitization
Core tip: Pain management in chronic pancreatitis often remains unsatisfactory. An overview of the current theories on pain generation in chronic pancreatitis is crucial in order to understand the complexity and limitations of current therapeutic approaches. Also, optimal treatment will only be achieved on the basis of a better understanding of these mechanisms. Furthermore, this review may serve as an inspiration for future research and development of methods that can determine the relative contribution of different mechanisms to the “collective” abdominal pain, before patients are subjected to more or less empirical treatment.
INTRODUCTION
Abdominal pain is the most significant symptom and a major clinical challenge in chronic pancreatitis (CP). It is present in up to 90% of the patients and the primary cause of hospitalization[1]. Pancreatic pain is characteristically described as a constant, severe, dull, epigastric pain that often radiates to the back and typically worsens after high-fat meals. However, many different pain patterns have been described, ranging from no pain to recurrent episodes of pain and pain free intervals, to constant pain with clusters of severe exacerbations[2,3]. In the often cited Zürich series, pain was reported to decrease over time, coinciding with the occurrence of exocrine insufficiency, which led to the “burn-out” hypothesis of pain in CP[3,4]. However, evidence against this hypothesis was subsequently provided in two large prospective studies[5,6], where no association between the duration of CP and the quality or frequency of pain was found. Today the “burn-out” hypothesis is regarded obsolete by most clinicians, and even though a few patients may experience spontaneously pain relief, pain in the majority of patients is unpredictable and has a substantial impact on physical and mental quality of life, employment and health care expenses[6-8].
The etiology of pain in CP is increasingly better understood and likely involves multiple mechanisms. Traditional theories focused on the role of local pathology within or in close proximity to the pancreas, as the main generator of pain, but since the late 1990’s there has been a shift towards a neurobiological understanding of pain in CP. Accordingly, there is solid histological and neurophysiological evidence for an abnormal pain processing in many of these patients[1,9]. Various mechanisms responsible for this altered pain processing have been proposed, among them sensitization of the peripheral and central nerves, reorganization of the cerebral cortex and alterations in pain control systems. In addition, local complications (such as pancreatic pseudocysts and duodenal and/or bile duct obstruction) and adverse effects to treatment also contribute to the complex symptomatology in many patients and is an often a neglected problem in the clinical settings. Also, it has been shown, that the clinical pain pattern varies with the etiology of CP, and hypothesized that different pathogenic mechanisms of pain are associated with or predominates depending on etiology[2,4], but so far it is not possible to correlate pain mechanism and clinical pain pattern to the etiology of CP uniformly.
The aim of this review is to highlight current theories regarding the pain mechanisms in CP. This is important in order to understand the complexity of this debilitating disease and why pain management is often insufficient. Also, optimal treatment will only be achieved on the basis of a better understanding of the mechanisms underlying pain in CP[10]. There is a variety of potential pain mechanisms that needs to be taken into consideration (Figure 1), but in this review we present for simplicity a schematic division of the pain mechanisms into five main categories as an easy-to-remember overview (Table 1).
Figure 1.
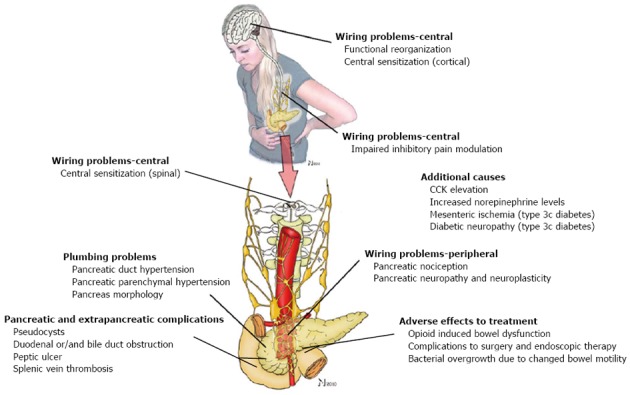
Pain mechanisms in chronic pancreatitis. The source of pain in chronic pancreatitis is complex and almost certainly multifactorial, and the relative contribution of different factors can be difficult to determine in the individual patient. See manuscript for further details. Modified from Demir et al[87]. CCK: Cholecystokinin.
Table 1.
Pain mechanisms in chronic pancreatitis
| Main category | Proposed mechanism/source | Ref. |
| Plumbing problems | Pancreatic duct hypertension | [14,16,20] |
| Pancreatic parenchymal hypertension | [21-23] | |
| Pancreas morphology | [29,30] | |
| Wiring problems | Peripheral nociception | [35-37] |
| Pancreatic neuropathy | [41,42,44] | |
| Central mechanisms of pain | [50,52,53,56] | |
| Pancreatic and extrapancreatic complications | Pseudocysts | [62,64] |
| Duodenal obstruction | [66] | |
| Bile duct obstruction | [66,67] | |
| Peptic ulcer | [71,72] | |
| Splenic vein thrombosis | [75,78] | |
| Adverse effects to treatment | Opioid induced bowel dysfunction | [79] |
| Complications to surgery and endoscopic therapy | [12] | |
| Bacterial overgrowth due to changed bowel motility | [81] | |
| Additional causes | CCK elevation | [82] |
| Increased norepinephrine levels | [85] | |
| Diabetic neuropathy (type 3c diabetes) | [86] | |
| Mesenteric ischemia1 |
Complications to and/or possible concomitant diseases. CCK: Cholecystokinin.
“PLUMBING” PROBLEMS
The mainstay of pain treatment in CP has for many years focused on the pancreatic gland based on the assumption, that pain is generated by increased pressure in the pancreatic duct or in the pancreatic parenchyma[11]. This mechanistic understanding of pain, termed the “the plumbing theory”, has been the most widely accepted theory regarding the cause of pain and it is the theoretical background of most interventions including surgical and endoscopic drainage procedures[12].
Pancreatic duct hypertension and pain
A direct relationship between pancreatic duct hypertension and pain was reported for the first time in a case report by White et al[13]. A patient undergoing open necrosectomy following acute necrotizing pancreatitis had a drainage catheter placed in a fistula tract communicating with the pancreatic duct. He reproducibly developed pain after saline infusion when the ductal pressure exceeded 25 mmHg[13]. Inspired by this finding, subsequent studies attempted to compare ductal pressure measurements in CP patients and other patient groups. Many of these studies were flawed by inappropriate comparison of intraoperative pressure measurements in CP patients to endoscopic pressure measurement in controls. Accordingly, only a few studies have compared ductal pressure using the same technique in patients and controls. Based on intraoperative measurement of pancreatic duct pressure, significantly increased pressures were evident in patients with CP undergoing a surgical pancreatic drainage procedure compared to patients undergoing surgery for gastric cancer[14]. Moreover, in a study comparing endoscopic manometry of the pancreatic duct, CP patients had evidence of ductal hypertension compared to controls[15]. Other studies based on endoscopic manometry have, however, not documented ductal hypertension in CP, and the only study comparing CP patients by the presence or absence of pain demonstrated no difference in pressure levels[16-20]. In addition, the pathological mechanism by which increased ductal pressure causes pain in CP is not clear and, taken together, the link between ductal hypertension and pain in CP remains hypothetical.
Pancreatic parenchymal hypertension and pain
Measurement of intrapancreatic pressure was pioneered by Ebbehøj et al[21], who developed a technique based on a needle probe inserted directly into the pancreatic parenchyma. Measurements were employed directly intraoperatively or by using a percutaneous procedure guided by ultrasonography[21]. In 39 patients with CP, they found that the intrapancreatic pressure was higher in patients with pain than in patients without pain, and this intrapancreatic hypertension was reversed following surgical drainage[22]. At one-year follow-up, patients with recurrent pain had a rebound of intrapancreatic hypertension, while patients who remained pain free continued to have normal intrapancreatic pressures[23]. Although intriguing, these findings were not reproduced by a subsequent study in which no relation between intrapancreatic pressure and pain was found[24].
The pathophysiological link between intrapancreatic hypertension and pain has been explained as a “compartment-like syndrome” induced by fibrosis of the pancreatic parenchyma and peripancreatic capsule[2]. During secretory stimulation in an animal model of CP, increased interstitial pressures, diminished blood flow and ensuing tissue acidosis were documented[25]. The latter was possibly secondary to ischemia, thus mimicking the pathophysiology underlying muscular compartment syndrome. Although no pain data were collected in this experiment, it is plausible that acidosis may have caused pain by activation of the transient receptor potential vanilloid-1, which is a nociceptor found to be up regulated in CP and other conditions characterized by neurogenic inflammation[26,27] (described further below). However, it must be highlighted that these findings have never been reproduced in a human study and needs confirmation.
Pancreas morphology and pain
In clinical practice, measurement of ductal or intrapancreatic pressure is not routinely performed and most decisions regarding surgery or endotherapy to relieve pain rely on morphological abnormalities of the pancreas[2,11,28]. However, the relationship between ductal or intrapancreatic pressure and pancreatic morphology is ambiguous[29]. Furthermore, in a very recent study by Frokjaer et al[30] magnetic resonance cholangiopancreatography including diffusion weighted imaging was obtained in 23 patients with painful CP and 17 controls. Compared to traditional imaging techniques (i.e., endoscopic retrograde cholangiopancreatography, magnetic resonance cholangiopancreatography, computed tomography, and endoscopic ultrasound), which mainly focus on atrophy and ductal pathology, this modality also provides a measure of fibrotic changes in the pancreatic gland[31]. Interestingly, the authors found no association between degree of pathological imaging (fibrosis, atrophy, and ductal pathology) and pain. However, pancreatic atrophy and ductal pathology were associated with diabetes (i.e., more severe atrophy in patients with high levels of glycated hemoglobin), and low levels of phosphate and hemoglobin.
Taken together, pathological pancreatic morphology does not necessarily reflect the burden of pain in CP. Nowhere is this observation made more obvious, than by the patient who undergoes total pancreatectomy for painful CP and still continues to suffer from pain[11].
“WIRING” PROBLEMS
Lesions to intrapancreatic nerves and their impact on the pathogenesis of pain in CP have been the focus of a substantial amount of research past 25 years. The International Association for the Study of Pain recently changed its definition of neuropathic pain to “pain caused by a lesion or disease of the somatosensory system”[32]. In the context of pain and CP there is emerging histological and neurophysiological evidence of such lesions to peripheral nerves in the pancreatic gland and coincident aberrant central pain processing. Three aspects of the neural basis of pain in CP are usually considered: Peripheral nociception, pancreatic neuropathy and central mechanisms of pain.
Peripheral nociception
A nociceptor is a sensory nerve cell capable of integrating and transducing nociceptive stimuli into pain signals[33]. Nociception begins with the primary afferent nerves, whose cell bodies lies in the dorsal root ganglia next to the spinal cord. They have two branches-one terminating in the target tissue and one that ends in the dorsal horn of the spinal cord. The peripheral nerve endings sense tissue injury or pain stimuli via a variety of receptors, which respond to physical or chemical factors in their surroundings[12]. Depending on the excitability of the neural membrane, the stimulus leads to an action potential, which travels to the spinal end of the nerves in the dorsal horn. Here it triggers the release of neurotransmitters, which cross the synapse and act on secondary neurons and transmit the stimulus to the brain through diverse pathways, ultimately resulting in the sensation of pain.
Currently, it is unknown what actually activates the intrapancreatic nociceptors in humans. However, increased expression of the transient receptor potential vanilloid-1 has been demonstrated in an animal model of CP[34], as well as in humans, although no correlation to pain was seen[35]. Furthermore, studies on nociceptive activation markers, i.e., neurotransmitters secreted by activated afferents, especially the large family of neurotrophic factors, are gaining increasing attention. Among others, both nerve growth factor and brain-derived neurotrophic factor have been shown to be upregulated in CP patients[36,37], as well as many other proinflammatory cytokines, and in some cases this upregulation has been associated with increased pain intensity and/or frequency (for a review see[2]).
These changes render the nociceptors more sensitive to further stimulation, so that there can be a reduction in the threshold for activation, an increase in the response to a given stimulus, or the appearance of spontaneous activity[38,39]. This sensitization, called peripheral sensitization, results in an increased barrage of pain signals to the spinal cord[40].
Taken together, several articles have demonstrated upregulation signaling molecules involved in inflammation and pronociceptive mediators, but also neurotrophic factors in CP. The trophic nature of the nociceptive activation markers further suggests a link between this altered nociception and the neuropathy of CP described below.
Pancreatic neuropathy
Increased neural density and hypertrophy, sprouting and neuritis of the intrapancreatic nerves, as well as activation of glia and immune cells have also been reported in pancreatic tissue from CP patients[41-43]. These changes, collectively known as neuropathy, have been strongly associated with clinical pain scores, and thus suggests to be an important factor in the pathogenesis of pain in CP[41,42,44]. Autonomic innervation, especially sympathetic, is also decreased[43], and has been correlated with abdominal pain.
Overall, substantial evidence supports the notion that pancreatic neuropathy leads to a remodeling of the intra-pancreatic innervation, a concept known as pancreatic neuroplasticity, which is likely an important factor in the pathogenesis of pain in CP.
Central mechanisms of pain
A sustained and increased peripheral nociceptive drive may result in an increased responsiveness of central pain transmitting neurons. This phenomenon is known as central sensitization, and refers to an increased synaptic efficiency established in sensory neurons in the dorsal horn of the spinal cord (and/or at supraspinal sites), following intense peripheral noxious stimuli, tissue injury, or nerve damage[45,46]. The end result is pain, which is no longer coupled to the presence, intensity, or duration of noxious peripheral stimuli[47]. Clinically these changes manifests as allodynia (a painful response to stimuli not normally painful), hyperalgesia (increased sensitivity to painful stimuli) and secondary hyperalgesia (a receptive field expansion that enables input from non-injured tissue to produce pain)[45].
Several studies have reported findings compatible with central sensitization in CP. Patterns of referred pain can be examined to assess central sensitization, as viscera-somatic convergence between peripheral nerves occurs at the spinal cord and higher levels of the central nervous system. Hence, visceral sensitivity of the upper gastrointestinal organs (sharing spinal innervation with the pancreatic gland) serves as a proxy of spinal sensitization[48,49]. In one study, increased areas of referred pain to electrical stimulation of the esophagus, stomach, and duodenum was reported in CP patients compared to controls[50]. Other studies reported decreased pain thresholds to visceral stimulation of the rectosigmoid as well as somatic stimulation of muscle and bone[51,52]. Taken together, these findings characterize a generalized hyperalgesic state of the pain system and likely mirrors widespread sensitization of the central nervous system as seen in many other chronic pain disorders[45].
From the spinal cord, visceral afferents are projected to cortical and subcortical structures of the brain. Experimental pain studies, based on somatic stimulation of the epigastric skin area (sharing spinal segmental innervation with the pancreatic gland) as well as visceral stimulation of the upper and lower gut with concomitant recording of evoked brain potentials and brain source localization, have indicated that chronic pain and hyperalgesia is associated with functional reorganization of the cerebral cortex[50,53-55]. Hence, compared to healthy controls CP patients show reorganization of the brain areas involved in visceral pain processing including the insula, secondary somatosensory cortex and cingulate cortex similar to what is seen in phantom pain[53]. Interestingly, insular reorganization was associated with clinical pain intensity. In addition to reorganization of the brain areas involved in visceral pain processing, the excitability of these neural networks is abnormal with evidence of impaired habituation to noxious stimuli, possibly reflecting a neuronal hyperexcitability (i.e., cortical sensitization)[55].
The structural correlate of functional cortical reorganization and hyperexcitability is found in studies based on advanced magnetic resonance imaging (MRI). In one study using diffusion weighted MRI, microstructural changes in the insular and frontal brain areas was associated with clinical pain intensity and functional scores[56]. Patients with a constant pain pattern demonstrated the most severe microstructural abnormalities compared to patients with an attack-wise pain pattern. This translates well to the clinic where patients with constant pain was recently reported to have the most reduced quality of life and functioning[6]. In another MRI study based on cortical volumetry, brain areas involved in visceral pain processing was shown to have a reduced thickness[57]. This finding suggests a central neurodegenerative response to severe and sustained pain.
The pain system has several inherent mechanisms whereby inflowing pain signals can be modulated. In particular, modulation of spinal pain transmission from the brainstem and higher cortical areas has been subject to increased interest during the last decades. Descending pain modulation where the brain can exert a downstream gating of the incoming spinal activity can lead to either an increase in the spinal transmission of pain impulses (facilitation) or a decrease in transmission (inhibition). The balance between these states ultimately determines the quality and strength of the pain signals perceived by the brain. Alterations in the state of descending modulation from inhibition towards facilitation have been implicated in the transition of acute into chronic and neuropathic pain. In the context of pain and CP, impaired descending inhibitory pain modulation has been reported in studies based on experimental human pain models[52,58]. In addition, brainstem facilitation has been reported to maintain pancreatic pain in an animal model of CP[59].
Taken together, several lines of evidence indicate that central pain processing is abnormal in CP. However, it is difficult to determine whether these central abnormalities are maintained by a sustained nociceptive drive from the pancreatic gland or whether they have become independent of peripheral input. In favor of the latter, a recent pilot project documented generalized hyperalgesia (a clinical measurable proxy of central sensitization) to be associated with failure of thoracoscopic splanchnic denervation[60]. The authors concluded that in patients with hyperalgesia and failure to denervation, the disease has advanced and the generation of pain becomes self-perpetuating and independent of the initial peripheral nociceptive drive. However, these findings need confirmation in larger and longitudinal studies.
PANCREATIC AND EXTRAPANCREATIC COMPLICATIONS
Although questions still remain, “plumbing” and “wiring” problems are probably important generators of pain in CP. Nevertheless, pain due to complications to the disease is also likely to contribute, and should not be overlooked, when evaluating the origin of abdominal pain in CP patients.
Pseudocysts
Pancreatic pseudocysts are relatively common in CP with estimated incidence rates of 20%-40%[61,62], however, there is a lack of precise data based on long term follow-up. Yet, due to the chronic nature of the disease, CP patients are at high risk of developing pseudocyst in the course of their disease[63]. The exact pathogenesis of pseudocyst formation in CP is not known, but it has been proposed, that blockage of the major branch of the main pancreatic duct, and ongoing pancreatic secretion proximal to the obstruction leads to a saccular dilatation of the duct. This is then filled with pancreatic juice resulting in a pseudocyst[62]. The range of symptoms is wide-from asymptomatic to severe abdominal pain dependent on etiology, localization, and size[64,65].
Duodenal and bile duct obstruction
Advanced CP is characterized by replacement of the pancreatic parenchyma with fibrous tissue. Due to the close anatomical relationship of the common bile duct and the second part of the duodenum with the head of the pancreas, fibrosis can exert extrinsic pressure on these structures. The end result is mechanical obstruction of the common bile duct and duodenum[66,67].
The clinical presentation of duodenal and bile duct obstruction is variable and can be asymptomatic[66,68]. Postprandial abdominal pain, early satiety and nausea are the most common symptoms of duodenal obstruction, while pain and abnormal liver function tests are suggestive of bile duct stenosis and cholangitis[69]. The mechanisms underlying such “obstructive pain” are unclear and one study concluded that bile duct obstruction without cholangitis is not the cause of pain in patients with CP[70].
Peptic ulcer
Upper abdominal pain is a prominent symptom in patients with peptic ulcer. Previous studies have demonstrated, that the prevalence of duodenal ulcer is high in patients with CP (ranges from 3.6% to 37.5%)[5,71,72]. The reason for the higher prevalence is unclear and many hypotheses have been suggested, including higher prevalence of Helicobacter pylori infection[71], increased gastric acid secretion[73,74], and insufficient pancreatic exocrine function which reduces bicarbonate secretion and decreases duodenal pH[72]. Furthermore, changes in gastric and intestinal blood flow due to previous acute pancreatitis may also be of importance.
As peptic ulcer can be asymptomatic, the high prevalence may be a result of a “detection bias” due to intensive search for the “origin” of epigastric pain in a selected group of patients. Patients with CP are more likely to undergo an upper gastrointestinal endoscopy, and therefore ulcers with atypical or no symptoms may be included, causing a higher prevalence than in the background population[72].
Under all circumstances, peptic ulcer may contribute to or be the reason for abdominal pain in CP patients and therefore diagnostic upper gastrointestinal endoscopy should be considered on wide indications.
Splenic vein thrombosis
The splenic vein runs along the posterior surface of the pancreas and may be affected by inflammation in the pancreatic gland, leading to thrombosis and abdominal pain. Because most patients are asymptomatic the incidence of splenic vein thrombosis in CP is not known[75], but incidence from 4%[76] to 45%[77] have been reported. Only few studies address the issue, but in one of the largest studies on splenic vein thrombosis in CP, 266 patients were investigated prospectively for a mean time of 8.2 years[78]. Splenic vein thrombosis was found in 22 patients (8.3%), but was only symptomatic in two patients. Hence, the role of splenic vein thrombosis in the pathogenesis of pain in CP is still questionable.
ADVERSE EFFECTS TO TREATMENT
In many CP patients, strong opioids are often necessary to relieve pain. Unfortunately, opioids have the potential to produce substantial gastrointestinal adverse effects, including constipation, reflux, nausea and abdominal pain-a phenomenon known as opioid-induced bowel dysfunction[79]. In a study on the prevalence of gastrointestinal symptoms in patients treated with opioids for non-cancerous diseases, chronic abdominal pain was reported in 58% of the patients[80]. Hence, opioid consumption may confuse the clinical picture in CP and worsen or contribute significantly to abdominal pain. Medications that change bowel motility may also contribute to the bacterial overgrowth that is reported in up to 40% of the patients[81]. This may manifest as abdominal distension and pain.
In addition to side effects to medical therapy, complications to surgical end endoscopic therapy may cause pain in a number of patients. These may include postoperative adhesions, ductal and parenchymal trauma, pancreatic and bile duct strictures, and complications to pancreatic stenting-all of which may result in abdominal pain. However, the relative contribution to abdominal pain due to surgical complications in CP is difficult to assess, and as far as the authors are aware of, no studies have examined this.
ADDITIONAL CAUSES FOR PAIN
The above mentioned theories and mechanisms of pain are considered the most common and well-founded by the authors, but other rarer, less well examined causes, and complications to concomitant and comorbid diseases may also play in role in pain generation. A thorough evaluation of all these plausible causes is beyond the scope of this review, but the reader is referred Table 1 for an overview.
However, elevated cholecystokinin (CCK) levels and increased sympathetic activity deserves to be mentioned separately as an additional cause of pain in CP. It has been shown, that CCK levels are elevated threefold in some early pancreatitis patients compared to controls[82]. This may generate pain by increasing the pressure in the pancreatic duct, but also through direct activation of nociceptive pathways in the central nervous system[83]. In one placebo-controlled, multicentre trial including 207 patients with chronic pancreatitis, an oral CCK receptor antagonist significantly decreased pain over placebo, although the placebo response was 30%[84]. However, further research into CCK antagonists has been hampered by concerns about induction of exocrine insufficiency and gallstones[11].
Although limited by a small number of patients, Buscher et al[85] showed significantly lower pain tolerance thresholds to pressure pain in dermatome T10 (pancreatic dermatome) in CP patients with increased norepinephrine levels compared to CP patients with normal norepinephrine levels, suggesting that sympathetic activity may play a role in these patients pain processing.
CONCLUSION
Intense abdominal pain is a dominant feature of CP and it is associated with poor mental and physical quality of life. Basic studies of pancreatic nerves and experimental human pain research have provided evidence that pain processing is abnormal in these patients and in many cases resembles that seen in neuropathic and chronic pain disorders. This neurobiological view of pain is somewhat in opposition to the traditional view of pain etiology in CP, where pain was assumed to arise from pathology in or in close proximity to the pancreatic gland. However, these theories are not mutually exclusive, and aspects of both may contribute in the generation and perpetuation of pain. In addition, adverse effects and complications to medical and interventional therapies may account for a substantial morbidity in many patients and should be considered as an additional source of pain. Therefore, it is important to consider the different mechanisms, when evaluating the origin of pain in CP patients (Figure 1), and it is plausible that the “collective” abdominal pain is a result of a complex interplay of several mechanisms. This novel and multifaceted understanding of pain etiology in CP requires a paradigm shift in pain management. Hence, modern mechanism based pain treatments taking into account especially altered pain processing are likely to increasingly replace traditional invasive therapies. In addition, the improved understanding of pain etiology will likely pave the way for new treatment modalities.
Footnotes
Supported by The Danish Council for Strategic Research
P- Reviewers: Farré A, Frulloni L, Seicean A S- Editor: Wen LL L- Editor: A E- Editor: Zhang DN
References
- 1.Pasricha PJ. Unraveling the mystery of pain in chronic pancreatitis. Nat Rev Gastroenterol Hepatol. 2012;9:140–151. doi: 10.1038/nrgastro.2011.274. [DOI] [PubMed] [Google Scholar]
- 2.Fasanella KE, Davis B, Lyons J, Chen Z, Lee KK, Slivka A, Whitcomb DC. Pain in chronic pancreatitis and pancreatic cancer. Gastroenterol Clin North Am. 2007;36:335–64, ix. doi: 10.1016/j.gtc.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Ammann RW, Muellhaupt B. The natural history of pain in alcoholic chronic pancreatitis. Gastroenterology. 1999;116:1132–1140. doi: 10.1016/s0016-5085(99)70016-8. [DOI] [PubMed] [Google Scholar]
- 4.Ammann RW, Buehler H, Muench R, Freiburghaus AW, Siegenthaler W. Differences in the natural history of idiopathic (nonalcoholic) and alcoholic chronic pancreatitis. A comparative long-term study of 287 patients. Pancreas. 1987;2:368–377. doi: 10.1097/00006676-198707000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Lankisch PG, Löhr-Happe A, Otto J, Creutzfeldt W. Natural course in chronic pancreatitis. Pain, exocrine and endocrine pancreatic insufficiency and prognosis of the disease. Digestion. 1993;54:148–155. doi: 10.1159/000201029. [DOI] [PubMed] [Google Scholar]
- 6.Mullady DK, Yadav D, Amann ST, O’Connell MR, Barmada MM, Elta GH, Scheiman JM, Wamsteker EJ, Chey WD, Korneffel ML, et al. Type of pain, pain-associated complications, quality of life, disability and resource utilisation in chronic pancreatitis: a prospective cohort study. Gut. 2011;60:77–84. doi: 10.1136/gut.2010.213835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amann ST, Yadav D, Barmada MM, O’Connell M, Kennard ED, Anderson M, Baillie J, Sherman S, Romagnuolo J, Hawes RH, et al. Physical and mental quality of life in chronic pancreatitis: a case-control study from the North American Pancreatitis Study 2 cohort. Pancreas. 2013;42:293–300. doi: 10.1097/MPA.0b013e31826532e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner TB, Kennedy AT, Gelrud A, Banks PA, Vege SS, Gordon SR, Lacy BE. Chronic pancreatitis and its effect on employment and health care experience: results of a prospective American multicenter study. Pancreas. 2010;39:498–501. doi: 10.1097/MPA.0b013e3181c5c693. [DOI] [PubMed] [Google Scholar]
- 9.Drewes AM, Krarup AL, Detlefsen S, Malmstrøm ML, Dimcevski G, Funch-Jensen P. Pain in chronic pancreatitis: the role of neuropathic pain mechanisms. Gut. 2008;57:1616–1627. doi: 10.1136/gut.2007.146621. [DOI] [PubMed] [Google Scholar]
- 10.Woolf CJ. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140:441–451. doi: 10.7326/0003-4819-140-8-200404200-00010. [DOI] [PubMed] [Google Scholar]
- 11.Lieb JG, Forsmark CE. Review article: pain and chronic pancreatitis. Aliment Pharmacol Ther. 2009;29:706–719. doi: 10.1111/j.1365-2036.2009.03931.x. [DOI] [PubMed] [Google Scholar]
- 12.Anaparthy R, Pasricha PJ. Pain and chronic pancreatitis: is it the plumbing or the wiring? Curr Gastroenterol Rep. 2008;10:101–106. doi: 10.1007/s11894-008-0029-4. [DOI] [PubMed] [Google Scholar]
- 13.White TT, Bourde J. A new observation on human intraductal pancreatic pressure. Surg Gynecol Obstet. 1970;130:275–278. [PubMed] [Google Scholar]
- 14.Sato T, Miyashita E, Yamauchi H, Matsuno S. The role of surgical treatment for chronic pancreatitis. Ann Surg. 1986;203:266–271. doi: 10.1097/00000658-198603000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okazaki K, Yamamoto Y, Ito K. Endoscopic measurement of papillary sphincter zone and pancreatic main ductal pressure in patients with chronic pancreatitis. Gastroenterology. 1986;91:409–418. doi: 10.1016/0016-5085(86)90576-7. [DOI] [PubMed] [Google Scholar]
- 16.Laugier R. Dynamic endoscopic manometry of the response to secretin in patients with chronic pancreatitis. Endoscopy. 1994;26:222–227. doi: 10.1055/s-2007-1008947. [DOI] [PubMed] [Google Scholar]
- 17.Novis BH, Bornman PC, Girdwood AW, Marks IN. Endoscopic manometry of the pancreatic duct and sphincter zone in patients with chronic pancreatitis. Dig Dis Sci. 1985;30:225–228. doi: 10.1007/BF01347888. [DOI] [PubMed] [Google Scholar]
- 18.Rolny P, Arlebäck A, Järnerot G, Andersson T. Endoscopic manometry of the sphincter of Oddi and pancreatic duct in chronic pancreatitis. Scand J Gastroenterol. 1986;21:415–420. doi: 10.3109/00365528609015156. [DOI] [PubMed] [Google Scholar]
- 19.Ugljesić M, Bulajić M, Milosavljević T, Stimec B. Endoscopic manometry of the sphincter of Oddi and pancreatic duct in patients with chronic pancreatitis. Int J Pancreatol. 1996;19:191–195. doi: 10.1007/BF02787367. [DOI] [PubMed] [Google Scholar]
- 20.Vestergaard H, Kruse A, Rokkjaer M, Frøbert O, Thommesen P, Funch-Jensen P. Endoscopic manometry of the sphincter of Oddi and the pancreatic and biliary ducts in patients with chronic pancreatitis. Scand J Gastroenterol. 1994;29:188–192. doi: 10.3109/00365529409090461. [DOI] [PubMed] [Google Scholar]
- 21.Ebbehøj N, Svendsen LB, Madsen P. Pancreatic tissue pressure: techniques and pathophysiological aspects. Scand J Gastroenterol. 1984;19:1066–1068. [PubMed] [Google Scholar]
- 22.Ebbehøj N, Borly L, Madsen P, Svendsen LB. Pancreatic tissue pressure and pain in chronic pancreatitis. Pancreas. 1986;1:556–558. doi: 10.1097/00006676-198611000-00015. [DOI] [PubMed] [Google Scholar]
- 23.Ebbehøj N, Borly L, Bülow J, Rasmussen SG, Madsen P. Evaluation of pancreatic tissue fluid pressure and pain in chronic pancreatitis. A longitudinal study. Scand J Gastroenterol. 1990;25:462–466. doi: 10.3109/00365529009095516. [DOI] [PubMed] [Google Scholar]
- 24.Manes G, Büchler M, Pieramico O, Di Sebastiano P, Malfertheiner P. Is increased pancreatic pressure related to pain in chronic pancreatitis? Int J Pancreatol. 1994;15:113–117. doi: 10.1007/BF02924661. [DOI] [PubMed] [Google Scholar]
- 25.Patel AG, Toyama MT, Alvarez C, Nguyen TN, Reber PU, Ashley SW, Reber HA. Pancreatic interstitial pH in human and feline chronic pancreatitis. Gastroenterology. 1995;109:1639–1645. doi: 10.1016/0016-5085(95)90654-1. [DOI] [PubMed] [Google Scholar]
- 26.Zhu Y, Colak T, Shenoy M, Liu L, Pai R, Li C, Mehta K, Pasricha PJ. Nerve growth factor modulates TRPV1 expression and function and mediates pain in chronic pancreatitis. Gastroenterology. 2011;141:370–377. doi: 10.1053/j.gastro.2011.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz ES, La JH, Scheff NN, Davis BM, Albers KM, Gebhart GF. TRPV1 and TRPA1 antagonists prevent the transition of acute to chronic inflammation and pain in chronic pancreatitis. J Neurosci. 2013;33:5603–5611. doi: 10.1523/JNEUROSCI.1806-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warshaw AL, Banks PA, Fernández-Del Castillo C. AGA technical review: treatment of pain in chronic pancreatitis. Gastroenterology. 1998;115:765–776. doi: 10.1016/s0016-5085(98)70157-x. [DOI] [PubMed] [Google Scholar]
- 29.Andrén-Sandberg A, Hoem D, Gislason H. Pain management in chronic pancreatitis. Eur J Gastroenterol Hepatol. 2002;14:957–970. doi: 10.1097/00042737-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 30.Frøkjær JB, Olesen SS, Drewes AM. Fibrosis, atrophy, and ductal pathology in chronic pancreatitis are associated with pancreatic function but independent of symptoms. Pancreas. 2013;42:1182–1187. doi: 10.1097/MPA.0b013e31829628f4. [DOI] [PubMed] [Google Scholar]
- 31.Balci NC, Perman WH, Saglam S, Akisik F, Fattahi R, Bilgin M. Diffusion-weighted magnetic resonance imaging of the pancreas. Top Magn Reson Imaging. 2009;20:43–47. doi: 10.1097/RMR.0b013e3181b48667. [DOI] [PubMed] [Google Scholar]
- 32.Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, Hansson P, Hughes R, Nurmikko T, Serra J. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008;70:1630–1635. doi: 10.1212/01.wnl.0000282763.29778.59. [DOI] [PubMed] [Google Scholar]
- 33.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 34.Xu GY, Winston JH, Shenoy M, Yin H, Pendyala S, Pasricha PJ. Transient receptor potential vanilloid 1 mediates hyperalgesia and is up-regulated in rats with chronic pancreatitis. Gastroenterology. 2007;133:1282–1292. doi: 10.1053/j.gastro.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 35.Hartel M, di Mola FF, Selvaggi F, Mascetta G, Wente MN, Felix K, Giese NA, Hinz U, Di Sebastiano P, Büchler MW, et al. Vanilloids in pancreatic cancer: potential for chemotherapy and pain management. Gut. 2006;55:519–528. doi: 10.1136/gut.2005.073205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friess H, Zhu ZW, di Mola FF, Kulli C, Graber HU, Andren-Sandberg A, Zimmermann A, Korc M, Reinshagen M, Büchler MW. Nerve growth factor and its high-affinity receptor in chronic pancreatitis. Ann Surg. 1999;230:615–624. doi: 10.1097/00000658-199911000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu ZW, Friess H, Wang L, Zimmermann A, Büchler MW. Brain-derived neurotrophic factor (BDNF) is upregulated and associated with pain in chronic pancreatitis. Dig Dis Sci. 2001;46:1633–1639. doi: 10.1023/a:1010684916863. [DOI] [PubMed] [Google Scholar]
- 38.Gebhart GF. Visceral pain-peripheral sensitisation. Gut. 2000;47 Suppl 4:iv54–iv5; discussion iv58. doi: 10.1136/gut.47.suppl_4.iv54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anand P, Aziz Q, Willert R, van Oudenhove L. Peripheral and central mechanisms of visceral sensitization in man. Neurogastroenterol Motil. 2007;19:29–46. doi: 10.1111/j.1365-2982.2006.00873.x. [DOI] [PubMed] [Google Scholar]
- 40.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 41.Bockman DE, Buchler M, Malfertheiner P, Beger HG. Analysis of nerves in chronic pancreatitis. Gastroenterology. 1988;94:1459–1469. doi: 10.1016/0016-5085(88)90687-7. [DOI] [PubMed] [Google Scholar]
- 42.Keith RG, Keshavjee SH, Kerenyi NR. Neuropathology of chronic pancreatitis in humans. Can J Surg. 1985;28:207–211. [PubMed] [Google Scholar]
- 43.Ceyhan GO, Demir IE, Rauch U, Bergmann F, Müller MW, Büchler MW, Friess H, Schäfer KH. Pancreatic neuropathy results in “neural remodeling” and altered pancreatic innervation in chronic pancreatitis and pancreatic cancer. Am J Gastroenterol. 2009;104:2555–2565. doi: 10.1038/ajg.2009.380. [DOI] [PubMed] [Google Scholar]
- 44.Ceyhan GO, Bergmann F, Kadihasanoglu M, Altintas B, Demir IE, Hinz U, Müller MW, Giese T, Büchler MW, Giese NA, et al. Pancreatic neuropathy and neuropathic pain--a comprehensive pathomorphological study of 546 cases. Gastroenterology. 2009;136:177–186.e1. doi: 10.1053/j.gastro.2008.09.029. [DOI] [PubMed] [Google Scholar]
- 45.Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152:S2–15. doi: 10.1016/j.pain.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuner R. Central mechanisms of pathological pain. Nat Med. 2010;16:1258–1266. doi: 10.1038/nm.2231. [DOI] [PubMed] [Google Scholar]
- 47.Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arendt-Nielsen L, Laursen RJ, Drewes AM. Referred pain as an indicator for neural plasticity. Prog Brain Res. 2000;129:343–356. doi: 10.1016/s0079-6123(00)29026-2. [DOI] [PubMed] [Google Scholar]
- 49.Drewes AM, Pedersen J, Liu W, Arendt-Nielsen L, Gregersen H. Controlled mechanical distension of the human oesophagus: sensory and biomechanical findings. Scand J Gastroenterol. 2003;38:27–35. [PubMed] [Google Scholar]
- 50.Dimcevski G, Sami SA, Funch-Jensen P, Le Pera D, Valeriani M, Arendt-Nielsen L, Drewes AM. Pain in chronic pancreatitis: the role of reorganization in the central nervous system. Gastroenterology. 2007;132:1546–1556. doi: 10.1053/j.gastro.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 51.Buscher HC, Wilder-Smith OH, van Goor H. Chronic pancreatitis patients show hyperalgesia of central origin: a pilot study. Eur J Pain. 2006;10:363–370. doi: 10.1016/j.ejpain.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 52.Olesen SS, Brock C, Krarup AL, Funch-Jensen P, Arendt-Nielsen L, Wilder-Smith OH, Drewes AM. Descending inhibitory pain modulation is impaired in patients with chronic pancreatitis. Clin Gastroenterol Hepatol. 2010;8:724–730. doi: 10.1016/j.cgh.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 53.Olesen SS, Frøkjær JB, Lelic D, Valeriani M, Drewes AM. Pain-associated adaptive cortical reorganisation in chronic pancreatitis. Pancreatology. 2010;10:742–751. doi: 10.1159/000321644. [DOI] [PubMed] [Google Scholar]
- 54.Olesen SS, Hansen TM, Graversen C, Steimle K, Wilder-Smith OH, Drewes AM. Slowed EEG rhythmicity in patients with chronic pancreatitis: evidence of abnormal cerebral pain processing? Eur J Gastroenterol Hepatol. 2011;23:418–424. doi: 10.1097/MEG.0b013e3283457b09. [DOI] [PubMed] [Google Scholar]
- 55.Olesen SS, Hansen TM, Graversen C, Valeriani M, Drewes AM. Cerebral excitability is abnormal in patients with painful chronic pancreatitis. Eur J Pain. 2013;17:46–54. doi: 10.1002/j.1532-2149.2012.00155.x. [DOI] [PubMed] [Google Scholar]
- 56.Frøkjær JB, Andersen LW, Brock C, Simrén M, Ljungberg M, Søfteland E, Dimcevski G, Yavarian Y, Gregersen H, Drewes AM. Altered brain microstructure assessed by diffusion tensor imaging in patients with diabetes and gastrointestinal symptoms. Diabetes Care. 2013;36:662–668. doi: 10.2337/dc12-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frøkjær JB, Bouwense SA, Olesen SS, Lundager FH, Eskildsen SF, van Goor H, Wilder-Smith OH, Drewes AM. Reduced cortical thickness of brain areas involved in pain processing in patients with chronic pancreatitis. Clin Gastroenterol Hepatol. 2012;10:434–8.e1. doi: 10.1016/j.cgh.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 58.Bouwense SA, Olesen SS, Drewes AM, Frøkjær JB, van Goor H, Wilder-Smith OH. Is altered central pain processing related to disease stage in chronic pancreatitis patients with pain? An exploratory study. PLoS One. 2013;8:e55460. doi: 10.1371/journal.pone.0055460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vera-Portocarrero LP, Xie JY, Kowal J, Ossipov MH, King T, Porreca F. Descending facilitation from the rostral ventromedial medulla maintains visceral pain in rats with experimental pancreatitis. Gastroenterology. 2006;130:2155–2164. doi: 10.1053/j.gastro.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 60.Bouwense SA, Buscher HC, van Goor H, Wilder-Smith OH. Has central sensitization become independent of nociceptive input in chronic pancreatitis patients who fail thoracoscopic splanchnicectomy? Reg Anesth Pain Med. 2011;36:531–536. doi: 10.1097/AAP.0b013e31822e0d4a. [DOI] [PubMed] [Google Scholar]
- 61.Boerma D, Obertop H, Gouma DJ. Pancreatic pseudocysts in chronic pancreatitis. Surgical or interventional drainage? Ann Ital Chir. 2000;71:43–50. [PubMed] [Google Scholar]
- 62.Andrén-Sandberg A, Dervenis C. Pancreatic pseudocysts in the 21st century. Part I: classification, pathophysiology, anatomic considerations and treatment. JOP. 2004;5:8–24. [PubMed] [Google Scholar]
- 63.Ammann RW, Akovbiantz A, Largiader F, Schueler G. Course and outcome of chronic pancreatitis. Longitudinal study of a mixed medical-surgical series of 245 patients. Gastroenterology. 1984;86:820–828. [PubMed] [Google Scholar]
- 64.Aghdassi A, Mayerle J, Kraft M, Sielenkämper AW, Heidecke CD, Lerch MM. Diagnosis and treatment of pancreatic pseudocysts in chronic pancreatitis. Pancreas. 2008;36:105–112. doi: 10.1097/MPA.0b013e31815a8887. [DOI] [PubMed] [Google Scholar]
- 65.Gouyon B, Lévy P, Ruszniewski P, Zins M, Hammel P, Vilgrain V, Sauvanet A, Belghiti J, Bernades P. Predictive factors in the outcome of pseudocysts complicating alcoholic chronic pancreatitis. Gut. 1997;41:821–825. doi: 10.1136/gut.41.6.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vijungco JD, Prinz RA. Management of biliary and duodenal complications of chronic pancreatitis. World J Surg. 2003;27:1258–1270. doi: 10.1007/s00268-003-7246-7. [DOI] [PubMed] [Google Scholar]
- 67.Abdallah AA, Krige JE, Bornman PC. Biliary tract obstruction in chronic pancreatitis. HPB (Oxford) 2007;9:421–428. doi: 10.1080/13651820701774883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kalvaria I, Bornman PC, Marks IN, Girdwood AH, Bank L, Kottler RE. The spectrum and natural history of common bile duct stenosis in chronic alcohol-induced pancreatitis. Ann Surg. 1989;210:608–613. doi: 10.1097/00000658-198911000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prinz RA, Aranha GV, Greenlee HB. Combined pancreatic duct and upper gastrointestinal and biliary tract drainage in chronic pancreatitis. Arch Surg. 1985;120:361–366. doi: 10.1001/archsurg.1985.01390270099017. [DOI] [PubMed] [Google Scholar]
- 70.Kahl S, Zimmermann S, Genz I, Schmidt U, Pross M, Schulz HU, Malfertheiner P. Biliary strictures are not the cause of pain in patients with chronic pancreatitis. Pancreas. 2004;28:387–390. doi: 10.1097/00006676-200405000-00006. [DOI] [PubMed] [Google Scholar]
- 71.Chebli JM, de Souza AF, Gaburri PD, Bastos KV, Ribeiro TC, Filho RJ, Chebli LA, Castro Ferreira LE. Prevalence and pathogenesis of duodenal ulcer in chronic alcoholic pancreatitis. J Clin Gastroenterol. 2002;35:71–74. doi: 10.1097/00004836-200207000-00015. [DOI] [PubMed] [Google Scholar]
- 72.Schulze S, Thorsgaard Pedersen N, Jørgensen MJ, Møllmann KM, Rune SJ. Association between duodenal bulb ulceration and reduced exocrine pancreatic function. Gut. 1983;24:781–783. doi: 10.1136/gut.24.9.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saunders JH, Cargill JM, Wormsley KG. Gastric secretion of acid in patients with pancreatic disease. Digestion. 1978;17:365–369. doi: 10.1159/000198129. [DOI] [PubMed] [Google Scholar]
- 74.Piubello W, Vantini I, Scuro LA, Novelli P, Benini L, Brocco G, Cavallini G. Gastric secretion, gastroduodenal histological changes, and serum gastrin in chronic alcoholic pancreatitis. Am J Gastroenterol. 1982;77:105–110. [PubMed] [Google Scholar]
- 75.Weber SM, Rikkers LF. Splenic vein thrombosis and gastrointestinal bleeding in chronic pancreatitis. World J Surg. 2003;27:1271–1274. doi: 10.1007/s00268-003-7247-6. [DOI] [PubMed] [Google Scholar]
- 76.ROSCH J, HERFORT K. Contribution of splenoportography to the diagnosis of diseases of the pancreas. II. Inflammatory diseases. Acta Med Scand. 1962;171:263–272. doi: 10.1111/j.0954-6820.1962.tb04188.x. [DOI] [PubMed] [Google Scholar]
- 77.Rignault D, Mine J, Moine D. Splenoportographic changes in chronic pancreatitis. Surgery. 1968;63:571–575. [PubMed] [Google Scholar]
- 78.Bernades P, Baetz A, Lévy P, Belghiti J, Menu Y, Fékété F. Splenic and portal venous obstruction in chronic pancreatitis. A prospective longitudinal study of a medical-surgical series of 266 patients. Dig Dis Sci. 1992;37:340–346. doi: 10.1007/BF01307725. [DOI] [PubMed] [Google Scholar]
- 79.Brock C, Olesen SS, Olesen AE, Frøkjaer JB, Andresen T, Drewes AM. Opioid-induced bowel dysfunction: pathophysiology and management. Drugs. 2012;72:1847–1865. doi: 10.2165/11634970-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 80.Tuteja AK, Biskupiak J, Stoddard GJ, Lipman AG. Opioid-induced bowel disorders and narcotic bowel syndrome in patients with chronic non-cancer pain. Neurogastroenterol Motil. 2010;22:424–30, e96. doi: 10.1111/j.1365-2982.2009.01458.x. [DOI] [PubMed] [Google Scholar]
- 81.Casellas F, Guarner L, Vaquero E, Antolín M, de Gracia X, Malagelada JR. Hydrogen breath test with glucose in exocrine pancreatic insufficiency. Pancreas. 1998;16:481–486. doi: 10.1097/00006676-199805000-00004. [DOI] [PubMed] [Google Scholar]
- 82.Garcés MC, Gómez-Cerezo J, Alba D, Codoceo R, Vázquez-Muñoz E, Arnalich F, Barbado FJ, Vázquez JJ. Relationship of basal and postprandial intraduodenal bile acid concentrations and plasma cholecystokinin levels with abdominal pain in patients with chronic pancreatitis. Pancreas. 1998;17:397–401. doi: 10.1097/00006676-199811000-00011. [DOI] [PubMed] [Google Scholar]
- 83.Xie JY, Herman DS, Stiller CO, Gardell LR, Ossipov MH, Lai J, Porreca F, Vanderah TW. Cholecystokinin in the rostral ventromedial medulla mediates opioid-induced hyperalgesia and antinociceptive tolerance. J Neurosci. 2005;25:409–416. doi: 10.1523/JNEUROSCI.4054-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shiratori K, Takeuchi T, Satake K, Matsuno S. Clinical evaluation of oral administration of a cholecystokinin-A receptor antagonist (loxiglumide) to patients with acute, painful attacks of chronic pancreatitis: a multicenter dose-response study in Japan. Pancreas. 2002;25:e1–e5. doi: 10.1097/00006676-200207000-00003. [DOI] [PubMed] [Google Scholar]
- 85.Buscher HC, van Goor H, Sweep CG, Lenders JW, Wilder-Smith OH. Increased sympathetic activity in chronic pancreatitis patients is associated with hyperalgesia. J Pain Palliat Care Pharmacother. 2010;24:362–366. doi: 10.3109/15360288.2010.519762. [DOI] [PubMed] [Google Scholar]
- 86.Chandrasekharan B, Srinivasan S. Diabetes and the enteric nervous system. Neurogastroenterol Motil. 2007;19:951–960. doi: 10.1111/j.1365-2982.2007.01023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Demir IE, Tieftrunk E, Maak M, Friess H, Ceyhan GO. Pain mechanisms in chronic pancreatitis: of a master and his fire. Langenbecks Arch Surg. 2011;396:151–160. doi: 10.1007/s00423-010-0731-1. [DOI] [PMC free article] [PubMed] [Google Scholar]