

Background: HCN2 channels are regulated by membrane potential and by the direct binding of cyclic nucleotides to their carboxyl-terminal region.
Results: The flavonoid fisetin potentiates HCN2 channel activation.
Conclusion: Fisetin acts as a partial agonist for HCN2 channels by binding to the channel's cyclic nucleotide-binding site.
Significance: HCN2 may be a target for the physiologic action of flavonoids such as fisetin.
Keywords: Cyclic AMP (cAMP), Electrophysiology, Flavonoids, Fluorescence Resonance Energy Transfer (FRET), Ion Channels, NMR, HCN, Fluorescence Anisotropy, Purine Ring
Abstract
The hyperpolarization-activated cyclic nucleotide-modulated (HCN) channels are pacemaker channels whose currents contribute to rhythmic activity in the heart and brain. HCN channels open in response to hyperpolarizing voltages, and the binding of cAMP to their cyclic nucleotide-binding domain (CNBD) facilitates channel opening. Here, we report that, like cAMP, the flavonoid fisetin potentiates HCN2 channel gating. Fisetin sped HCN2 activation and shifted the conductance-voltage relationship to more depolarizing potentials with a half-maximal effective concentration (EC50) of 1.8 μm. When applied together, fisetin and cAMP regulated HCN2 gating in a nonadditive fashion. Fisetin did not potentiate HCN2 channels lacking their CNBD, and two independent fluorescence-based binding assays reported that fisetin bound to the purified CNBD. These data suggest that the CNBD mediates the fisetin potentiation of HCN2 channels. Moreover, binding assays suggest that fisetin and cAMP partially compete for binding to the CNBD. NMR experiments demonstrated that fisetin binds within the cAMP-binding pocket, interacting with some of the same residues as cAMP. Together, these data indicate that fisetin is a partial agonist for HCN2 channels.
Introduction
With their unique gating and direct regulation by cyclic nucleotides, the hyperpolarization-activated, cyclic nucleotide-modulated (HCN)3 channels fill specialized physiologic roles. For example, HCN channels give rise to the “funny” current of the heart, which underlies the pacemaking activity of the sinoatrial node and cardiac rhythmicity (1). In the brain, HCN currents contribute to long term potentiation (2) and play an essential role in chronic pain (3). Moreover, HCN dysfunction is implicated in various diseases including epilepsy (4, 5) and Parkinson disease (6).
Four mammalian HCN genes have been identified (HCN1–4) and encode for ion channels that are members of the voltage-gated K+ channel superfamily (7). Like other voltage-gated K+ channels, HCN channels are composed of four subunits arranged around a centrally located, ion-conducting pore (8, 9). Each channel subunit contains six transmembrane segments (S1–S6), including a voltage-sensing domain (S1–S4) and a re-entrant pore-forming loop between S5 and S6. Unlike other members of this superfamily, HCN channels are only weakly K+-selective, activated by hyperpolarizing voltages, and regulated by the direct binding of cyclic nucleotides to their intracellularly located, carboxyl-terminal region. This carboxyl-terminal region includes a cyclic nucleotide-binding domain (CNBD) and a C-linker that connects the domain to the pore (7, 10). The x-ray crystal structure of the C-linker/CNBD bound to cyclic nucleotides has been solved for several HCN channels (11–14). These nearly identical structures revealed that the C-linker is composed of six α-helices (designated A′–F′), and the CNBD includes four α-helices and an eight-stranded β-roll. Cyclic nucleotide binding to this domain potentiates the activation of HCN channels to various degrees depending on the isoform. For HCN2, cAMP binding stabilizes the open state, speeds activation, and shifts the voltage dependence toward more positive potentials (15).
Recently, we reported that fisetin (2-(3,4-dihydroxyphenyl)-3,7-dihydroxychromen-4-one) and structurally related flavonoids potentiate EAG1 channels (16). EAG1 channels are encoded by the KCNH1 gene and, like HCN channels, are members of the voltage-gated K+ channel superfamily. Unlike HCN channels, EAG1 channels open in response to depolarizing voltages and are highly selective for K+ (17). All KCNH channels, including EAG1, contain a cyclic nucleotide-binding homology domain (CNBHD) in their carboxyl-terminal region that shares sequence and structural similarity with the CNBD of HCN channels (18–20). The CNBHD in KCNH family channels does not bind cyclic nucleotides, and these channels are not directly regulated by cAMP or cGMP (18, 21). However, we found that flavonoids such as fisetin bind to the CNBHD of EAG1 to stabilize the channel's open state (16, 22).
Based on structural similarity between HCN and KCNH channels, we hypothesized that fisetin may also bind to the CNBD of HCN2 channels and regulate gating. Here, we demonstrate that fisetin potentiates HCN2 channels in a manner similar to cAMP. Using inside-out patch clamp recordings, we found that fisetin and cAMP regulated gating in a nonadditive fashion. Further, fisetin did not potentiate channels lacking a CNBD. Two fluorescence-based binding assays, as well as NMR, reported that fisetin bound to the purified HCN2 CNBD. Additionally we found that fisetin and cAMP partially competed for binding, and NMR indicated that fisetin bound within or proximal to the cAMP-binding pocket. Together, these results indicate that fisetin is a partial agonist for HCN2.
EXPERIMENTAL PROCEDURES
Materials
8-Fluo-cAMP was purchased from Axxora (Farmingdale, NY). All other chemicals were purchased from Sigma.
Electrophysiology
The cDNA encoding two versions of the murine HCN2 channels, wild-type HCN2 and HCN2V526stop, in the pGHE vectors, were kindly provided by Steve Siegelbaum and Bina Santoro (Columbia University, New York, NY). The cRNA for these channels was transcribed using the T7 MEGAscript kit (Ambion) and expressed in Xenopus laevis oocytes that were defolliculated and injected with the cRNA as described previously (23). The vitelline membranes were manually removed, and currents were recorded in the inside-out patch clamp configuration (24) with an EPC-10 patch clamp amplifier (HEKA Elektronik). Following excision, we observed a slow decrease in the HCN2 current that required 20–30 min to stabilize. This run-down in current is thought to be mediated by a depletion of phosphatidylinositol 4,5-bisphosphate in the patch membrane (25, 26). Therefore, all electrophysiology experiments were performed at least 30 min following excision once the current level had reached steady state. Patch pipettes were pulled from borosilicate glass and had resistances of 0.40–0.6 megaohms after fire polishing. Various solutions were applied to patches with the RSC-100 solution changer (BioLogic). Both intracellular (bath) and extracellular (pipette) solutions contained 130 mm KCl, 10 mm HEPES, 0.2 mm EDTA, pH 7.2. Patches were held at 0 mV, and the HCN2 currents were elicited by applying a series of 5-s voltage pulses (ranging from −140 to −70 mV in 10-mV increments) followed by a 1-s voltage pulse to −40 mV. Currents were not leak-subtracted. Data were acquired with Pulse software (HEKA Elektronik) and analyzed with Igor (WaveMetrics, Inc).
To measure the conductance-voltage relationships, peak tail current amplitudes at −40 mV were normalized to the peak tail current following a step to −140 mV. Plots of the normalized conductance versus the test voltage were fit with a Boltzmann function
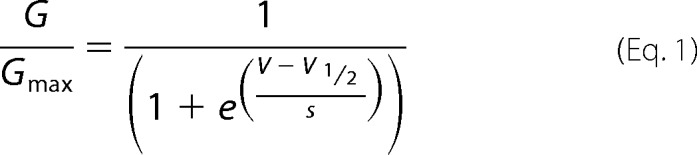 |
where V represents the test voltage, V½ is the midpoint activation voltage, and s is the slope of the relation.
To determine the concentration-response relationship, a plot of the change in the V½ (ΔV½) versus free fisetin concentration was fit with a Hill equation
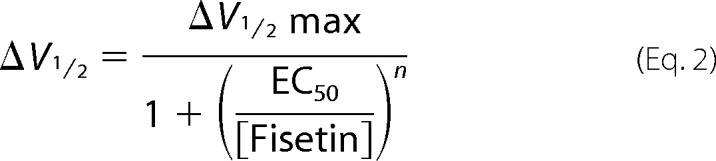 |
where ΔV½max represents the maximum shift in the V½, EC50 represents the half-maximal concentration, and n represents the Hill coefficient.
Protein Expression and Purification
Two versions of the HCN2 carboxyl-terminal region were purified for binding assays and NMR experiments. A shorter version was used for fluorescence anisotropy and NMR experiments. This protein (HCN2CNBD) is composed of residues 487–640 and includes the C- and D-helices of the C-linker and the entire CNBD. For protein expression, the DNA encoding HCN2CNBD was subcloned into the pETM11 vector. A longer protein composed of the entire C-linker and CNBD was used for fluorescence resonance energy transfer (FRET) experiments. This protein (HCN2C-linker/CNBD-L586W) included residues 443–645 and a tryptophan substituted for a leucine at position 586 (21). For protein expression, the DNA encoding HCN2C-linker/CNBD-L586W was subcloned into the pETGQ vector (27).
For protein purification, the appropriate DNA was transformed into BL21 (DE3) cells, grown at 37 °C, and induced with 1 mm isopropyl-1-thio-β-d-galactopyranoside at an optical density of 0.6–0.8. For NMR experiments, the bacteria were induced in minimal media containing 13C-labeled d-glucose (Cambridge Isotopes). After incubating overnight at 18 °C, the BL21 (DE3) cells were lysed in an Emulsiflex-C3 (Avestin), and the lysate was cleared by sedimentation for 45 min at 131,000 × g at 4 °C. The protein was then purified with Ni2+-nitrilotriacetic acid chromatography. The hexahistidine tag was cleaved with either tobacco etch virus protease for HCN2CNBD or thrombin protease (Calbiochem) for HCN2C-linker/CNBD-L586W. The HCN2CNBD protein was then purified with ion exchange chromatography and used within 3 days of purification for fluorescence anisotropy experiments. Fluorescence anisotropy experiments with HCN2CNBD were performed in the following solution: 150 mm KCl, 30 mm HEPES, pH 7.0. For NMR, HCN2CNBD was additionally purified with size exclusion chromatography on a Superdex 200 column (Amersham Biosciences) and used within 5 days of purification. Following the Ni2+-nitrilotriacetic acid chromatography, the HCN2C-linker/CNBD-L586W was then further purified by size exclusion chromatography on a Superdex 200 column equilibrated with the buffer used for the FRET experiments (150 mm KCl, 10% glycerol, 1 mm tris(2-carboxyethyl)phosphine, 30 mm HEPES; pH 7.5). Purified HCN2C-linker/CNBD-L586W was stored in small aliquots at −80 °C and thawed immediately before experimentation. Protein concentration was determined by absorbance at 280 nm.
Fluorescence Measurements
Fluorescence intensity and anisotropy were each recorded in a 100-μm quartz cuvette with a Fluorolog 3 spectrophotometer and FluorEssence software (both from HORIBA Jobin Yvon). For fluorescence anisotropy measurements, the spectrophotometer was outfitted with Glan-Thompson polarizers. The fluorescence anisotropy was recorded from 5 μm fisetin in solution with varying concentrations of HCN2CNBD. Fluorescence anisotropy measurements were made with 420 nm excitation and 490 nm emission light and 13-nm slit widths as described previously (16, 28). Each experiment was repeated three times, and the data were plotted as mean ± S.E.
To estimate the apparent binding affinity, plots of the fluorescence anisotropy versus the total protein concentration were fit with the following equations (29)
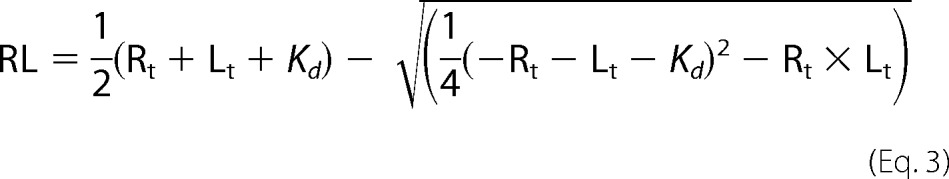 |
where RL is the concentration of the free receptor-ligand complex; Rt and Lt are total receptor and ligand concentrations, respectively; and Kd is the apparent binding affinity. The fluorescence anisotropy, A, was then calculated from the following equation
where A0 is the fluorescence anisotropy of unbound fisetin, and A∞ is the fluorescence anisotropy of fisetin at saturating concentrations of receptor. Data analysis and plot fittings were performed in Igor.
For tryptophan-to-fisetin FRET, the fluorescence spectra of 4 μm HCN2C-linker/CNBD-L586W or 4 μm free tryptophan were recorded in the absence and presence of 100 μm fisetin. Samples were excited with 295-nm light, and emission spectra were recorded from 300 to 500 nm with 5-nm slit widths. To account for the decrease in excitation and emission intensities due to the optical density, observed fluorescence intensities of the sample were corrected for the inner filter effect according to the equation (30)
where Fci and Foi represent the corrected and observed fluorescence intensities at wavelength i nm, and A295 and Ai are the absorbance measured at wavelengths 295 nm and i nm, respectively. Each experiment was repeated at least three times, and the data were plotted as means. Data analysis and plot fittings were performed in Origin (OriginLab Corp.).
Modeling Simultaneous Binding
To explore whether fisetin and cAMP can simultaneously bind the CNBD, we created a four-state model to fit our binding data. Fluorescence anisotropy-based binding experiments and Equation 4 were used to determine the fisetin-to-CNBD binding affinity in 0 or 1 mm cAMP. Igor was then used to calculate the roots of the following equations
 |
 |
 |
where R, L, and A represent the concentrations of free HCN2CNBD, free fisetin, and free cAMP respectively. RT, LT, and AT, represent the total concentrations of HCN2CNBD, fisetin, and cAMP respectively. [RL] is the concentration of HCN2CNBD bound to fisetin, [RA] is the concentration of HCN2CNBD bound to cAMP, and [RLA] is the concentration of HCN2CNBD bound to both fisetin and cAMP. These fits were scaled to the maximum and minimum fluorescence anisotropy values determined experimentally, 0.25 and 0.0656, respectively.
NMR Spectroscopy
NMR spectra were recorded on a Bruker Avance III spectrometer equipped with a cryoprobe and operating at 600 MHz. Spectra were recorded at 25 °C in 30 mm HEPES, 150 mm KCl, 10% D2O, 1% dimethyl sulfoxide (DMSO), pH 7.5. All NMR spectra were collected on samples containing 175 μm of either the wild-type or the M572T versions of the HCN2CNBD protein. The HCN2CNBD were measured alone or in titrations with 175 μm fisetin or 175 μm cAMP. All titration data sets were collected using 1H,13C HSQC experiments. Data were processed using NMRPipe/NMRDraw (31) and visualized using NMRView (32).
RESULTS
Fisetin Potentiated HCN2 Channels
To determine whether fisetin regulates HCN2, we measured its effects on the kinetic and steady-state properties of the channels (Fig. 1). For these experiments, HCN2 channels were expressed in X. laevis oocytes and studied in the inside-out configuration of the patch clamp technique. Currents were elicited by voltage steps to hyperpolarizing potentials ranging from −70 to −140 mV, in −10-mV increments. Fig. 1A depicts representative recordings of HCN2 currents before (black) and during (red) application of 30 μm fisetin. The conductance-voltage relationship was measured from tail currents recorded at −40 mV. In 13 separate experiments, 30 μm fisetin significantly shifted the V½ of the conductance-voltage relationship from −126.8 ± 1.6 mV to −117.9 ± 1.6 mV (p < 0.01, Student's paired t test), reflecting a facilitation of channel opening (Fig. 1D). To study HCN2 kinetics, the rate of activation was quantified by fitting single exponentials to the slow component of the currents evoked by steps to −140 mV. 30 μm fisetin sped the time constant of activation (τ) at −140 mV from 1.08 ± 0.08 to 0.73 ± 0.05 s (n = 9) (p = 0.01, Student's paired t test) (Fig. 1D). To determine the concentration dependence of fisetin on HCN2 channels, the change in the V½ of the conductance-voltage relationship was plotted versus fisetin concentration and fit with a Hill equation (Fig. 1C). Fisetin potentiated HCN2 currents with an EC50 of 1.8 ± 0.3 μm and a Hill coefficient of 1.9 ± 0.3 (n = 3). Together, these results indicate that fisetin potently and reversibly potentiated HCN2 channels.
FIGURE 1.
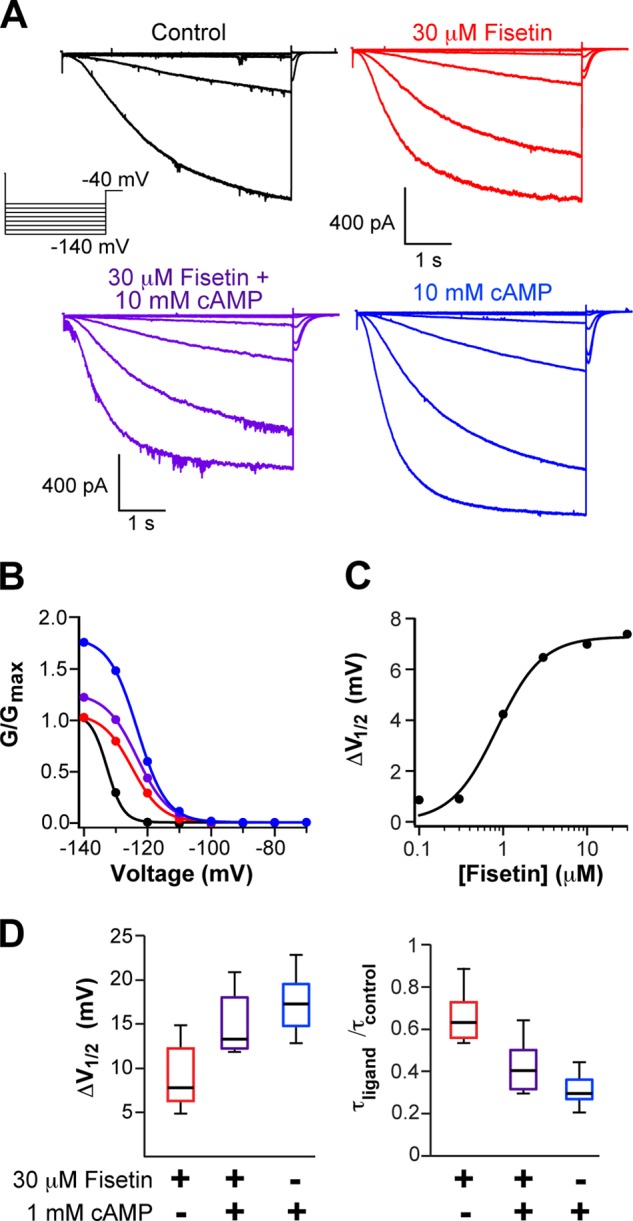
Fisetin potentiated HCN2 and was nonadditive with cAMP. A and B, representative current traces (A) and conductance-voltage relationships (B) of HCN2 channels recoded in the inside-out patch configuration in the following conditions: control (black), with 30 μm fisetin (red), with 10 mm cAMP alone (blue), or with 30 μm fisetin and 10 mm cAMP (purple). C, representative plot of the concentration response of fisetin application to an excised inside-out patch and fit with Equation 2. The EC50 for this patch was 1.8 μm. D, box plot distributions of the change in the V½ of the conductance-voltage relationship and the -fold change in the time constant for the rate of activation at −140 mV (n = 8–13).
Fisetin and cAMP Had Nonadditive Effects on HCN2 Currents
Fisetin potentiation of HCN2 channels resembled the channel's regulation by cAMP, thereby suggesting that fisetin and cAMP may use similar mechanisms to stabilize the channel's open state. We reasoned that if the regulators share a common mechanism, they may have nonadditive effects. Conversely, if fisetin and cAMP act independently to promote channel opening, they would have additive effects on the current. To discern between these two possibilities, we applied 0 or 30 μm fisetin with 0 or 10 mm cAMP to inside-out patches expressing HCN2 channels and recorded their hyperpolarization-evoked currents (Fig. 1A). Each channel regulator shifted the V½ of the conductance-voltage relationship; the V½ was shifted by 8.9 ± 0.9 mV for fisetin and by 17.5 ± 1.1 mV for cAMP (Fig. 1, B and D). Together, however, fisetin and cAMP yielded an intermediate shift of 14.9 ± 1.2 mV, n = 8, which was not significantly different from the shift evoked by cAMP alone (p = 0.33, Student's paired t test) (Fig. 1, B and D). Similarly, fisetin and cAMP individually sped the rate of activation at −140 mV by 1.5-fold for fisetin, to 0.73 ± 0.05 s, and by 2.8-fold for cAMP, to 0.38 ± 0.07 s, n = 7–9 (Fig. 1, B and D). When applied together, however, fisetin and cAMP again produced an intermediate acceleration of the rate of activation by 2.2-fold, to 0.49 ± 0.08 s (Fig. 1D). Again, this intermediate acceleration was not significantly different from the cAMP-evoked acceleration of channel activation (p = 0.31, Student's paired t test). Combined, these data indicate that fisetin and cAMP had nonadditive effects on HCN2 channels. These findings are consistent with the hypothesis that cAMP and fisetin potentiate HCN2 channel gating through a common mechanism. Interestingly, fisetin inhibited the current in the presence of 10 mm cAMP (Fig. 1, A and B), suggesting that fisetin inhibition results from binding to a second site that is noncompetitive with cAMP.
Fisetin Did Not Potentiate HCN2 Channels Lacking Their CNBD
One possible explanation for the nonadditive potentiation of HCN2 by fisetin and cAMP is that fisetin acts via the CNBD. To directly test this hypothesis, we asked whether the flavonoid could potentiate HCN2 channels lacking their CNBD (HCN2V526stop). This previously characterized channel is truncated in the carboxyl-terminal region immediately before the CNBD and is not regulated by the direct binding of cAMP (15). Fig. 2A shows representative recordings from the HCN2V526stop channels before, during, and after washout of 30 μm fisetin. Fisetin application did not shift the V½ of the conductance-voltage relationship, which was −120.6 ± 1.1 mV without and −120.0 ± 2.0 mV with 30 μm fisetin (n = 6) (p = 0.78, Student's paired t test) (Fig. 2C). Furthermore, fisetin did not speed the rate of activation of HCN2V526stop at −130 mV (Fig. 2C), which had a time constant of 1.41 ± 0.53 s in the absence of and 1.43 ± 0.50 s in the presence of 30 μm fisetin (n = 6) (p = 0.24, Student's paired t test). These data demonstrate that the CNBD is required for fisetin potentiation of HCN2 channels.
FIGURE 2.

Fisetin did not potentiate HCN2 channels lacking their CNBD. A and B, representative current traces (A) and conductance-voltage relationships (B) of HCN2V526stop channels recorded in the inside-out patch clamp configuration before (black), with 30 μm fisetin (red), or after washout (gray). C, scatter plots of V½ values of the conductance-voltage relationship or the time constants of activation at −130 mV, with 30 μm fisetin (n = 6).
The HCN2V526stop currents, however, were markedly reduced in the presence of fisetin (Fig. 2A). Specifically, 30 μm fisetin reduced the maximum current at −130 mV by an average of 45.6 ± 3.7%, n = 6. This inhibition was reminiscent of the inhibition seen in wild-type HCN2 channels in the presence of cAMP (Fig. 2, A and B). We hypothesize that, in addition to potentiation mediated by the CNBD, fisetin can also act as inhibitor or blocker at a site outside the CNBD.
Fisetin Bound to Purified HCN2 CNBD
The inability of fisetin to potentiate HCN2V526stop indicated that the CNBD is required for the fisetin regulation of gating. Experiments on this mutant channel, however, did not provide information on the location of the fisetin-binding site. To explore the possibility that fisetin binds to the CNBD, we employed two fluorescence-based binding assays. The first binding assay was based on FRET, which reports the proximity of two fluorophores and is a reliable indicator of molecular interactions (30). Fisetin is a fluorescent flavonoid whose absorbance spectrum overlaps with the emission spectrum of tryptophan (16), indicating that FRET between these two fluorophores could report binding of the flavonoid to a tryptophan-containing protein. However, the HCN2 C-linker/CNBD has no endogenous tryptophan residues. Previously, we engineered an HCN2 construct containing a tryptophan in the cAMP-binding pocket and used this protein to report cAMP binding to the isolated CNBD (HCN2C-LINKER/CNBD-L586W) (21). Here, the tryptophan fluorescence of the purified HCN2C-LINKER/CNBD-L586W protein was recorded in the absence (black trace) and presence of 100 μm fisetin (red trace) (Fig. 3A). With fisetin, the tryptophan fluorescence intensity of HCN2C-linker/CNBD-L586W at 348 nm decreased by ∼20%, thereby reporting that fisetin bound to HCN2C-LINKER/CNBD-L586W. Consistent with tryptophan-to-fisetin FRET, we also found that the fluorescence intensity of fisetin at 500 nm greatly increased (Fig. 3A). By contrast, 100 μm fisetin had nominal effects on the fluorescence of free tryptophan (Fig. 3B), indicating that changes in tryptophan fluorescence were induced by fisetin binding and were not the result of nonspecific interactions.
FIGURE 3.
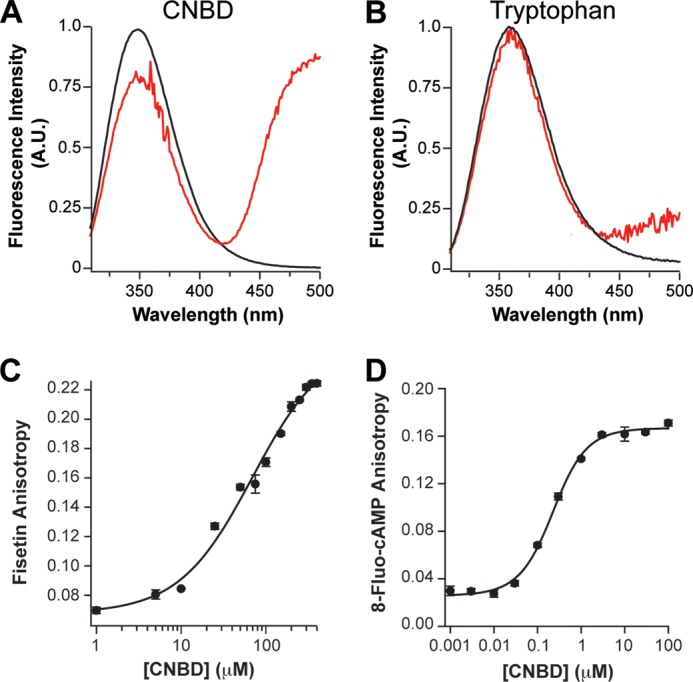
Fisetin bound to the purified HCN2 CNBD. A and B, the inner filter-corrected and background-subtracted emission spectra of HCN2C-LINKER/CNBD-L586W, recorded in the absence (black) or presence (red) of 100 μm fisetin (A) and of 4 μm free tryptophan in solution recorded in the absence (black) or presence 100 μm fisetin (red) (B). A. U., absorbance units. C and D, fluorescence emission spectra were normalized to the maximum fluorescence in the absence of fisetin. The averaged fluorescence anisotropy of fisetin (C) or 8-fluo-cAMP (D) plotted versus total concentration of purified HCN2CNBD (n = 3). Error bars represent the S.E. These data were fit with Equation 4 to give an apparent binding affinity of 63 μm for fisetin, and 176 nm for 8-fluo-cAMP.
In addition to FRET, we used a second binding assay that measured changes in fluorescence anisotropy of the fisetin. Fluorescence anisotropy reports the tumbling rate of a fluorescent molecule in solution. When a small fluorophore binds a much larger protein, the fluorophore will have a slower rate of tumbling, measured as increased fluorescence anisotropy. Being both small and fluorescent, fisetin is an ideal candidate for a fluorescence anisotropy-based binding assay (16). The fluorescence anisotropy of 5 μm fisetin was measured in the presence of various concentrations of purified HCN2CNBD, ranging from 1 to 400 μm. As expected for a ligand-receptor interaction, the fluorescence anisotropy of fisetin increased as the concentration of HCN2CNBD increased. The steady-state fluorescence anisotropy was plotted against the total protein concentration and fit with Equation 4 (see “Experimental Procedures”) (Fig. 3C). From this calculation, we found that fisetin bound the purified HCN2CNBD with an apparent affinity of 63 μm (n = 3). Together results from these binding experiments indicate that fisetin binds to the CNBD and are consistent with the hypothesis that fisetin binding to the CNBD mediates the potentiation of HCN2 channels.
To determine the affinity of the CNBD for cAMP, we measured the anisotropy of the fluorescent cyclic nucleotide 8-fluo-cAMP with varying concentrations of HCN2CNBD ranging from 1 nm to 100 μm (Fig. 3D). Fitting a plot of the steady-state anisotropy versus the HCN2CNBD concentration with Equation 4 yielded an apparent affinity of 176 nm, consistent with previous measurements (12, 13, 33, 34).
Fisetin and cAMP Partially Competed for CNBD Binding
The data thus far indicate that cAMP and fisetin both bind to the CNBD to potentiate HCN2 channel gating. We next sought to determine whether fisetin competes with cAMP for binding to the purified CNBD. To do so, we used fluorescence anisotropy to determine whether cAMP altered the affinity of fisetins for the CNBD. The fluorescence anisotropy of 5 μm fisetin was measured with 1 mm cAMP and varying concentrations of the HCN2CNBD ranging from 1 to 400 μm (Fig. 4B, closed circles). Fitting these data with Equation 4 yielded a dissociation constant of 210 μm (not shown): an over 3-fold increase from the 63 μm Kd determined in the absence of cAMP (Figs. 3C and 4B, open circles). These data indicate that the binding of cAMP inhibits the binding of fisetin, suggesting competition between the two agonists.
FIGURE 4.
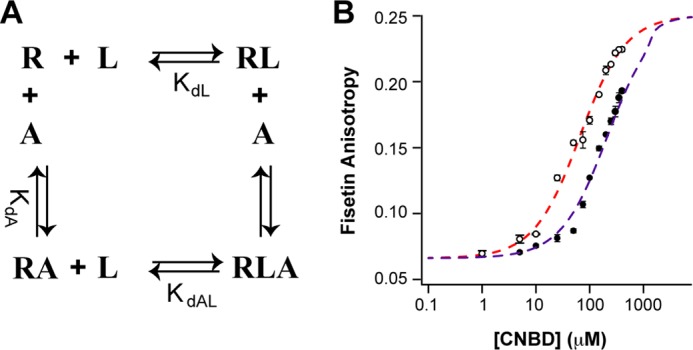
Fisetin and cAMP partially competed for binding to the HCN2 CNBD. A, a four-state model for partial competition between binding of fisetin (L) and cAMP (A) to the HCN2CNBD (R). B, the averaged fluorescence anisotropy of 5 μm fisetin measured in the presence of 0 (empty circles) or 1 mm cAMP (solid circles) and plotted against the total concentration of purified HCN2CNBD. The dashed lines are fits of the model in A for 0 (red) or 1 mm (purple) cAMP. The parameters used in the model fits are as follows: LT = 5 μm, AT = 0 or 1 mm, KdL = 63 μm, KdA = 176 nm, KdAL = 210 μm.
Although the affinity was decreased, apparently fisetin was still able to bind the CNBD even in the presence of 1 mm cAMP (about 1000 times the affinity for cAMP (Fig. 3D) (12, 13, 33, 34)). Two possible mechanisms could explain fisetin binding to the CNBD in the presence of cAMP. The cAMP could bind the CNBD with a similar or lower affinity than fisetin. We believe that this is not the case as we have found the affinity of cAMP for the isolated CNBD to be 176 nm. Alternatively, the finding that fisetin may bind the CNBD even in the presence of saturating cAMP could indicate that both regulators bind the protein simultaneously. One way they could both bind is shown in Fig. 4A. This four-state model assumes only partial competition between cAMP and fisetin binding to the CNBD. Although both cAMP and fisetin can bind simultaneously, the binding of fisetin is lower affinity when cAMP is bound (and likewise the binding of cAMP is lower affinity when fisetin is bound). Using the apparent binding affinities of fisetin for the CNBD determined by fluorescence anisotropy (63 μm in the absence of cAMP, and 210 μm with 1 mm cAMP), and a Kd of 176 nm for cAMP, the partial competition model can explain the decreased binding affinity of fisetin in the presence of cAMP (Fig. 4B).
Fisetin and cAMP Bound to the Same Site
Our data indicate that fisetin and cAMP partially compete for binding to the CNBD. This partial competition could result either directly from fisetin and cAMP binding to overlapping sites or indirectly from cAMP inducing a conformational change in the fisetin-binding site. To determine whether the two regulators bind overlapping sites, we used 1H,13C HSQC NMR spectroscopy to monitor and compare the binding sites for the two ligands. Spectra derived from these two-dimensional experiments contain a peak for each carbon-hydrogen bonded pair in the protein. The peaks have predictable properties depending on the bond pair they originate from (35). We found that at equimolar concentrations of the protein and ligands, cAMP and fisetin induced overlapping chemical shift perturbations in the HCN2CNBD spectrum (Fig. 5A). When compared with cAMP, fisetin evoked smaller perturbations, which was expected given the lower affinity of fisetin and the differences between the chemical structures of the two compounds.
FIGURE 5.

Fisetin bound within the cAMP-binding pocket. A, representative 13C HSQC spectra of methyl groups in HCN2CNBD. Unbound HCN2CNBD is shown in black (both panels). HCN2CNBD after the addition of 175 μm fisetin is shown in red (top panel). Shown in green is HCN2CNBD after the addition of 175 μm cAMP (bottom panel). B, selected Met-ϵ resonances from the previous panel indicate similar perturbations from the addition of fisetin (top panel) and cAMP (middle panel). The structure of HCN2C-linker/CNBD with cAMP (3BPZ) and Met-572 is highlighted. C, similar to A but with HCN2CNBD-M572T. D, the top panel is similar to B but with HCN2CNBD-M572T. The bottom panel contains a structural overlay of HCN2 (lighter gray, red methionine) and HCN4 (darker gray, green threonine) indicating similarities in the binding pockets and position of the affected methionine residue.
Taking advantage of the fact that methyl groups from methionines have resonances in a region that is distinct in a 1H,13C HSQC spectrum, we could monitor methionine resonances without having to assign the spectrum. The HCN2CNBD protein contains six methionines, including a single methionine residue (Met-572) positioned at the entrance of the cAMP-binding pocket (Fig. 5B). Because of its proximity to and hydrophobic interaction with the cAMP purine ring (14, 36), we predicted that the Met-572 methyl resonance would be perturbed by the presence of a ligand in the binding pocket. Indeed, we found that cAMP caused a significant chemical shift in one resonance within the methionine region of the spectrum. Notably, this resonance was completely absent in the spectra of HCN2CNBD with the M572T mutation, which was previously shown not to alter cAMP affinity (Fig. 5D) (13). Otherwise, the spectra of M572T HCN2CNBD in the absence and presence of cAMP were nearly identical to those of wild-type HCN2CNBD (Fig. 5C). These data indicate that cAMP interacts with Met-572 as predicted by the x-ray crystal structure (14).
Remarkably, the Met-572 resonance was also shifted with fisetin (Fig. 5B). The smaller chemical shift perturbation observed for fisetin is consistent with the lower affinity of the CNBD for fisetin when compared with cAMP. The behavior of Met-572 in these titration experiments indicates that cAMP and fisetin both interact with Met-572 and therefore occupy partially overlapping sites within the known binding pocket for cAMP. Thus, we conclude that fisetin binds within or proximal to the cAMP-binding pocket.
DISCUSSION
Here, we report that the flavonoid fisetin regulates HCN2 channels. Like cyclic nucleotides, fisetin speeds channel activation and shifts their voltage dependence toward more depolarizing potentials. Fisetin bound to the purified HCN2CNBD and did not potentiate channels lacking this domain. We also show that fisetin partially competes with cAMP for binding to the cAMP-binding pocket and use NMR spectroscopy to show that that fisetin binds within the cAMP-binding pocket. These results suggest that fisetin binds to the CNBD and potentiates activation, similar to cAMP. Deletion of the CNBD, however, revealed that in addition to potentiating channel gating, fisetin also inhibits or blocks HCN channels at a site distinct from the CNBD.
Fisetin acts as a partial agonist for HCN2 channels. Partial agonists have the ability to bind to and activate a receptor, but are unable to elicit the full response. Like cAMP, fisetin shifts the conductance-voltage relationship toward more depolarizing potentials and speeds channel activation, but fisetin elicits only half of the shift of cAMP in the V½ or speeding of the rate of activation. We hypothesize that this difference in potentiation is due to the fact that fisetin does not interact with all of the HCN2 residues that coordinate cAMP. Therefore, the energy of fisetin/CNBD binding may not be sufficient to fully evoke the change in conformation evoked by cAMP binding to the same site. Alternatively, the inability of fisetin to elicit a complete potentiation of HCN2 currents could be explained by fisetin binding to both the cAMP pocket to fully potentiate and a secondary site on the channel to inhibit the current. Indeed, fisetin inhibits wild-type HCN2 channels in the presence of saturating cAMP and mutant HCN2V526stop channels, thereby suggesting that fisetin is able to bind a second site on the channel, outside the CNBD, to reduce the current. However, the fisetin-induced reduction in HCN2V526stop current was not accompanied by any shift in the conductance-voltage relationship nor a change in the rate of channel activation as seen in the wild-type channel. Thus, this inhibition would not explain the difference in the shift of the conductance-voltage relationship or rate of activation of the wild-type channel elicited by fisetin when compared with cAMP. Rather, it seems likely that fisetin acts as a true partial agonist on HCN2 channels.
A fluorescence anisotropy-based binding assay reported that fisetin bound to the CNBD in a concentration-responsive manner. However, we observed a significant disparity in the affinity measured by fluorescence anisotropy, 63 μm (Fig. 3C), when compared with the potency determined by electrophysiology, 1.8 μm (Fig. 1D). This discrepancy between affinity and potency could indicate that the binding site of the purified CNBD is not the binding site that potentiates channel gating. However, it seems more likely that the higher potency of fisetin on the intact channel reflects gating and cooperativity in the intact tetrameric channel not present in the isolated monomeric CNBD (37). Indeed, similar discrepancies have been reported for the affinity of cAMP. The affinity of cAMP for the isolated HCN2 C-linker/CNBD has been measured by various methods including fluorescence anisotropy with fluorescent cAMP analogs and isothermal titration calorimetry, with Kd values ranging from 100 nm to 3 μm (12, 13, 33, 34). Here, we report the Kd of the HCN2CNBD for 8-fluo-cAMP to be 176 nm. This affinity of cAMP for the isolated C-linker/CNBD is ∼1–30-fold lower than the 100 nm EC50 determined with electrophysiology (13, 33, 34, 38, 39). Therefore, despite differences in affinity and potency, the data presented here implicate the CNBD as the fisetin-binding site for HCN2 potentiation.
Our data suggest that the binding sites in the CNBD for fisetin and cAMP partially overlap. Therefore, we were surprised to detect fisetin binding to the CNBD in the presence of a saturating concentration of cAMP (Fig. 4B). We expected that in the presence of 1 mm cAMP (about 1000× the Kd) and 5 μm fisetin, there would not be any fisetin binding to the purified CNBD. The finding that fisetin still bound the CNBD even in the presence of saturating cAMP (albeit with a lower affinity) could suggest that fisetin binds to a secondary binding site, distinct from the pocket where cAMP binds. However, there were virtually no resonances in the NMR spectra that were perturbed by fisetin that were not also perturbed by cAMP. Thus, we favor an alternative hypothesis that the cAMP-binding pocket can simultaneously accommodate both ligands. Because fisetin interacts with M572, we predict that fisetin interacts with the HCN2 residues that typically interact with the purine ring of cAMP. With fisetin occupying the purine portion of the binding site, the cyclic phosphate portion may still interact with the ribose and cyclic phosphate portion of cAMP.
Fisetin is not the first plant compound shown to potentiate HCN2 channels. Another abundant plant metabolite, genistein, regulates HCN2 (40). A well known inhibitor of tyrosine kinase (41), genistein is an isoflavone and accordingly has a phenyl group substituted at position 3 of the pyrone ring instead of position 2 in flavonoids (42). Like fisetin, genistein shifts the V½ of the conductance-voltage relationship of HCN2 to more depolarizing potentials. Genistein, however, potentiated HCN2V526stop channels, whereas fisetin did not. Because genistein did not require the CNBD to regulate HCN2 channels, we predict that it acts via a distinct mechanism.
To our knowledge, this is the first report of flavonoid binding to a cyclic nucleotide-binding pocket. However, this is not the first instance of flavonoids interacting with proteins that bind purine ring-containing second messengers. Several flavonoids have been identified as competitive inhibitors for ATP- and adenosine-binding sites (42, 43). For example, many of the cyclin-dependent kinases (CDKs) are inhibited by flavonoids, and CDK6 has been crystallized bound to fisetin (44). Comparing the fisetin-occupied ATP-binding site of CDK6 with the ATP-occupied site of the closely related CDK2 reveals that fisetin interacts with the CDK residues that typically coordinate the purine ring of ATP (44, 45). However, the purine ring-binding sites in the ATP-binding pocket of CDK and the cAMP-binding pocket of HCN are not conserved. We predict that a conserved binding domain is not required, rather that shared chemical properties between fisetin and the purine rings of cAMP and ATP allow them to bind to the same structures. Accordingly, we predict that fisetin will more broadly bind receptors for other purine ring-containing compounds such as caffeine, NAD+, and/or coenzyme A.
The results presented here characterize fisetin as a novel partial agonist for HCN2 channels. Moreover, these results may shed light on the physiologic targets of flavonoids in general and fisetin in particular. Flavonoids are secondary plant metabolites synthesized by all vascular plants (42). A flavonoid-rich diet has been correlated with several measures of health including improved learning and memory (46, 47), protection against Alzheimer and Parkinson diseases (48, 49), reduced mortality from coronary heart disease (50), and a reduced risk of cancer (51). Although the molecular mechanisms that account for many of these physiologic effects has yet to be determined, HCN2 channels should be included in the growing list of potential targets of flavonoids.
Acknowledgments
We thank S. Camp and S. Cunnington for excellent technical assistance. We also thank the members of the Zagotta and Klevit laboratories for helpful discussions and T. Aman, J. R. Bankston, H. A. DeBerg, Y. Haitin, and M. C. Puljung for critical review of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 EY010329 (to W. N. Z.), F32 HL095241 (to A. E. C.), 5T32CA080416-14 (to J. C. R.), and R01 EY017370 (to R. E. K.).
- HCN
- hyperpolarization-activated, cyclic nucleotide-modulated
- CNBD
- cyclic nucleotide-binding domain
- CNBHD
- cyclic nucleotide-binding homology domain
- 8-fluo-cAMP
- 8-[[2-[(fluoresceinylthioureido)amino]ethyl]thio]adenosine-3′,5′-cyclic monophosphate sodium salt
- HSQC
- heteronuclear single quantum correlation
- CDK
- cyclin-dependent kinase.
REFERENCES
- 1. Biel M., Wahl-Schott C., Michalakis S., Zong X. (2009) Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89, 847–885 [DOI] [PubMed] [Google Scholar]
- 2. Fan Y., Fricker D., Brager D. H., Chen X., Lu H. C., Chitwood R. A., Johnston D. (2005) Activity-dependent decrease of excitability in rat hippocampal neurons through increases in Ih. Nat. Neurosci. 8, 1542–1551 [DOI] [PubMed] [Google Scholar]
- 3. Emery E. C., Young G. T., Berrocoso E. M., Chen L., McNaughton P. A. (2011) HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 333, 1462–1466 [DOI] [PubMed] [Google Scholar]
- 4. DiFrancesco J. C., Barbuti A., Milanesi R., Coco S., Bucchi A., Bottelli G., Ferrarese C., Franceschetti S., Terragni B., Baruscotti M., DiFrancesco D. (2011) Recessive loss-of-function mutation in the pacemaker HCN2 channel causing increased neuronal excitability in a patient with idiopathic generalized epilepsy. J. Neurosci. 31, 17327–17337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lewis A. S., Chetkovich D. M. (2011) HCN channels in behavior and neurological disease: too hyper or not active enough? Mol. Cell. Neurosci. 46, 357–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Good C. H., Hoffman A. F., Hoffer B. J., Chefer V. I., Shippenberg T. S., Bäckman C. M., Larsson N. G., Olson L., Gellhaar S., Galter D., Lupica C. R. (2011) Impaired nigrostriatal function precedes behavioral deficits in a genetic mitochondrial model of Parkinson's disease. FASEB J. 25, 1333–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Robinson R. B., Siegelbaum S. A. (2003) Hyperpolarization-activated cation currents: from molecules to physiological function. Annu. Rev. Physiol. 65, 453–480 [DOI] [PubMed] [Google Scholar]
- 8. Ludwig A., Zong X., Jeglitsch M., Hofmann F., Biel M. (1998) A family of hyperpolarization-activated mammalian cation channels. Nature 393, 587–591 [DOI] [PubMed] [Google Scholar]
- 9. Santoro B., Liu D. T., Yao H., Bartsch D., Kandel E. R., Siegelbaum S. A., Tibbs G. R. (1998) Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell 93, 717–729 [DOI] [PubMed] [Google Scholar]
- 10. Craven K. B., Zagotta W. N. (2006) CNG and HCN channels: two peas, one pod. Annu. Rev. Physiol. 68, 375–401 [DOI] [PubMed] [Google Scholar]
- 11. Flynn G. E., Black K. D., Islas L. D., Sankaran B., Zagotta W. N. (2007) Structure and rearrangements in the carboxy-terminal region of SpIH channels. Structure 15, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lolicato M., Nardini M., Gazzarrini S., Möller S., Bertinetti D., Herberg F. W., Bolognesi M., Martin H., Fasolini M., Bertrand J. A., Arrigoni C., Thiel G., Moroni A. (2011) Tetramerization dynamics of C-terminal domain underlies isoform-specific cAMP gating in hyperpolarization-activated cyclic nucleotide-gated channels. J. Biol. Chem. 286, 44811–44820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu X., Vysotskaya Z. V., Liu Q., Zhou L. (2010) Structural basis for the cAMP-dependent gating in the human HCN4 channel. J. Biol. Chem. 285, 37082–37091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zagotta W. N., Olivier N. B., Black K. D., Young E. C., Olson R., Gouaux E. (2003) Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 425, 200–205 [DOI] [PubMed] [Google Scholar]
- 15. Wainger B. J., DeGennaro M., Santoro B., Siegelbaum S. A., Tibbs G. R. (2001) Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411, 805–810 [DOI] [PubMed] [Google Scholar]
- 16. Carlson A. E., Brelidze T. I., Zagotta W. N. (2013) Flavonoid regulation of EAG1 channels. J. Gen. Physiol. 141, 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warmke J. W., Ganetzky B. (1994) A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. U.S.A. 91, 3438–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brelidze T. I., Carlson A. E., Sankaran B., Zagotta W. N. (2012) Structure of the carboxy-terminal region of a KCNH channel. Nature 481, 530–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brelidze T. I., Gianulis E. C., DiMaio F., Trudeau M. C., Zagotta W. N. (2013) Structure of the C-terminal region of an ERG channel and functional implications. Proc. Natl. Acad. Sci. U.S.A. 110, 11648–11653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marques-Carvalho M. J., Sahoo N., Muskett F. W., Vieira-Pires R. S., Gabant G., Cadene M., Schönherr R., Morais-Cabral J. H. (2012) Structural, biochemical, and functional characterization of the cyclic nucleotide binding homology domain from the mouse EAG1 potassium channel. J. Mol. Biol. 423, 34–46 [DOI] [PubMed] [Google Scholar]
- 21. Brelidze T. I., Carlson A. E., Zagotta W. N. (2009) Absence of direct cyclic nucleotide modulation of mEAG1 and hERG1 channels revealed with fluorescence and electrophysiological methods. J. Biol. Chem. 284, 27989–27997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brelidze T. I., Carlson A. E., Davies D. R., Stewart L. J., Zagotta W. N. (2010) Identifying regulators for EAG1 channels with a novel electrophysiology and tryptophan fluorescence based screen. PloS One 5, e12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zagotta W. N., Hoshi T., Aldrich R. W. (1989) Gating of single Shaker potassium channels in Drosophila muscle and in Xenopus oocytes injected with Shaker mRNA. Proc. Natl. Acad. Sci. U.S.A. 86, 7243–7247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391, 85–100 [DOI] [PubMed] [Google Scholar]
- 25. Pian P., Bucchi A., Robinson R. B., Siegelbaum S. A. (2006) Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J. Gen. Physiol. 128, 593–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zolles G., Klöcker N., Wenzel D., Weisser-Thomas J., Fleischmann B. K., Roeper J., Fakler B. (2006) Pacemaking by HCN channels requires interaction with phosphoinositides. Neuron 52, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 27. Chen G. Q., Gouaux E. (1997) Reduction of membrane protein hydrophobicity by site-directed mutagenesis: introduction of multiple polar residues in helix D of bacteriorhodopsin. Protein Eng. 10, 1061–1066 [DOI] [PubMed] [Google Scholar]
- 28. Rossi A. M., Taylor C. W. (2011) Analysis of protein-ligand interactions by fluorescence polarization. Nat. Protoc. 6, 365–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cukkemane A., Grüter B., Novak K., Gensch T., Bönigk W., Gerharz T., Kaupp U. B., Seifert R. (2007) Subunits act independently in a cyclic nucleotide-activated K+ channel. EMBO Rep. 8, 749–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, Third Ed., pp. 56 and 550, Springer, New York, NY [Google Scholar]
- 31. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 32. Johnson B. A., Blevins R. A. (1994) NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 33. Chow S. S., Van Petegem F., Accili E. A. (2012) Energetics of cyclic AMP binding to HCN channel C terminus reveal negative cooperativity. J. Biol. Chem. 287, 600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu S., Vysotskaya Z. V., Xu X., Xie C., Liu Q., Zhou L. (2011) State-dependent cAMP binding to functioning HCN channels studied by patch-clamp fluorometry. Biophys J. 100, 1226–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cavanagh J., Fairbrother W. J., Palmer A. G., III, Skelton N. J., Rance M. (2005) Protein NMR Spectroscopy: Principles and Practice, 2nd Ed., pp. 540, Elsevier Academic Press, Burlington, MA [Google Scholar]
- 36. Craven K. B., Olivier N. B., Zagotta W. N. (2008) C-terminal movement during gating in cyclic nucleotide-modulated channels. J. Biol. Chem. 283, 14728–14738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Colquhoun D. (1998) Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 125, 924–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang J., Chen S., Siegelbaum S. A. (2001) Regulation of hyperpolarization-activated HCN channel gating and cAMP modulation due to interactions of COOH terminus and core transmembrane regions. J. Gen. Physiol. 118, 237–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou L., Siegelbaum S. A. (2007) Gating of HCN channels by cyclic nucleotides: residue contacts that underlie ligand binding, selectivity, and efficacy. Structure 15, 655–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rozario A. O., Turbendian H. K., Fogle K. J., Olivier N. B., Tibbs G. R. (2009) Voltage-dependent opening of HCN channels: Facilitation or inhibition by the phytoestrogen, genistein, is determined by the activation status of the cyclic nucleotide gating ring. Biochim. Biophys. Acta 1788, 1939–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Akiyama T., Ishida J., Nakagawa S., Ogawara H., Watanabe S., Itoh N., Shibuya M., Fukami Y. (1987) Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 262, 5592–5595 [PubMed] [Google Scholar]
- 42. Middleton E., Jr., Kandaswami C., Theoharides T. C. (2000) The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 52, 673–751 [PubMed] [Google Scholar]
- 43. Jacobson K. A., Moro S., Manthey J. A., West P. L., Ji X. D. (2002) Interactions of flavones and other phytochemicals with adenosine receptors. Adv. Exp. Med. Biol. 505, 163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu H., Chang D. J., Baratte B., Meijer L., Schulze-Gahmen U. (2005) Crystal structure of a human cyclin-dependent kinase 6 complex with a flavonol inhibitor, fisetin. J. Med. Chem. 48, 737–743 [DOI] [PubMed] [Google Scholar]
- 45. Echalier A., Cot E., Camasses A., Hodimont E., Hoh F., Jay P., Sheinerman F., Krasinska L., Fisher D. (2012) An integrated chemical biology approach provides insight into Cdk2 functional redundancy and inhibitor sensitivity. Chem. Biol. 19, 1028–1040 [DOI] [PubMed] [Google Scholar]
- 46. Galli R. L., Shukitt-Hale B., Youdim K. A., Joseph J. A. (2002) Fruit polyphenolics and brain aging: nutritional interventions targeting age-related neuronal and behavioral deficits. Ann. N.Y. Acad. Sci. 959, 128–132 [DOI] [PubMed] [Google Scholar]
- 47. Haque A. M., Hashimoto M., Katakura M., Tanabe Y., Hara Y., Shido O. (2006) Long-term administration of green tea catechins improves spatial cognition learning ability in rats. J. Nutr. 136, 1043–1047 [DOI] [PubMed] [Google Scholar]
- 48. Inanami O., Watanabe Y., Syuto B., Nakano M., Tsuji M., Kuwabara M. (1998) Oral administration of (−)catechin protects against ischemia-reperfusion-induced neuronal death in the gerbil. Free Radic. Res. 29, 359–365 [DOI] [PubMed] [Google Scholar]
- 49. Spencer J. P. (2008) Flavonoids: modulators of brain function? Br. J. Nutr. 99, E Suppl. 1, ES60–ES77 [DOI] [PubMed] [Google Scholar]
- 50. Hertog M. G., Feskens E. J., Hollman P. C., Katan M. B., Kromhout D. (1993) Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet 342, 1007–1011 [DOI] [PubMed] [Google Scholar]
- 51. Cui Y., Morgenstern H., Greenland S., Tashkin D. P., Mao J. T., Cai L., Cozen W., Mack T. M., Lu Q. Y., Zhang Z. F. (2008) Dietary flavonoid intake and lung cancer–a population-based case-control study. Cancer 112, 2241–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]