

Abstract
Purpose of review
The present review summarizes recent knowledge on polycystic liver diseases (PCLDs), mechanisms of hepatic cystogenesis and potential therapies for these conditions.
Recent findings
PCLD may be classified as cholangiociliopathies. In PCLD associated with polycystic kidney disease, cell proliferation is one of the major mechanisms of cystogenesis, whereas in isolated PCLD (autosomal dominant polycystic liver disease), disrupted cell adhesion may be more important in cyst progression. In cystic cholangiocytes, overexpression of ion transporters and water channels facilitates fluid secretion into the cystic lumen, and growth factors, estrogens and cytokines promote cholangiocyte proliferation. With age, cholangiocytes lining liver cysts acquire features of mesenchymal cells contributing to hepatic fibrocystogenesis. A novel mechanism of liver cyst expansion in PCLD involves microRNA regulatory pathways. Hyperproliferation of cystic cholangiocytes is linked to abnormalities in cell cycle progression and microRNA expression. Decreased levels of miR-15a are coupled to upregulation of its target – the cell cycle regulator, Cdc25A. Cholangiocyte cilia in liver cysts are structurally abnormal. Somatostatin analogues and sirolimus reduce liver cyst volume in PCLD patients.
Summary
Clarification of molecular mechanisms of hepatic cystogenesis provides an opportunity for the development of targeted therapeutic options in PCLD.
Keywords: cholangiociliopathies, fluid secretion, hepatic cystogenesis, proliferation
Introduction
The polycystic liver diseases (PCLDs) represent a group of genetic disorders inherited in dominant or recessive mode and occur alone or in association with polycystic kidney diseases (PKDs) [1•]. To date, one form of isolated PCLD (characterized by the presence of cysts only in the liver) is known as autosomal dominant polycystic liver disease (ADPLD). The list of PKD-associated PCLD includes but is not limited to autosomal dominant PKD (ADPKD), autosomal recessive PKD (ARPKD), Meckel–Gruber syndrome (MKS), Bardet–Biedl syndrome (BBS), nephronophthisis (NPHP) and Joubert syndrome (JBTS). The most common symptoms and complications in patients with PCLD include hypertension, back pain, abdominal distension and discomfort, dyspnea, gastroesophageal reflux, cyst hemorrhage, infection and/or rupture. No curative or preventive therapies of PCLD currently exist [1•].
In PCLD, mutations of genes (Table 1) lead to hepatic cytogenesis or hepatic fibrosis or both. All proteins encoded by these genes (except two proteins implicated in development of ADPLD) are localized to primary cilia and many of them interact with each other forming functional complexes and converge into similar signaling cascades. Mutations in genes encoding ciliary-associated proteins result in aberrantly formed and malfunctional cilia leading to development of various forms of polycystic kidney and liver disease that have been categorized as ciliopathies [2•,3•]. We proposed to call cilia-related PCLDs the cholangiociliopathies [1•] because in the liver, cilia are present only in cholangiocytes. Liver cysts are derived from cholangiocytes and their progressive expansion is associated with cholangiocyte hyperproliferation, cell cycle dysregulation, enhanced fluid secretion, cell-matrix remodeling, neovascularization and structural and functional ciliary abnormalities.
Table 1.
Polycystic liver diseases
| Disease (frequency) | Gene
|
Protein
|
Liver disorder | |||
|---|---|---|---|---|---|---|
| Name | Locus | Name | Function | Localization | ||
| ADPLD 1 : 100 000 | PRKCSH | 19p13.2 | Hepatocystin (Glucosidase II B subunit) Sec63 |
Protein translocation | ER | Liver cysts |
| sec63 | 6q21 | |||||
| ADPKD 1 : 800 | PKD1 | 16p133 | Polycystin1 | Cell cycle regulation, [Ca2+]i signaling | Primary cilia, cell junctions, desmosomes | Liver cysts |
| PKD2 | 4q212 | Polycystin2 | ||||
| PKD3? | [Ca2+]i signaling | Primary cilia, ER, centrosomes | ||||
| ARPKD 1 : 20 000 | PKHD1 | 6p21–23 | Fibrocystin/polyductin | [Ca2+]i signaling | Primary cilia, apical membrane | CHF, biliary dysgenesis, Caroli’s disease |
| MKS 1 : 135 000 | MKS1 | 17q21–24 | MKS1 | Ciliogenesis | Centrosome | Biliary dysgenesis |
| MKS2 | 11q13 | Meckelin | Primary cilia | |||
| MKS3 | 8q24 | |||||
| NPHP 1 : 50 000 | NPHP1 | 2q13 | Nephrocystin1 | Ciliogenesis | Cell junctions, focal adhesion primary cilia, centrosome | CHF, bile duct proliferation, |
| NPHP2/Inv | 9q31 | Inversin | Cell cycle, Wnt signaling | |||
| NPHP3 | 3q22.1 | Nephrocystin3 | Ciliary function | |||
| NPHP4 | 1p36.22 | Nephrocystin4 | Signal transduction, cell adhesion | |||
| NPHP5/IQCB1 | 3q13.33; 3q21.1 | Nephrocystin5 | ||||
| NPHP6/CEP290 | 12q21.32 | Nephrocystin6 | Ciliary function | |||
| NPHP7 | 16p13.3 | Nephrocystin7 | ||||
| NPHP8/RPGRIP1L | 16q12.2 | Nephrocystin8 | ||||
| NEK8 | 17q11.1 | |||||
| Joubert syndrome 1 : 100 000 | JBTS1 | 9q34.3 | Jouberin | Protein transport | Primary cilia, cell junctions centrosomes, | Biliary dysgenesis, liver fibrosis |
| JBTS2 | 11p12-q13.3 | Nephrocystin1 | ||||
| JBTS3/AHI1 | 6q23.3 | CEP290 | ||||
| JBTS4/NPHP1 | 2q13 | |||||
| JBTS5/NPHP6/CEP290 | 12q21.32 | |||||
| MKS3 | 16q12.2 8q24 |
|||||
| BBS 1 : 100 000 | BBS1 | 11g13.1 | BBS1 | Basal body and centrosome function | Centrosome (BBS1-9) Primary cilia (BBS4, 8) Cytoskeleton (BBS11) | |
| BBS2 | 16q21 | BBS2 | ||||
| BBS3/ARL6 | 3q11.2 | ARL6 | ||||
| BBS4 | 15q22.3-q23 | BBS4 | ||||
| BBS5 | 2q31 | BBS5 | ||||
| BBS6/MKKS | 20p12 | BBS6/MKKS | ||||
| BBS7 | 4q27 | BBS7 | ||||
| BBS8/TTC8 | 14g31.3 | BBS8/TTC8 | ||||
| BBS9 | 7p14 | PTHB1 | ||||
| BBS10 | 12q21.2 | BBS10 | ||||
| BBS11/TRIM32 | 9q31.1 | TRIM32 | ||||
| BBS12 | 4q27 | BBS12 | ||||
Question mark after PKD3 means gene is suspected but not yet identified. ADPKD, autosomal dominant polycystic kidney disease; ADPLD, autosomal dominant polycystic liver disease; ARPKD, autosomal recessive polycystic kidney disease; BBS, Bardet–Biedl syndrome; CHF, congenital hepatic fibrosis; ER, endoplasmic reticulum; MKS, Meckel–Gruber syndrome; NPHP, nephronophthisis.
Isolated polycystic liver diseases
ADPLD is characterized by the presence of cysts only in the liver [4•,5]. The exact prevalence of ADPLD is unknown but estimated at 1 : 100 000. The disease is the result of mutations in two genes: PRKCSH that encodes the protein, hepatocystin (protein kinase C substrate 80K-H); and Sec63 that encodes the protein, Sec63 (Table 1). Hepatocystin (~59 kDa) is expressed in a variety of organs and is localized in endoplasmic reticulum being associated with the α-subunit of glucosidase II [5,6]. In cultured HeLa and Madin–Darby canine kidney (MDCK) cells, hepatocystin is secreted into the medium; however, no protein was detected in ADPLD cyst fluid samples [7•]. This observation is consistent with that in a previous study showing that in patients with truncated mutations in PRKCSH, hepatocystin was not detected in hepatic cystic epithelia [7•]. Sec63 protein (~83 kDa) is a component of the protein translocation machinery in the endoplasmic reticulum and is involved in oligosaccharide processing of newly synthesized glycoproteins [6].
Polycystic kidney disease-related polycystic liver disease: cholangiociliopathies
ADPKD is the most common inherited renal cystic disease that affects the liver in more than 85% of PKD patients. The prevalence and number of hepatic cysts in patients with ADPKD increase with age, female sex, severity of renal cystic disease and degree of renal dysfunction. ADPKD is caused by mutations in two genes, PKD1 and PKD2; existence of a third gene, PKD3, is suspected (Table 1). Mutations in PKD1 are more frequent and severe and account for 85–90% of cases, whereas mutations in PKD2 affect approximately 10–15% of ADPKD patients. The patients with PKD1 mutations have more cysts of larger size than patients with PKD2 mutations [8•].
The gene products of PKD1 and PKD2, polycystin-1 (PC-1) and polycystin-2 (PC-2), respectively, are both transmembrane proteins known to form a functional complex. PC-1 (~460 kDa) is expressed in renal, biliary, pancreatic and intestinal epithelial cells. At the subcellular level, PC-1 is localized to both apical and basolateral plasma membranes, at the site of cell–cell contact, and in primary cilia. PC-2 (~110 kDa) is expressed in many tissues, including kidney, liver, heart, ovary, testicles, smooth muscle and small intestine. At the subcellular level, PC-2 is present at the plasma membrane, endoplasmic reticulum, primary cilia, in mitotic spindles and centrosomes [6,8•]. Both PC-1 and PC-2 are involved in a variety of cell functions, including cell–cell interactions, cell cycle control, [Ca2+]i regulation and Wnt signaling [9].
ARPKD is less common than ADPKD and is linked to mutations in a single gene, PKHD1, which encodes the protein, fibrocystin/polyductin [10,11,12•]. ARPKD is a significant cause of morbidity and mortality –approximately 30% of affected neonates die as a result of respiratory difficulties due to enlarged kidneys. In surviving patients, hepatic disease becomes progressively more severe with age. Liver involvement is characterized by biliary dysgenesis, congenital hepatic fibrosis (CHF), intrahepatic bile duct dilatation, cholangiocyte proliferation and/or cyst development [12•,13]. Recently, a renal-hepatobiliary morbidity pattern (i.e., patients exhibit exclusively renal or liver phenotype, or both) in ARPKD has been proposed [14]. This observation is consistent with the notion that isolated CHF or Caroli’s disease in some cases is associated with mutations in PKHD1. Despite the disruption of different Pkhd1 exons (i.e., exons 3–4, exon 4 and exon 40), three animal models of ARPKD are characterized by liver phenotype without any or significantly delayed renal disease [15•]. On the other hand, deletion of exon 1–3 of Pkhd1 results in severe renal cystic disease and classical ductal plate malformation observed in human ARPKD patients [16•], further suggesting that the site of mutation might define the pathological phenotype.
Fibrocystin (~447 kDa) is a type-I integral, transmembrane protein expressed in kidney, liver, pancreas, testis and lung. At the subcellular level, fibrocystin is found in primary cilia (where it is often absent in pathological conditions), centrosome, apical plasma membrane and cytoplasm. On the basis of its homology to D86 (a mouse protein secreted from the lymphocytes), a secreted form of fibrocystin was predicted [12•].
MKS is an autosomal recessive disorder characterized by renal cystogenesis, polydactyly and occipital encephalocele. Biliary dysgenesis is typical and hepatic fibrosis is a constant finding. Three genes linked to the development of MKS are described: MSK1, MKS2 and MKS3 (Table 1). MKS1 encodes the protein MKS1, which is predicted to be a cytoplasmic protein. MKS3 encodes the protein meckelin, which is predicted to be a receptor. Both proteins are expressed in many different tissues being more abundant in brain, liver and kidney. Meckelin is expressed in primary cilia in cultured cholangiocytes and renal epithelial cells. In contrast, MKS1 is detected in cholangiocyte centrosomes [2•,17,18,19•].
NPHP is a rare autosomal recessive disease with three (defined by the age of onset) forms – infantile, juvenile and adolescent [20•]. The most notable phenotype in NPHP includes kidney cysts and renal fibrosis, retinal pigmentosa, situs invertus and central nervous system malformation. Liver involvement in NPHP is characterized by hepatomegaly, hepatic fibrosis, bile duct enlargement and proliferation [20•]. At least nine genes, mutations of which can cause NPHP, have been identified (Table 1). NPHP genes encode respective proteins, nephrocystins. All of them are found in cilia and basal bodies [20•,21]. Mutations in several NPHP genes are also associated with JBTS and MKS [2•].
JBTS is a rare pleiotropic disorder inherited in recessive mode. JBTS causes abnormal eye development, malformation of the central nervous system, autism, polydactyly, duodenal atresia and liver fibrosis. JBTS is genetically heterogeneous with several genes identified (Table 1). Some JBTS proteins, that is, CEP290, are required for ciliogenesis [22•]. The JBTS phenotype overlaps with several other disorders such as NPHP, MKS and BBS [2•,20•,23,24•].
BBS is a pleiotropic multiorgan genetic disorder associated with obesity, retinitis pigmentosa, polydactyly, mental retardation, renal cyst formation and hepatic fibrosis. Twelve genes involved in disease development have been cloned (Table 1). The products of BBS genes, the BBS proteins, are all localized to the basal bodies and cilia. Recent data suggest that BBS proteins are organized in a large complex, that is, ‘BBSome’, which is thought to be responsible for transporting intracellular vesicles to the base of the cilia and thus playing an important role in ciliary function [2•,19•].
Cholangiocyte proliferation
Cholangiocyte proliferation is considered one of the major contributors to hepatic cystogenesis. Over time, many different factors have been implicated in this process [25]. Recent data suggest that the hepatic cystic epithelium of ADPKD patients is characterized by overexpression of estrogen receptors and insulin growth factor 1 receptors (IGF1-Rs), and cystic fluid is enriched in IGF1 and 17-β-estradiol [26••]. IGF1-R appears to be mislocalized to the apical membrane of cystic cholangiocytes [26••]. In cultured cells derived from liver cysts of ADPKD patients, both estrogens and IGF1 significantly increase the rate of cell proliferation. In addition, estrogen also promotes secretion of IGF1 by cystic epithelium [26••].
Cystic fluid of ADPKD patients contains a significant amount of CXCR2 receptor agonists such as IL-8, epithelial neutrophil-activating peptide (ENA-78) and growth-related oncogene-α (GRO-α) [27••]. Cultured cholangiocytes derived from the Pkd2 (WS25-) mutant mice, an animal model of ADPKD, secrete CXCR2 agonists both apically and basolaterally, suggesting that they may simultaneously influence functions of epithelial and endothelial cells. Indeed, one of the CXCR2 agonists, IL-8, induces proliferation of HMEC-1 cells (i.e., human microvascular endothelial cell line) and Mz-ChA1 cells (a human cholangiocyte cell model) [27••].
Whereas in PKD-associated PCLD cholangiocyte proliferation is one of the major mechanisms underlying hepatic cystogenesis, in isolated PCLD (i.e., ADPLD) this does not appear to be the case. None of the hepatic cysts from ADPLD patients were positive for cell proliferation marker, Ki67 [28••]. Cell adhesion markers such as E-cadherin and Ep-CAM were also lost in ADPLD cystic epithelia. It is likely that disturbed cell adhesion but not cholangiocyte proliferation promotes hepatic cystogenesis in this form of PCLD [28••].
Fluid secretion
Fluid secretion is known to play an important role in progressive expansion of hepatic cysts [1•,25]. This process is strongly linked to overexpression of ion transporters [i.e., cystic fibrosis transport regulator (CFTR) and anion exchanger isoform 2 (AE2)] and water channels [i.e., aquaporin 1 (AQP1)] in cholangiocytes of liver cysts in the polycystic kidney (PCK) rat, an animal model of ARPKD [29••]. The subcellular localization of these proteins appears to be perturbed in cystic cholangiocytes. In normal rats, these proteins are preferentially localized at the apical plasma membrane, whereas in the PCK cholangiocytes CFTR, AQP1 and AE2 are overexpressed at the basolateral domains [29••]. This basolateral over-expression may facilitate ion and water movement from the basolateral to apical membrane and thus may account for enhanced fluid secretion into the cyst lumen.
Hepatic fibrosis
Most forms of PCLD (i.e., ARPKD, BBS, NPHP, JBTS and MKS) are associated with hepatic fibrosis. Hepatic fibrosis may occur in response to hepatocyte damage and subsequent necrosis involving activation of hepatic stellate cells and myofibroblasts [30]. However, in PCLD patients, the hepatic parenchyma appears to be well preserved suggesting that other mechanisms contribute to hepatic fibrogenesis. Recent data have demonstrated that cystic cholangiocytes have the ability to undergo morphological transition from an epithelial to a mesenchymal phenotype. Whereas in young PCK rats cystic cholangiocytes are cuboidal in appearance, in aged animals the majority of cholangiocytes become flat-shaped and characterized by reduced levels of the epithelial cell marker, CK-19, and increased levels of the mesenchymal markers, vimentin and fibronectin [31]. Thus, cystic cholangiocytes acquired mesenchymal features in response to transforming growth factor-β-1 (TGF-β-1), in this way contributing to progressive hepatic fibrosis [31]. Activation of Hedgehog signaling may facilitate this epithelial-mesenchymal transition of cholangiocytes [32•].
Cell cycle, miRNA and hepatic cystogenesis
Hyperproliferation of cholangiocytes is associated with cell cycle dysregulation. Progression of cells through the cell cycle is controlled, in particular, by a family of dual-specificity phosphatases, Cdc25, that activate cyclin-dependent kinases (Cdks) [33]. In PCLD patients and PCK rats, Cdc25A protein (one of the three known isoforms of Cdc25) is overexpressed in cholangiocytes lining liver cysts. Cdc25A upregulation in diseased livers is accompanied by downregulation of one of the miRNAs (i.e., miR-15a). MicroRNAs are small noncoding RNAs that posttranscriptionally inhibit target messenger RNA transcripts via sequence-specific base paring [34••]. miR-15a has a conserved complementarity to Cdc25A mRNA. Overexpression of miR-15a in cystic cholangiocytes inhibits G1-S cell cycle transition, cholangiocyte proliferation and cyst growth [34••]. In contrast, downregulation of miR-15a levels in normal cholangiocytes increases cell proliferation, Cdc25A expression and cyst growth. The data suggest that the miR-15a/Cdc25A complex is involved in dysregulation of the cell cycle and subsequent growth of liver cysts in PCLD [34••]. The potential role of abnormal cilia as a driving force generating dysregulation of miR-15a/Cdc25A complex is possible but needs to be proven (Fig. 1) [35•].
Figure 1. Role of miR-15a, Cdc25A and abnormal cilia in hepatic cystogenesis.
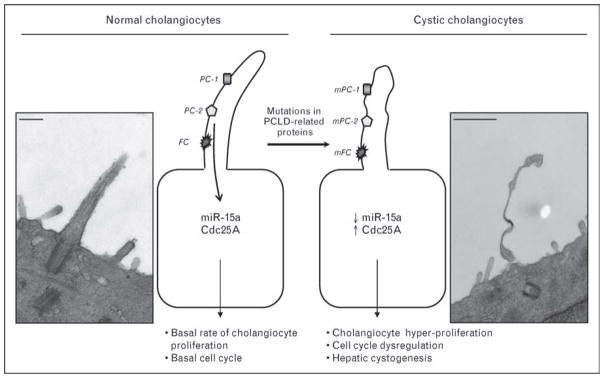
Under normal conditions, cholangiocyte primary cilia express polycystic liver disease (PCLD)-related proteins, in particular, polycystin-1 (PC-1), polycystin-2 (PC-2) and fibrocystin, which are involved in regular cell function. miR-15a and cell cycle protein Cdc25A are expressed at the basal level resulting in basal rate of cholangiocyte proliferation and normal progression through the cell cycle. Due to mutations in genes encoding PCLD-related proteins, cholangiocyte cilia become functionally and structurally abnormal. These abnormalities affect cholangiocyte functions and lead to dysregulation of miR-15a/Cdc25A complex subsequently inducing cholangiocyte proliferation, cell cycle dysregulation and, finally, hepatic cystogenesis. Scanning electron microscopy (SEM) images show cholangiocyte cilia in normal (left) and PCK (right) rats. Scale bars, 1 μm. mFC, mutated fibrocystin; mPC-1, mutated polycystin-1; mPC-2, mutated polycystin-2. SEM images published with permission.
Cholangiocyte primary cilia in polycystic liver disease
Cholangiocyte primary cilia, long tubular appendages extending from the apical domain of bile duct epithelial cells, function as mechanosensor [36], osmosensor [37] and chemosensors [38•] under normal conditions. The connection between hepatic cystogenesis and primary cilia was proposed several years ago [1•,25]. There are now substantial data describing functional (i.e., abnormal expression of ciliary-associated disease-related proteins in these organelles) and structural (i.e., shortened and malformed cilia) ciliary abnormalities in different animal models of PCLD [1•,39]. Detailed analysis of the liver in the BALB/c-cpk mouse revealed the presence of hepatic cysts, substantial fibrosis and much shorter cilia compared with normal cholangiocytes [40•]. It was also reported that primary cilia are abnormal in cholangiocytes lining liver cysts of ADPKD patients [26••,41•]. In small cysts (less than 1 cm), primary cilia appear to be normal; in medium-sized cysts (1–3 cm), they are short and aberrantly formed; in large cysts (more than 3 cm in diameter), these organelles are absent [26••,41•]. These observations further support the ciliary hypothesis of cystogenesis.
Therapies of polycystic liver disease
Due to advances in the understanding of the molecular mechanisms underlying hepatic and renal cystogenesis, as well as the availability of animal models for PKD and PCLD, potential treatments have emerged. The therapeutic options in the management of PKD include, in particular, the targeting of epidermal growth factor receptor (EGFR), the vasopressin receptor type 2 (V2R), Cdks, caspases and mammalian target of rapamycin (mTOR) pathway [42•,43,44•]. Although the effects of many of these inhibitors on hepatic cystogenesis are unknown, administration of a V2R inhibitor has no effect on hepatic cystogenesis due to the absence of V2R in cholangiocytes [43].
To date, available treatment strategies for PCLDs have been limited to surgical procedures mainly intended to reduce liver volume. However, some progress toward designing alternative therapies has recently been made. Octreotide, a somatostatin analogue that decreases elevated cAMP levels in cystic cells, attenuates hepatic and renal cyst volume and fibrosis in PCK rats [45]. Based on these animal studies, a randomized clinical trial of a somatostatin analogue is now underway at Mayo Clinic [45]. A recent publication suggested that intravenous administration of octreotide to one patient with severe PCLD resulted in a significant reduction in liver (by ~38.3%) and kidney (by ~18%) volumes; a second patient who received lanreotide (another somatostatin analogue) also showed significant improvement [46••]. Sirolimus, a drug that suppresses cell proliferation by targeting the mTOR pathway, also had a positive effect on liver cystogenesis (i.e., liver cyst volume was reduced by ~11%) in seven PCLD patients probably by preventing activation of mTOR in cystic cholangiocytes [47••].
Conclusion
Current progress in the identification of the genes that cause PCLD, increased understanding of the mechanisms underlying hepatic cystogenesis, recognition of the intracellular signaling pathways involved in disease progression and the availability of animal models, all provide an opportunity for the development of curative or preventive therapies for the cholangiociliopathies.
Acknowledgments
The authors have no conflicts of interest.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 297).
- 1•.Masyuk AI, Masyuk TV, LaRusso NF. Cholangiocyte primary cilia in liver health and disease. Dev Dyn. 2008;237:2007–2012. doi: 10.1002/dvdy.21530. Functions and role of cholangiocyte cilia in normal and pathological conditions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2•.Adams M, Smith UM, Logan CV, Johnson CA. Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. J Med Genet. 2008;45:257–267. doi: 10.1136/jmg.2007.054999. Role of cilia in development of ciliopathies. [DOI] [PubMed] [Google Scholar]
- 3•.Pan J. Cilia and ciliopathies: from Chlamydomonas and beyond. Sci China C Life Sci. 2008;51:479–486. doi: 10.1007/s11427-008-0071-3. Overview of various cilia-related disorders. [DOI] [PubMed] [Google Scholar]
- 4•.Hoevenaren IA, Wester R, Schrier RW, et al. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver Int. 2008;28:264–270. doi: 10.1111/j.1478-3231.2007.01595.x. Clinical features in ARPKD and ADPLD. [DOI] [PubMed] [Google Scholar]
- 5.Everson GT, Taylor MR, Doctor RB. Polycystic disease of the liver. Hepatology. 2004;40:774–782. doi: 10.1002/hep.20431. [DOI] [PubMed] [Google Scholar]
- 6.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 7•.Waanders E, Lameris AL, Op den Camp HJ, et al. Hepatocystin is not secreted in cyst fluid of hepatocystin mutant polycystic liver patients. J Proteome Res. 2008;7:2490–2495. doi: 10.1021/pr8000282. ADPLD protein, hepatocystin, is lost from cystic fluid. [DOI] [PubMed] [Google Scholar]
- 8•.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2008 doi: 10.1146/annurev.med.60.101707.125712. Epub ahead of print. Heterogeneity of PKD, animal models, therapies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ong AC, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int. 2005;67:1234–1247. doi: 10.1111/j.1523-1755.2005.00201.x. [DOI] [PubMed] [Google Scholar]
- 10.Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 11.Adeva M, El-Youssef M, Rossetti S, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD) Medicine (Baltimore) 2006;85:1–21. doi: 10.1097/01.md.0000200165.90373.9a. [DOI] [PubMed] [Google Scholar]
- 12•.Al-Bhalal L, Akhtar M. Molecular basis of autosomal recessive polycystic kidney disease (ARPKD) Adv Anat Pathol. 2008;15:54–58. doi: 10.1097/PAP.0b013e31815e5295. Molecular basis of hepatic and renal cystogenesis in ARPKD. [DOI] [PubMed] [Google Scholar]
- 13.Menezes LF, Onuchic LF. Molecular and cellular pathogenesis of autosomal recessive polycystic kidney disease. Braz J Med Biol Res. 2006;39:1537–1548. doi: 10.1590/s0100-879x2006001200004. [DOI] [PubMed] [Google Scholar]
- 14.Bergmann C, Kupper F, Schmitt CP, et al. Multiexon deletions of the PKHD1 gene cause autosomal recessive polycystic kidney disease (ARPKD) J Med Genet. 2005;42:e63. doi: 10.1136/jmg.2005.032318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15•.Gallagher AR, Esquivel EL, Briere TS, et al. Biliary and pancreatic dysgenesis in mice harboring a mutation in Pkhd1. Am J Pathol. 2008;172:417–429. doi: 10.2353/ajpath.2008.070381. Disruption of exon 4 of Pkhd1 results in biliary cystogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16•.Williams SS, Cobo-Stark P, James LR, et al. Kidney cysts, pancreatic cysts, and biliary disease in a mouse model of autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2008;23:733–741. doi: 10.1007/s00467-007-0735-4. Disruption of exons 1–3 of Pkhd1 results in renal and biliary phenotype. [DOI] [PubMed] [Google Scholar]
- 17.Bisceglia M, Galliani CA, Senger C, et al. Renal cystic diseases: a review. Adv Anat Pathol. 2006;13:26–56. doi: 10.1097/01.pap.0000201831.77472.d3. [DOI] [PubMed] [Google Scholar]
- 18.Dawe HR, Smith UM, Cullinane AR, et al. The Meckel–Gruber syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16:173–186. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- 19•.Klysik M. Ciliary syndromes and treatment. Pathol Res Pract. 2008;204:77–88. doi: 10.1016/j.prp.2007.10.013. Liver disorder in different ciliopathies. [DOI] [PubMed] [Google Scholar]
- 20•.Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatr Nephrol. 2008 doi: 10.1007/s00467-008-0840-z. [Epub ahead of print]. Genetics and clinical features in NPHP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hildebrandt F, Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet. 2005;6:928–940. doi: 10.1038/nrg1727. [DOI] [PubMed] [Google Scholar]
- 22•.Kim J, Krishnaswami SR, Gleeson JG. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet. 2008;17:3796–3805. doi: 10.1093/hmg/ddn277. CEP290, protein that causes MKS and JBST, is required for ciliogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harris PC, Torres VE. Understanding pathogenic mechanisms in polycystic kidney disease provides clues for therapy. Curr Opin Nephrol Hypertens. 2006;15:456–463. doi: 10.1097/01.mnh.0000232888.65895.e7. [DOI] [PubMed] [Google Scholar]
- 24•.Brancati F, Travaglini L, Zablocka D, et al. RPGRIP1L mutations are mainly associated with the cerebellorenal phenotype of Joubert syndrome-related disorders. Clin Genet. 2008;74:164–170. doi: 10.1111/j.1399-0004.2008.01047.x. Molecular basis of JBTS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masyuk T, LaRusso N. Polycystic liver disease: new insights into disease pathogenesis. Hepatology. 2006;43:906–908. doi: 10.1002/hep.21199. [DOI] [PubMed] [Google Scholar]
- 26••.Alvaro D, Onori P, Alpini G, et al. Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. Am J Pathol. 2008;172:321–332. doi: 10.2353/ajpath.2008.070293. Cholangiocyte cilia are abnormal in cystic epithelia of ADPKD patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27••.Amura CR, Brodsky KS, Gitomer B, et al. CXCR2 agonists in ADPKD liver cyst fluids promote cell proliferation. Am J Physiol Cell Physiol. 2008;294:C786–C796. doi: 10.1152/ajpcell.00457.2007. Agonists of CXCR2 promote cholangiocyte proliferation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28••.Waanders E, Van Krieken JH, Lameris AL, Drenth JP. Disrupted cell adhesion but not proliferation mediates cyst formation in polycystic liver disease. Mod Pathol. 2008;21:1293–1302. doi: 10.1038/modpathol.2008.115. Cell adhesion appears to play a major role in hepatic cystogenesis in ADPLD. [DOI] [PubMed] [Google Scholar]
- 29••.Banales JM, Masyuk TV, Bogert PS, et al. Hepatic cystogenesis is associated with abnormal expression and location of ion transporters and water channels in an animal model of autosomal recessive polycystic kidney disease. Am J Pathol. 2008;173:1637–1646. doi: 10.2353/ajpath.2008.080125. Overexpression of ion transporters and water channels is involved in hepatic cystogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson CA, Gissen P, Sergi C. Molecular pathology and genetics of congenital hepatorenal fibrocystic syndromes. J Med Genet. 2003;40:311–319. doi: 10.1136/jmg.40.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato Y, Harada K, Ozaki S, et al. Cholangiocytes with mesenchymal features contribute to progressive hepatic fibrosis of the polycystic kidney rat. Am J Pathol. 2007;171:1859–1871. doi: 10.2353/ajpath.2007.070337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32•.Omenetti A, Porrello A, Jung Y, et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest. 2008;118:3331–3342. doi: 10.1172/JCI35875. Hedgehog signaling may contribute to epithelial-mesenchymal cholangiocyte transition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karlsson-Rosenthal C, Millar JB. Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006;16:285–292. doi: 10.1016/j.tcb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 34••.Lee SO, Masyuk T, Splinter P, et al. MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J Clin Invest. 2008;118:3714–3724. doi: 10.1172/JCI34922. Novel mechanism of hepatic cystogenesis that involved microRNAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35•.Chu AS, Friedman JR. A role for microRNA in cystic liver and kidney diseases. J Clin Invest. 2008;118:3585–3587. doi: 10.1172/JCI36870. Many miRNAs may participate in pathogenesis of liver and kidney cyst development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masyuk AI, Masyuk TV, Splinter PL, et al. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–920. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gradilone SA, Masyuk AI, Splinter PL, et al. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc Natl Acad Sci U S A. 2007;104:19138–19143. doi: 10.1073/pnas.0705964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38•.Masyuk AI, Gradilone SA, Banales JM, et al. Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors. Am J Physiol Gastrointest Liver Physiol. 2008;295:G725–G734. doi: 10.1152/ajpgi.90265.2008. Cholangiocyte cilia function as a chemosensor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer E, Gresh L, Reimann A, Pontoglio M. Cystic kidney diseases: learning from animal models. Nephrol Dial Transplant. 2004;19:2700–2702. doi: 10.1093/ndt/gfh533. [DOI] [PubMed] [Google Scholar]
- 40•.Muchatuta MN, Gattone VH, Witzmann FA, Blazer-Yost BL. Structural and functional analyses of liver cysts from the BALB/c-cpk mouse model of polycystic kidney disease. Exp Biol Med (Maywood) 2008;234:17–27. doi: 10.3181/0807-RM-215. Cystic cholangiocyte cilia are abnormal in animal model of PCLD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41•.Alvaro D, Mancino MG. New insights on the molecular and cell biology of human cholangiopathies. Mol Aspects Med. 2008;29:50–57. doi: 10.1016/j.mam.2007.09.007. Cholangiocyte disorder in PCLD. [DOI] [PubMed] [Google Scholar]
- 42•.Chang MY, Ong AC. Autosomal dominant polycystic kidney disease: recent advances in pathogenesis and treatment. Nephron Physiol. 2008;108:1–7. doi: 10.1159/000112495. Recent advances in the development of PKD therapies. [DOI] [PubMed] [Google Scholar]
- 43.Torres VE, Harris PC. Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Intern Med. 2007;261:17–31. doi: 10.1111/j.1365-2796.2006.01743.x. [DOI] [PubMed] [Google Scholar]
- 44•.Edelstein CL. Mammalian target of rapamycin and caspase inhibitors in polycystic kidney disease. Clin J Am Soc Nephrol. 2008;3:1219–1226. doi: 10.2215/CJN.05611207. Therapies of PKD. [DOI] [PubMed] [Google Scholar]
- 45.Masyuk TV, Masyuk AI, Torres VE, et al. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology. 2007;132:1104–1116. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 46••.van Keimpema L, de Man RA, Drenth JP. Somatostatin analogues reduce liver volume in polycystic liver disease. Gut. 2008;57:1338–1339. doi: 10.1136/gut.2008.155721. Octreotide is a potential therapeutic agent for the treatment of hepatic cystogenesis. [DOI] [PubMed] [Google Scholar]
- 47••.Qian Q, Du H, King BF, et al. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol. 2008;19:631–638. doi: 10.1681/ASN.2007050626. Sirolimus treatment is associated with reduced liver volumes in patients with PCLD. [DOI] [PMC free article] [PubMed] [Google Scholar]