

Abstract
Anti-apoptotic Bcl-2 family proteins are important oncology therapeutic targets. To date, BH3 mimetics that abrogate anti-apoptotic activity have largely been directed at Bcl-2 and/or Bcl-xL. One observed mechanism of resistance to these inhibitors is increased Mcl-1 levels in cells exposed to such therapeutics. For this reason, and because Mcl-1 is important in the onset of lymphoid, myeloid, and other cancers, it has become a target of great interest. However, small molecule inhibitors displaying potency and selectivity for Mcl-1 are lacking. Identifying such compounds has been challenging due to difficulties in translating the target selectivity observed at the biochemical level to the cellular level. Herein we report the results of an HTS strategy coupled with directed hit optimization. Compounds identified have selective Mcl-1 inhibitory activity with greater than 100-fold reduced affinity for Bcl-xL. The selectivity of these compounds at the cellular level was validated using BH3 profiling, a novel personalized diagnostic approach. This assay provides an important functional biomarker that allows for the characterization of cells based upon their dependencies on various anti-apoptotic Bcl-2 proteins. We demonstrate that cells dependent on Mcl-1 or Bcl-2/Bcl-xL for survival are commensurately responsive to compounds that genuinely target those proteins. The identification of compound 9 with uniquely validated and selective Mcl-1 inhibitory activity provides a valuable tool to those studying the intrinsic apoptosis pathway and highlights an important approach in the development of a first-in-class cancer therapeutic.
1. Introduction
Modulation of apoptosis has long been of interest to the oncology community, as a primary mechanism of cancer cell survival by evading programmed cell death.1 Bcl-2 family proteins are central to the regulation of the intrinsic, or mitochondrial, apoptosis pathway2,3 as family members interactions result in heterodimer formation that modulates the activity of the multidomain pro-apoptotic proteins Bax and Bak.4 Oligomerization of Bax and Bak results in mitochondrial outer membrane permeabilization (MOMP) and release of apoptosis-promoting proteins, including Smac/DIABLO and cytochrome c, which in turn promote caspase activation and result in cell death.5 Myeloid cell factor 1 (Mcl-1) has been identified as an important therapeutic target for the treatment of non-solid tumor6,7,8,9,10,11,12,13 as well as solid tumor malignancies14,15,16 largely owing to its role as a critical node in intrinsic apoptotic susceptibility.17 Recently, a study of mutation analyses from 3,131 cancer specimens identified mutations surrounding Mcl-1 as being among the most significant causal factors.18
Inhibition of anti-apoptotic Bcl-2 family proteins has been validated as a therapeutic strategy by the clinical advancement of the Bcl-2 inhibitors Navitoclax19 and ABT-199.20 These small molecules bind to the hydrophobic groove in Bcl-2 and/or Bcl-xl and mimic the pro-apoptotic “BH3-only” proteins, thereby promoting activation of Bax and Bak. Cell lines found to be refractory to these compounds regained sensitivity when Mcl-1 was down-regulated.21,22 These findings strongly support the notion that Mcl-1 is a key resistance factor to Bcl-2/Bcl-xL targeted therapies and underscore the importance of developing an Mcl-1 targeted therapy. Two other purported MCL-1 inhibitors, obatoclax (GX15-070)23 and gossypol (AT-101)24, have each displayed significant off-target activities suggesting that their efficacy is largely not derived from Mcl-1 inhibition but rather from cytotoxicity in a Bax-Bak independent fashion and induced caspase-9 independent cell death.25,26 Further, inhibition of certain Bcl-2 family proteins can display adverse clinical consequences. For instance, thrombocytopenia has been observed following treatment with the Bcl-2/Bcl-xL inhibitor Navitoclax, halting its clinical development.27 In that case, the activity against Bcl-xL impacted platelet survival.28 Recent efforts have focused on development of selective compounds with diminished Bcl-xL activity, such as ABT-199, with limited platelet toxicity. Avoiding inhibition of other anti-apoptotic proteins may be preferred in some cases for patients comprising a specific malignant disease. A small molecule inhibitor that is selective for Mcl-1 would provide an important chemical probe to define the therapeutic potential of Mcl-1 inhibition, elucidating the significance of Mcl-1 in cancer and determining if tumor cells characterized by elevated Mcl-1 activity can be selectively targeted.
Efforts to develop effective Mcl-1 inhibitors have been slowed by frequent coincident and pronounced off-target activity. Our strategy of BH3 profiling addresses selectivity by providing a functional biomarker, allowing for identification of the mechanism of action of BH3 mimetics within a cellular context. This approach quantifies mitochondrial response to any one or any class of BH3 peptides and indicates a particular dependence upon an anti-apoptotic Bcl-2 family protein. For example, Noxa binds with high affinity only to Mcl-1, Bad binds to Bcl-xL and Bcl-2 but only weakly to Mcl-1, and Puma binds strongly to all three targets.29 Each cell line may therefore be characterized by its extent of “priming” with respect to a particular Bcl-2 family member, such as Mcl-1, Bcl-2, or Bcl-xL.29
2. Experimental
2.1. High Throughput Screening
A high throughput screen (HTS), fluorescence polarization (FP) assay,30 was performed at Scripps Research Institute Molecular Screening Center (SRIMSC) by peptide binding quantitation. The NIH screening library (315,100 compounds) was provided by the National Institutes of Health. FITC-Bim BH3-only peptide (FITC-AHA-MRPEIWIAQELRRIGDEFNA-[NH2]) was synthesized at the Tufts University Core Facility. Human Mcl-1, and Bcl-xL -GST (Glutathione-S-Transferase) fusion proteins, with deleted transmembrane regions, were cloned into pGEX 4T-1. Proteins were expressed in BL21 strain and purified using Amersham Hitrap Glutathione column on an ACTA-FPLC.
FP was performed in assay buffer (Dulbecco’s PBS buffer, pH 7.2, 0.001% v/v Brij 35) containing either GST-Mcl-1 or GST-Bcl-xL dispensed into 1,536-well microtiter plates (Corning). Test compound, unlabeled Bim control peptide, or DMSO was added to the appropriate test or control wells. Bim-FITC in assay buffer was dispensed into wells, and plates were centrifuged and incubated at 25°C for 20 min. Fluorescence polarization was read by Viewlux (PerkinElmer). Percent inhibition for each compound was calculated using a ratio of the test compound mP to median value of positive control mP, correcting each with the median value of the negative control mP. Following primary screening, potential active compounds were confirmed by repeat of the assay in triplicate at 10 M. The same assays were then performed using a ten-point, three-fold titration with compound concentrations from 100 M to 0.5 nM. The resulting response curves were used to calculate IC50 values for both Mcl-1 and Bcl-xL. Detailed protocols for these assays are found at the NIH’s MLPCN PubChem website (http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=2090).
2.2. Analog Preparation
Compounds were prepared at the Scripps Research Institute, Jupiter, FL or where purchased from commercial vendors. Methods utilized for synthesis are described in the supplementary material and described in detail for compound 9, a simple one-step procedure. Identity and purity of all new compounds were determined by NMR (Bruker Instruments) and LCMS (Agilent); purity was confirmed to be greater than 95% in each case. LCMS, HPLC, and NMR data for novel compound 9 and an analysis of its properties, and general SAR trends in the series are also shown in supplementary material. Vendor information for previously undescribed but commercially available compounds 3–7 are provided as well. Compound 8 was internally synthesized by the methods used for compound 9 and its confirmatory LCMS data is shown in supplementary material. Functional assays of compounds 1–9 were performed using racemic mixtures, therefore stereochemistry has not been indicated at chiral centers.
2.3. Computational Chemistry
Docking was performed using FlexX in LeadIT version 2.1.2 (BiosolveIT, St. Augustin, Germany). Compound structure was drawn in MDL ISIS Draw and minimized coordinates were generated in PipelinePilot. The binding site was prepared using the coordinates of Mcl-1 (PDB 2NLA) and defined as the amino acids within a 20Å radius of Arg263. An essential interaction was defined such that the side-chain amine of Asn260 was required to donate a hydrogen bond to all docked poses. The Hybrid Approach was used for base placement in the docking runs, which generated 200 solutions per iteration and fragmentation. Docked poses were exported in mol2 format for visualization in PyMOL.
2.4. Cell lines and growth inhibition assays
The mouse leukemia derived cell lines Mcl1-1780 and Bcl2-186331 have been licensed from the Dana Farber Cancer Institute. The human lymphoid SUDHL-10, SUDHL-6, myeloid NCI-H929, acute promyelocytic leukemia HL-60, erythroleukemia TF-1, malignant melanoma A-375, carcinoma A-549 and NCI-H187, and pancreactic carcinoma PANC-1 cell lines were obtained from American Type Culture Collection (ATCC), Manassas, VA. Suspension lines SUDHL-6, SUDHL-10, NCI-H929, MOLM13, ML2, HL-60, TF-1, and NCI-H187 were grown in RPMI-1640 with 10% fetal bovine serum (Life Technologies). Suspension lines Mcl1-1780 and Bcl2-1863 were grown in the same media with 3.6µL/L β-mercaptoethanol (BME). Adherent cell lines A-375, A-549 and PANC-1 were grown in DMEM with 10% fetal bovine serum (Life Technologies).
Cells were resuspended at 2.5 × 105 cells/mL and plated on 384-well plates with seven 2-fold dilutions of either DMSO (high concentration: 0.2%) or test compound (high concentration: 5 µM or 20 µM). Control wells of cells and media only were also included. Plates were incubated at 37°C for 48 hours and analyzed using Presto Blue (Invitrogen) on a Tecan Infinite F200 Pro Plate Reader. Background (media alone) was subtracted from all data. Percent viability was normalized to the reading obtained for wells containing only cells (considered 100% viability).
2.5. Analysis of cell lines by BH3 profiling
Peptides (Bim, Puma, Noxa, Bad) were from New England Peptide. Cells were seeded into a 384 well plate in duplicate with 100µM peptide, 2.5 × 104 cells, 1µM JC-1, and 0.005% digitonin,. Fluorescence was by continuous read every 5 min for 180 min on a Tecan Infinite F200 Pro plate reader and data was analyzed using GraphPad Prism (La Jolla, CA) to determine area under the curve (AUC). Percent priming was calculated using the AUC measurements as compared to the positive (carbonyl cyanide m-chlorophenyl hydrazone, CCCP) and negative (DMSO) controls.
2.6. Cytochrome c release determination
Cells were collected, washed in PBS, re-suspended at a concentration of 2 × 106 cells/ml, and incubated with test and control compounds (0.2% DMSO; 20µM EU-5346) or peptides (100µM Bim; 100µM Puma) for 2 hours at room temperature. Samples were fixed with 4% Formaldehyde in PBS for 20 minutes, washed once in PBS, blocked for 15 min with 2% FBS 0.5%TritonX-100 in PBS, then re-suspended in blocking buffer with 1:250 anti-cytochrome c-Alexa488 (Beckton-Dickinson) and incubated for 2 hours at 4°C. Samples were washed once with blocking buffer and mounted on slides using Vectashield Mounting Media with DAPI (Vector laboratories). A minimum of 100 DAPI positive cells per treatment were visualized by fluorescence microscopy and scored as positive or negative for cytochrome c loss. DMSO was calculated as 0% cytochrome c loss; Bim response for SUDHL-6 was used to determine 100% cytochrome c loss.
2.7. Nuclear Apoptosis Determination
Cell lines were seeded at 2 × 105 cells/ml in culture media and incubated with DMSO (0.1%), Navitoclax (5, 2.5, 1.25, and 0.625 µM) or compound 9 (1000, 500, 250, and 125 nM) for 24 hours at 37°C. Following treatment, samples were fixed in 100% methanol on ice for 10 min, washed once with PBS, and mounted on slides using Vectashield/DAPI. A minimum of 100 cells per treatment were visualized by fluorescence microscopy. Nuclei were scored as normal (round, normal sized, intact nuclei) or apoptotic (pyknotic, fragmented). DMSO treatment provided the negative control. Navitoclax at 5 M treatment on cell line Bcl2-1863 provided the positive control.
2.8. Annexin V Apoptosis Assay
Apoptosis was detected by Annexin V and propidium iodide staining. Mcl1-1780 and Bcl2-1863 were seeded at 1.0 × 106 cells/well in a 24 well plate in culture media containing a range of concentrations of compound 9 from 0.15 µM-40 µM and incubated for 16 hours. Control wells containing DMSO were also included. Cells were stained with BD Biosciences (San Jose, CA) FITC Annexin V Apoptosis Detection Kit II according to the manufacturer’s protocol. 1.0 × 105 cells were resuspended in 100ul of 1× Binding buffer along with 5ul of Annexin V FITC and 2ul of propidium iodide. Cells were mixed and incubated for 15 min at room temperature in the dark. 400ul of 1× Binding Buffer was added to each tube and analyzed on a FACSCanto II (BD Biosciences). A minimum of 20,000 cells per sample were analyzed using FACSDiva software (BD Biosciences).
3. Results
3.1. High-throughput screening identified novel, selective Mcl-1 inhibitors
An FP assay provided the primary Mcl-1 biochemical screen. A second FP assay utilizing Bcl-xL provided the counter screen. Both assays were used to screen the NIH Molecular Libraries Small Molecule Repository (MLSMR); 315,000 compounds; 2,141 compounds showing greater than 40% inhibition of Mcl-1 were triaged as possible hits (0.68% rate) Elimination of compounds with considerable Bcl-xL binding reduced the hit set to 1,720 compounds (0.54% rate). Confirmation of both Mcl-1 activity and lack of Bcl-xL activity, resulted in identification of 179 validated hits which were advanced to dose response studies. 52 compounds demonstrated IC50 ≤10 µM against Mcl-1; 24 of these small molecules met selectivity criteria of reduced Bcl-xL inhibitory activity (IC50 ≥10 µM against Bcl-xL).
An assessment of chemical tractability, as well as the potential for target specificity as indicated by their activity in other HTS campaigns, resulted in the selection of the substituted 7-hydroxyquinoline 1 (Figure 1a) for additional study. The favorable physicochemical properties expected for compound 1 (solubility, lipophilicity), its reasonable molecular weight (436), lack of any apparent toxicophores, and low hit rate in other HTS assays made this compound a promising candidate. Compound 1 demonstrated good Mcl-1 inhibition (IC50 = 2.4 µM) with no appreciable inhibition of Bcl-xL at 100 µM.
Figure 1.

(a) Structure of compound 1, a 7-hydroxyquinoline identified by high-throughput screening as possessing Mcl-1-selective inhibitory activity relative to the related Bcl-2 family protein Bcl-xL; (b) Structure of the piperazine-substituted hydroxyquinoline 9, which displays selective inhibition of Mcl-1 relative to Bcl-xL as indicated by fluorescence polarization assay.
Upon structural analysis of compound 1, two functional groups were identified as desirable for elimination or modification; the carboxylic acid moiety, which could contribute to poor cellular permeability, and the 4-chloro group, which typically raises cLogP and reduces aqueous solubility. Examination of a small set of analogs lacking these groups demonstrated that Mcl-1 binding affinity was maintained upon their removal. Therefore, subsequent efforts focused upon the generation of derivatives lacking these moieties.
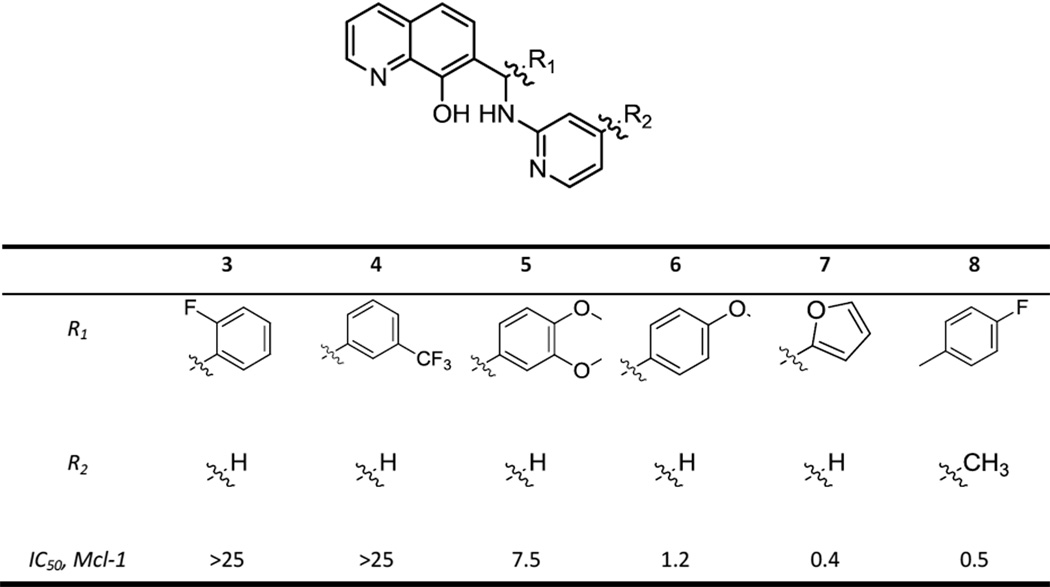
A review of the literature indicated that hydroxyquinoline 2 had previously been disclosed as an Mcl-1 inhibitor, though without a reported IC50 value.32 We explored group and substituent effects in the phenyl region of hit 1 by preparing approximately 100 related analogs. This work resulted in the conclusion that both the quinoline nitrogen atom and the 8-hydroxyl group are essential. Representative compounds that demonstrate key SAR trends are shown in Figures 1a and Table 1. Ortho- and meta- substituted compounds were much weaker Mcl-1 inhibitors, whereas a bis-substituted analog also showed reduced potency (3–5 Table 1). Para-substitution (methoxy derivative 6) and 5-membered heterocycles (furan 7) enhanced potency. An electron-withdrawing F group at the para position (analog 8), which might be expected to enhance metabolic stability, was five-fold more potent than the initial hit.
Table 1.
Structures and Mcl-1 inhibitory properties of a series of aminopyridine-substituted hydroxyquinoline derivatives;
 |
The next stage of analog efforts focused upon modification of the aminopyridine ring. Conversion of the aminopyridine to a secondary amine such as a piperidine, morpholine, or piperazine, resulted in compounds which displayed excellent potency against Mcl-1 and maintained or enhanced selectivity over Bcl-xL. This change from an aromatic to a non-aromatic, basic amine also served to reduce LogD and enhance predicted water solubility, as desired. The combination of this finding with the benefit to Mcl-1 binding conferred by a para substituent on the phenyl group (compound 8) led to the identification of lead compound 9. This analog demonstrated IC50s of 0.31 µM and 40 µM for Mcl-1 and Bcl-xL, respectively (Figure 1b). Moreover, compound 9 possesses moderate molecular weight (415), hydrophobicity (cLogD7.4 = 3.7) and lacks inherent toxicophores. The para-CF3 group and piperazine ring contribute to a moderately high liver microsome stability for compound 9 (half-life 55 minutes in human microsomes, 40 min, in mouse microsomes) and IC50’s for inhibition of 4 isoforms of CYP450 tested (1A2, 2D6, 3A4, 2C9) each greater than 10 M.
Computational chemistry studies were performed to obtain greater understanding of the nature of the interaction between hydroxyquinoline 9 and Mcl-1. X-ray crystal structures indicate a binding site for Mcl-1 and the BH3-only peptides that consist of a long, hydrophobic region which accommodates four lipophilic side chains of the BH3-only peptide.33 Our studies indicate that compound 9 fits compactly within this known Mcl-1 binding groove (Figure 2). Modeling of the R-configuration of compound 9 suggests that a hydrogen bond formed between the Mcl-1 residue Asn260 and the hydroxyquinoline moiety of 9 may serve to position the N-ethylpiperazine moiety to occupy one of the four primary lipophilic pockets typically filled by side chains of BH3-only peptides.
Figure 2.

Calculated binding mode of compound 9 in Mcl-1. Modeling suggests a hydrogen bond between the phenol of the hydroxyquinoline and the carbonyl of Asn260 may orient the ethylpiperazine group into one of the four major lipophilic pockets found in the Mcl-1/BH3-only peptide binding groove.
3.2. Potent activity in cell-based assays and selectivity correlation with BH3 profiling
The ability of hydroxyquinoline 9 to induce cell death was assessed in cellular proliferation assays. We focused on cell lines derived from hematologic malignancies where Mcl-1 has been shown to play a significant role. Initial screening demonstrated that compound 9 displayed low micromolar efficacy in the mouse lymphoma cell lines Mcl1-1780 and Bcl2-1863, the human lymphoid cell line SUDHL-6, and the human multiple myeloma line NCIH929 (Figure 3a).
Figure 3.

(a) EC50 values from cell proliferation assay experiments (viability determined by PrestoBlue staining) for the Mcl-1 inhibitor 9; Results indicated are the mean of two independent cell growth inhibition experiments conducted in triplicate (n=6) (b) Results of BH3 profiling assay on the lymphoid cell lines SU-DHL6 and SU-DHL10, myeloma cell line NCI-H929, and mouse leukemia cell lines Bcl2-1863 and Mcl1-1780. These cell lines display a range of mitochondrial depolarization upon exposure to various BH3-only peptides (Bim, Puma, Noxa, Bad). For example, SU-DHL6 is highly “primed” for all BH3 peptides tested but SU-DHL10 has low levels of mitochondrial priming. Priming values are derived from two independent assays conducted in triplicate (n=6); (c) Correlation between antiproliferative activity of 9 (EC50 represented in M) and BH3 profiling response (% mitochondrial depolarization, Bim peptide assayed at 0.3 M). An excellent correlation is observed between priming state and antiproliferative response observed in these compound 9 –treated cell lines (R2=0.81).
While these results provided evidence that compounds were cytotoxic, we sought to validate the precise mechanism of action using BH3 profiling. This assay provides a means of measuring the extent to which a cell relies on the function of any or all of the anti-apoptotic Bcl-2 family proteins for survival. BH3 profiling utilizes a panel of BH3- domain peptides (derived from the BH3-only proteins).34,35 The extent of MOMP observed following treatment of semi-permeablized cells with a specific BH3-only protein indicates that the cell is near the apoptotic threshold with respect to that binding partner and is mitochondrially “primed”.34,35
As determined by BH3 profiling, the lymphoma-derived cell line SUDHL-6 displays significant priming for a variety of BH3-only peptides (Figure 3b).28 In contract, the related SUDHL-10 line demonstrates low levels of priming for all Bcl-2 members. The readout of the BH3 profiling assay in cell lines provides a valuable functional biomarker for compounds that mimic the BH3 peptides used in the assay. Cell lines that demonstrate high levels of priming would be expected to display the greatest response to BH3 mimetics. We determined response to compound 9 in 11 human cell lines of varying origin, and with varying dependencies on the different Bcl-2 family proteins as determined by the BH3 profiling assay (Table 2). As shown in Figure 3c, a strong correlation was observed between the degree of mitochondrial priming with respect to the BH3-only peptide Bim.
Table 2.
EC50 values determined in cell proliferation assays for Mcl-1 inhibitor 9 in a diverse panel of human-derived cancer cell lines. Data represent the average of three replicate experiments;
| Cell Line | EC50 (µM) |
|---|---|
| Mcl1-1780 | 0.3 |
| Bcl2-1863 | 1.1 |
| SUDHL-6 | 3.3 |
| NCI-H929 | 1.6 |
| SUDHL-10 | 25 |
| HL60 | 6.3 |
| ML2 | 5.2 |
| MOLM13 | 7.0 |
| TF1 | 4.8 |
| A375 | 7.1 |
| A549 | 14.1 |
| H187 | 9.5 |
| PANC1 | 14.5 |
3.3. Cytochrome c release is correlative to mitochondrial priming state
A hallmark of on-target BH3 mimetics is their ability to promote timely release of cytochrome c compared to competing BH3 peptides. We analyzed the ability of Mcl-1 inhibitor 9 to promote release of cytochrome c, as compared to the Bim and PUMA BH3 peptides, in semi-permeablized cells (Figure 4a). Treatment of a series of lymphoma and leukemia cell lines with Mcl-1 inhibitor 9 resulted in significant cytochrome c release at 2 hours. The greatest extent of cytochrome c release was observed in the highly mitochondrially primed SUDHL-6 cell line. The release of cytochrome c from mitochondria in the poorly primed SUDHL-8 cell line and the Bax/Bak deficient SUDHL-10 cell line was significantly reduced. In the mouse-derived lymphoma cell lines Mcl1-1780 and Bcl2-1863, cytochrome Ccrelease was observed to be significantly greater in the former cell line (Figure 4a), which are selectively dependent on either Bcl-2 or Mcl-1 for survival.31 These two cell lines are derived from the same mouse lineage and are selectively dependent on either Mcl-1 or Bcl-2/Bcl-xL for survival, as determined by the Noxa (Mcl-1 restricted) or Bad (Bcl-2 restricted) signal by BH3 profiling (Figure 3b).
Figure 4.

(a) Compound 9 promotes cytochrome c release in a fashion that may be correlated with the extent of mitochondrial priming in a series of human-derived lymphoid cell lines (SU-DHL6, SU-DHL8, SU-DHL10) and two mouse leukemia cell lines (Bcl2-1863 and Mcl1-1780); (b) Cell viability of mouse leukemia cell lines Bcl2-1863 and Mcl1-1780 following treatment with ABT-263 or compound 9 as measured by DAPI assay; (c) Percentage of cells that demonstrate apoptosis, as detected by Annexin V –positive staining, following treatment with compound 9 at the indicated concentrations. Results depicted are representative of two independent experiments and are consistent for dose-response trends.
3.4. Compound 9 promotes apoptosis as determined by DAPI and Annexin V staining
Using standard methods we examined Mcl-1 inhibitor 9 for ability to induce apoptosis; this study was performed in the Mcl1-1780 and Bcl2-1863 cell lines (see Figure 4b). The Bcl-2/Bcl-xL inhibitor Navitoclax (ABT-263) was also examined in this study. This compound shows a distinct preference in inducing apoptosis in the highly Bcl-2/Bcl-xL primed cell line Bcl2-1863, and displays only minimal effectiveness in the highly Mcl1 primed cell line. In contrast, Mcl-1 inhibitor 9 displays an inverse pattern of apoptotic induction ability.
The extent of apoptosis conferred by analog 9 was determined using Annexin V staining. The percent of Annexin V positive cells displayed dose-dependency and was greater in Mcl1-1780 than in Bcl2-1863, providing further evidence of Mcl-1 inhibition driven apoptosis that correlates with the extent of Mcl-1 mediated mitochondrial priming (figure 4c).
4. Discussion
In this work we utilized a novel approach to select compounds that have activity against specific members of the Bcl-2 family proteins. We have identified a series of small molecule inhibitors of Mcl-1 through a high throughput screening effort and validated these compounds utilizing BH3 profiling. This platform technology provides a functional biomarker which was utilized to assess the on-target mechanism of these inhibitors in a cell-based context.
The primary screen was designed and implemented with the aim of identifying Mcl-1 inhibitors which displayed selectivity over Bcl-xL. The initial hit (1, Figure 1) demonstrated affinity for Mcl-1 (2.4 M with greater than 40-fold selectivity over Bcl-xL. However, this analog displayed only modest antiproliferative activity and therefore required structural modification. An analog effort allowed for the identification of a pharmacophore for Mcl-1 inhibition. Incorporation of electron-withdrawing groups on the aryl ring, and a piperazine replacement for the aminopyridine group, enhanced Mcl-1 inhibition and selectivity over Bcl-xL and resulted in Mcl-1 inhibitor 9. A proposed binding mode was identified using computational chemistry methods. The interactions between Mcl-1 and its various BH3-only peptide binding partners (such as Bim, Puma, NoxaA) are stabilized through interactions between four hydrophobic side chains of the BH3-only binding partner and corresponding hydrophobic pockets in the binding groove of Mcl-1. Docking and SAR studies suggest that hydrogen bonding between the quinoline C7-hydroxyl group of Mcl-1 inhibitor 9 with Asn260 of Mcl-1 places the lipophilic alkyl moiety of the piperazine group within a lipophilic pocket of the protein (Figure 2).
The antiproliferative activity of Mcl-1 inhibitor 9 was studied in several cell lines derived from various human malignancies. This lead compound displayed significant activity in a number of cell lines, with EC50 values in the range of 0.3–15 M. We then sought to further define the mechanism of action of this agent, and did so by application of BH3 profiling. Using this assay we found a strong correlation between the degree of priming, with respect to Bim BH3 peptide, and responsiveness of these cell lines to treatment with compound 9 (Figure 3c). These results demonstrate the utility of BH3 profiling as a drug discovery tool to validate the mechanism of action of BH3 mimetics.
We then examined the ability of our lead compound to induce cytochrome c release in cells that were characterized by the BH3 profiling assay. In three leukemia-derived cell lines possessing various priming states (SUDHL-10, -8, and -6; Figure 4a), we observed response nearly identical to that seen upon treatment with the peptides comprising the PUMA BH3 domain. Cytochrome c release was observed upon treatment of the highly primed SUDHL-6 cell line with both compound 9 and PUMA, whereas the poorly primed SUDHL-8 and SUDHL-10 cell lines did not produce such a response upon exposure to these agents. This indicated that priming was required for cell response. We also examined the activity of Mcl-1 inhibitor 9 in the mouse leukemia cell lines BCL2-1863 and MCL1-1780. These two cell lines have been characterized as mitochondrial-primed with respect to Bcl-2 or Mcl-1 (by determination of the BH3 profiling readout in response to treatment with the Bad or Noxa BH3-only proteins, respectively, Figure 3b). In contrast, compound 9 displays selective activation of cytochrome c in MCL1-1780. This result provides strong functional support for compound selectivity in a cellular context.
To further validate the on-target mechanism and selectivity of our lead analog, we determined the extent to which Mcl-1 inhibitor 9, as well as the Bcl-2/Bcl-xL inhibitor Navitoclax, were able to promote nuclear apoptosis. Navitoclax demonstrates the ability to selectively promote cell death in the BCL2-1863 cell line, which is characterized as Bcl-2 primed (Figure 4b). This finding clearly demonstrates the utility of BH3 profiling as correlative with selectivity in a cellular context, given the well-validated Bcl-2/Bcl-xL selectivity of this agent. The response observed for Mcl-1 inhibitor 9 is consistent with its Mcl-1 selectivity; the compound reduces viability with a marked preference for the MCL1-1780 cell line. We utilized Annexin V staining to provide additional confirmation of the ability of this compound to produce apoptosis. In this study (Fig 4c), we observed that the extent of apoptotic induction was greater in Mcl1-1780 relative to Bcl2-1863, a trend which mirrored the EC50 values determined for these cell lines in an antiproliferative assay (0.3 µM and 1.1 µM, respectively, Table 2).
Conclusion
Here we have described the discovery and characterization of a selective inhibitor of the Mcl-1 protein, a target central to intrinsic apoptosis that has been implicated in a cancer. We demonstrated that Mcl-1 inhibitor 9 displays efficacy in promoting death in a panel of cell lines derived from a variety of malignancies. Importantly, we highlighted the utility of the BH3 profiling assay in confirming the mechanism of action of agents that modify the Bcl-2 pathway. We have showed that Mcl-1 inhibitor 9 functions in the cellular context as a BH3 mimetic and that antiproliferative response in a number of cell lines may be correlated with the extent of mitochondrial priming. We also demonstrated that the Mcl-1 selectivity observed in biochemical assays may be translated to cell-based assays by correlating Mcl-1 and Bcl-2 priming with the degree of cytochrome c release and apoptosis induction. Compounds of this class represent promising chemical probes for the interrogation of Mcl-1 biology and we anticipate such tools will be of great utility to the chemical biology community.
Identification of relevant companion diagnostics at the preclinical research stage has been recognized as a critical component of facilitating the development of personalized cancer therapeutics. In addition to providing utility as a discovery tool by guiding SAR, BH3 profiling may be utilized as a companion diagnostic test for Mcl-1 inhibitors and other compounds which modulate the intrinsic apoptosis pathway.
Supplementary Material
Acknowledgements
This work was supported by NCI-SBIR grant #2R44CA135915-02A1. T.B., C.E.,M.K., Y.H., D.M., J.M. and P.H. were supported by NIH MLPCN grant #MH084512.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict-of-interest disclosure
David Richard, William Pierceall, Nicole Carlson, Ryan Lena, Noel Blake, and Michael Cardone are employees of Eutropics, Inc. The remaining authors declare no conflicts-of-interest.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 3.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 4.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 6.Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010;1:e40. doi: 10.1038/cddis.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002;99:1885–1893. doi: 10.1182/blood.v99.6.1885. [DOI] [PubMed] [Google Scholar]
- 8.Cho-Vega JH, Rassidakis GZ, Admirand JH, Oyarzo M, Ramalingam P, Paraquya A, et al. MCL-1 expression in B-cell non-Hodgkin's lymphomas. Hum. Pathol. 2004;35:1095–1100. doi: 10.1016/j.humpath.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W, Konopleva M, Ruvolo VR, McQueen T, Evans RL, Bornmann WG, et al. Sorafenib induces apoptosis of AML cells via Bim-mediated activation of the intrinsic apoptotic pathway. Leukemia. 2008;22:808–818. doi: 10.1038/sj.leu.2405098. [DOI] [PubMed] [Google Scholar]
- 10.Hussain SA, Cheney CM, Johnson AJ, Lin TS, Grever MR, Caligiuri MA, et al. Mcl-1 is a relevant therapeutic target in acute and chronic lymphoid malignancies: down-regulation enhances rituximab-mediated apoptosis and complement-dependent cytotoxicity. Clin. Cancer Res. 2007;13:2144–2150. doi: 10.1158/1078-0432.CCR-06-2294. [DOI] [PubMed] [Google Scholar]
- 11.Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012;26:120–125. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoue S, Walewska R, Dyer MJS, Cohen GM, et al. Downregulation of Mcl-1 potentiates HDACi-mediated apoptosis in leukemic cells. Leukemia. 2008;22:819–825. doi: 10.1038/leu.2008.1. [DOI] [PubMed] [Google Scholar]
- 13.Pepper C, Lin TT, Pratt G, Hewamana S, Brennan P, Hiller L, et al. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood. 2008;112:3807–3817. doi: 10.1182/blood-2008-05-157131. [DOI] [PubMed] [Google Scholar]
- Cavarretta IT, Neuwirt H, Zaki MH, Steiner H, Hobisch A, Nemeth JA, et al. Mcl-1 is regulated by IL-6 and mediates the survival activity of the cytokine in a model of late stage prostate carcinoma. Adv. Exp. Med. Biol. 2008;617:547–555. doi: 10.1007/978-0-387-69080-3_56. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Guttikonda S, Roberts L, Uziel T, Semizarov D, Elmore SW, et al. Mcl-1 is critical for survival in a subgroup of non-small-cell lung cancer cell lines. Oncogene. 2011;30:1963–1968. doi: 10.1038/onc.2010.559. [DOI] [PubMed] [Google Scholar]
- 16.Boisvert-Adamo K, Longmate W, Abel EV, Aplin AE. Mcl-1 is required for melanoma cell resistance to anoikis. Mol. Cancer Res. 2009;7:549–556. doi: 10.1158/1541-7786.MCR-08-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou P, Qian L, Kozopas KM, Craig RW. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89:630–643. [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 21.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bax/Bak if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulated MCL-1 and BFL-1. Blood. 2010;114:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien SM, Claxton DF, Crump M, Faderl S, Kipps T, Keating MJ, et al. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood. 2009;113:299–305. doi: 10.1182/blood-2008-02-137943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliver CL, Miranda MB, Shangary S, Land S, Wang S, Johnson DE, et al. Gossypol acts directly on the mitochondria to overcome Bcl-2- and Bcl-X(L)-mediated apoptosis resistance. Mol. Cancer Ther. 2005;4:23–31. [PubMed] [Google Scholar]
- 25.McCoy F, Hurwitz J, McTavish N, Paul I, Barnes C, O’Hagan B, et al. Obatoclax induces Atg7-dependent autophagy independent of beclin-1 and BAX/BAK. Cell Death Disease. 2010;1:e108. doi: 10.1038/cddis.2010.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJS, et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009;16:1030–1039. doi: 10.1038/cdd.2009.48. [DOI] [PubMed] [Google Scholar]
- 27.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu YL, Cui Y, Busman T, Elmore SW, Rosenberg SH, Krivoshik AP, Enschede SH, Humerickhouse RA. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, Preusser LC, Reinhart GA, Smith ML, Rosenberg SH, Elmore SW, Tse C. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14:943–951. doi: 10.1038/sj.cdd.4402081. 2007. [DOI] [PubMed] [Google Scholar]
- 29.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 30.Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, et al. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat. Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 31.Ryan JA, Burnelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc. Nat. Acad. Sci. USA. 2010;107:12895–12900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nikolovska-Coleska Z, Qiu S, Long J, Yang C-Y, Larsen M, Tomita Y, et al. Discovery and characterization of new small-molecule inhibitors of Mcl-1 by high-throughput screening. AACR Annual Meeting; April, 2008; San Diego, CA. Abstract 4148. [Google Scholar]
- 33.Acoca S, Cui Q, Shore GC, Purisima EO. Molecular dynamics study of small molecule inhibitors of the Bcl-2 family. Proteins: Struct., Funct., Bioinf. 2011;79:2624–2636. doi: 10.1002/prot.23083. [DOI] [PubMed] [Google Scholar]
- 34.Ni Chonghaile T, Sarosiek KA, Vo T, Ryan JA, Tammareddi A, Moore VDG, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vo T, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151:344–355. doi: 10.1016/j.cell.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.