

Significance
The negative-strand RNA viruses comprise several significant human, animal, and plant pathogens that have considerable health and economic impact globally. During infection, replication of the single-stranded negative-sense RNA genome occurs through a complementary RNA intermediate, which is believed to complex with viral proteins to form a complementary ribonucleoprotein (cRNP). The isolation of these complexes from infected cells has never been accomplished, greatly hampering our understanding of genome replication. We report a technological advance for the isolation of this elusive but essential component of the influenza A virus replication machine. Structural and functional characterization of the influenza A virus cRNP has led to the proposal of a model of genome replication that relies on a trans-activating viral RNA-dependent RNA polymerase.
Abstract
Negative-strand RNA viruses represent a significant class of important pathogens that cause substantial morbidity and mortality in human and animal hosts worldwide. A defining feature of these viruses is that their single-stranded RNA genomes are of opposite polarity to messenger RNA and are replicated through a positive-sense intermediate. The replicative intermediate is thought to exist as a complementary ribonucleoprotein (cRNP) complex. However, isolation of such complexes from infected cells has never been accomplished. Here we report the development of an RNA-based affinity-purification strategy for the isolation of cRNPs of influenza A virus from infected cells. This technological advance enabled the structural and functional characterization of this elusive but essential component of the viral RNA replication machine. The cRNP exhibits a filamentous double-helical organization with defined termini, containing the viral RNA-dependent RNA polymerase (RdRp) at one end and a loop structure at the other end. In vitro characterization of cRNP activity yielded mechanistic insights into the workings of this RNA synthesis machine. In particular, we found that cRNPs show activity in vitro only in the presence of added RdRp. Intriguingly, a replication-inactive RdRp mutant was also able to activate cRNP-templated viral RNA synthesis. We propose a model of influenza virus genome replication that relies on the trans-activation of the cRNP-associated RdRp. The described purification strategy should be applicable to other negative-strand RNA viruses and will promote studies into their replication mechanisms.
Negative-strand RNA viruses include several major human and animal pathogens that cause substantial morbidity and deaths worldwide, as well as several important plant pathogens that are responsible for plant diseases with considerable economic impact. Notable examples of human pathogens are influenza, rabies, mumps, measles, and respiratory syncytial virus, as well as zoonotic, hemorrhagic fever-causing Ebola and Lassa viruses. A defining feature of these viruses is that their single-stranded RNA genomes are of opposite polarity to messenger RNA and are replicated through a cRNA intermediate (1). The genomic viral RNA (vRNA) molecules of negative-strand RNA viruses are assembled into viral ribonucleoprotein particles (vRNPs) containing an oligomer of nucleoprotein (NP) and the RNA-dependent RNA polymerase (RdRp), which is responsible for transcription and replication of the viral genome. High-resolution structures of the genomic ribonucleoproteins of negative-strand RNA viruses have been determined by means of EM, the best-studied example of which is the vRNP of influenza A virus (2, 3).
The genome of influenza A virus is segmented into eight individual vRNP complexes that display a double-helical arrangement resembling a large loop twisted into a helical filament. One copy of the 250-kDa heterotrimeric RdRp, containing the polymerase basic 1 (PB1), polymerase basic 2 (PB2), and polymerase acidic (PA) subunits, binds at one end of the filament interacting with both the 5′ and the 3′ ends of the vRNA (2, 3). Influenza viruses initiate infection by delivering these highly specialized RNA synthesis machines into the nucleus of a host cell for the expression of viral genes. Upon entry into the nucleus, the vRNP-associated RdRp is responsible for transcription of the parental vRNAs into capped and polyadenylated mRNAs using 5′-capped RNA primers snatched from host pre-mRNAs (4, 5). The capped RNA primers are generated by the viral RdRp. Specifically, the PB2 subunit of the RdRp binds to the cap structure of host pre-mRNAs by sandwiching the 7-methyl guanine between two aromatic amino acid residues, whereas the PA subunit endonucleolytically cleaves the capped RNA 9–15 nt downstream of the cap. Subsequently, these primers are extended by condensation of nucleoside triphosphates by the RdRp catalytic domain within the PB1 subunit. Polyadenylation occurs by stuttering of the RdRp on a stretch of uridine residues ∼16 nt from the 5′ end of the vRNA template. Replication of the viral genome is also catalyzed by the viral RdRp, first by synthesizing full-length positive polarity cRNA de novo, which is believed to assemble with NP and the RdRp into a complementary ribonucleoprotein (cRNP). This cRNP then serves as a replication intermediate for the synthesis of multiple copies of progeny vRNPs, which subsequently serve for secondary rounds of transcription, genome amplification, or are exported from the nucleus for packaging into progeny virions (6). Although a cRNP complex has long been assumed to serve as a replicative intermediate for all negative-strand RNA viruses, such complexes have never been isolated. This shortcoming can be attributed to the technical difficulties of separating cRNPs from vRNPs during purification from infected cells and the low ratio of cRNPs to vRNPs in infected cells (7, 8). Consequently, cRNP complexes remain uncharacterized both structurally and functionally, greatly hampering our understanding of an essential aspect in the replication process of negative-strand RNA virus genomes.
To understand the overall molecular architecture of the influenza virus ribonucleoprotein complexes and the basic molecular requirements of influenza virus genome replication, we set out to isolate the replicative cRNP intermediate from infected cells using an RNA tag and affinity purification. Electron microscopy and single particle analysis revealed that the cRNPs isolated from infected cells exist as individual particles that exhibit a double-helical arrangement consisting of cRNA, NP, and the RdRp. Unexpectedly, it was shown in vRNA synthesis assays that cRNP activity requires a trans-activating RdRp for initiation and processivity by the resident RdRp. By using a replication-inactive mutant of the RdRp, we show that the role of this trans-activating RdRp is not enzymatic. These findings allow us to propose a model for genomic vRNA synthesis by the influenza virus replicative intermediate cRNP.
Results
RNA-Affinity Tag-Based Strategy for cRNP and vRNP Purification.
To isolate influenza virus cRNPs and vRNPs separately from infected cells we sought to use an affinity purification of RNPs based on an RNA tag. We took advantage of the translational operator RNA hairpin from the Pseudomonas aeruginosa bacteriophage PP7 (PP7) that binds with high affinity (Kd = ∼1 nM) to the coat protein of PP7 (PP7CP) (9, 10). We generated recombinant influenza A/WSN/33 viruses by reverse genetics (11) that encode the PP7 RNA tag in the neuraminidase (NA) gene allowing the isolation of either cRNA or vRNA, depending on the orientation of the tag (Fig. 1A). The RNA-tag was inserted into the region of the NA gene that encodes the NA stalk (12). Analysis of wild-type and recombinant viruses revealed that virus growth was unaffected by the presence of the RNA tag (Fig. 1 B and C), demonstrating that heterologous RNA secondary structures can be introduced into the influenza virus genome without impairing genome encapsidation. Furthermore, the corresponding insertion in the NA protein did not hamper virus release from the host cells in cell culture conditions, consistent with previous reports showing that insertions and deletions in the NA stalk can be tolerated by the virus (13–16).
Fig. 1.

Generation of recombinant influenza A viruses containing PP7 RNA tag in the NA. (A) PP7 RNA was inserted into the NA gene of influenza A/WSN/33 virus to allow RNA-affinity purification of cRNA or vRNA. (Left) Sequence of the PP7 RNA tag. (Right) Position of the PP7 RNA tag in the NA cRNA and vRNA. Untranslated terminal sequences (UTR), the NA ORF (black box), and the PP7 RNA tag are shown. (B) Growth curves of wild-type [(–) control] and tagged cRNA-PP7 and vRNA-PP7 viruses in MDBK cells. (C) Plaque phenotype of wild-type [(–) control] and tagged cRNA-PP7 and vRNA-PP7 viruses on MDBK cells infected for 72 h before staining with Coomassie Brilliant Blue.
Purification of Influenza A Virus cRNP and vRNP Separately from Infected Cells.
To purify PP7-tagged RNA, we established a stable HEK 293T cell line that expresses PP7CP. Alternatively, expression was achieved transiently by transfection. Infection of cells expressing PP7CP resulted in endogenous assembly of viral RNP complexes containing PP7CP for affinity purification. Target viral NA cRNPs and vRNPs were separated with high specificity using RNA-based affinity chromatography and density gradient centrifugation (Fig. 2 and Figs. S1 and S2). Analysis of RNA and protein revealed that the cRNP and vRNP contained RdRp, NP, and cRNA or vRNA, respectively (Fig. 2). On average, 20.0% of the total cRNA and 11.8% of the total vRNA from cell lysates could be purified. However, only 3.2% of total mRNA could be recovered in the purification process (Fig. 2B). These data demonstrate that an RNA tag-based affinity purification allows the separate isolation of influenza virus cRNPs and vRNPs and that sufficient flexibility exists within the cRNPs and vRNPs for secondary RNA structures to form that are not eliminated as a result of encapsidation by oligomeric NP.
Fig. 2.

Isolation of a cRNP replicative intermediate and vRNP from influenza virus-infected cells. (A) Primer extension analysis of total and purified NA-specific RNA from HEK 293T-PP7CP cells infected with wild-type (–), cRNA-PP7, or vRNA-PP7 viruses. Viral RNAs were analyzed by primer extension using primers specific for positive sense (mRNA and cRNA) and negative sense (vRNA) NA-specific viral RNAs that generate cDNAs with an expected size of 169–177 nt from mRNA (dependent on the length of the capped mRNA primer), 160 nt from cRNA, and 129 nt from vRNA. Primer extension analysis of 5S rRNA was used as a control producing a cDNA product of 100 nt. Ten times more sample of purified RNA was analyzed relative to total RNA. (B) Quantitation of primer extension analysis of purified RNA. RNA levels represent the mean, and the error bars represent SDs from three biological repeats. (C) Analysis of purified cRNP and vRNP fractions from RNA-affinity purifications and density glycerol gradients by SDS/PAGE and staining with silver. Note that lower amounts of cRNPs were purified compared with vRNPs owing to a lower accumulation of cRNA in infected cells (7, 8). In the cRNP lane the intensity of the PB1 band appears stronger than that of the PB2/PA bands owing to the presence of a contaminating cellular protein that migrates at the same position (Fig. S2). (D) Analysis of PB2 and NP in purified RNPs by Western blot.
Influenza Virus cRNP Exhibits a Double-Helical Arrangement Consisting of cRNA, NP, and the RdRp.
Negative-stain EM of NA cRNPs revealed filamentous particles (Fig. 3A) with a median length of 72 nm (n = 231), in agreement with the length of influenza virus vRNP segments of intermediate length in nucleotides (17). A dataset of 2,456 cRNP images was obtained, revealing that the filaments were often highly flexible (Fig. S3). This flexibility resulted in disorder in the 2D class averages of the particles, especially at the termini (Fig. 3B). To gain insight into the molecular architecture of the central filament and the termini, the corresponding subimages were classified independently. The central filament class averages revealed repeating triangular structural motifs, consistent with a double-helical arrangement of NP (Fig. 3C). We classified the termini and then split them into two groups, comprising either arrowhead-shaped (Fig. 3D) or looped structures (Fig. 3E), and these were interpreted to correspond to side views of the RdRp and NP-RNA loops, respectively. The RdRp structure exhibited a large and a small polymerase domain and adopted different angles relative to the central filament (Fig. 3D). The NP-RNA loops displayed varying diameters (∼14–23 nm) corresponding to approximately three to seven NP molecules, suggesting a degree of flexibility of the loop structure.
Fig. 3.

Structural organization of the influenza virus cRNP replicative intermediate. (A) One cRNP particle, extracted from an electron micrograph, is shown. NP-RNA loop end (L), central helical filament (C), and RdRp end (P) are labeled. (Scale bar, 20 nm.) (B) Class averages of cRNP particles. The two termini (T and T′) were not resolved in initial class averages. Further classification concentrated on subimages of the termini and the central filament (C). (C) Class averages of the central filament reveal variation due to variable curvature of the filament and different orientations around the filament long axis. Depending on the orientation, repeating triangular structural motifs were visible (triangle). (D) Class averages of the termini representing RdRp side views. Two globular densities, putatively corresponding to a large (white triangle) and small (black triangle) structural feature of RdRp are indicated. (E) Class averages of the NP-RNA loop side views reveal variation in loop diameter. The scale of B–E is the same as in A.
Purified vRNPs but Not cRNPs Are Active in Vitro.
The separation of cRNP and vRNP from infected cells enabled us to determine and compare the basic molecular requirements for viral RNA synthesis between these viral RNP complexes in in vitro activity assays. Viral RNAs were detected by performing a primer extension assay by using two radiolabeled primers that analyze the 5′ end of the template and product, one specific for NA mRNA and cRNA and the other specific for vRNA (7, 18). As demonstrated by the synthesis of de novo initiated cRNA, vRNPs were able to replicate (Fig. 4A). Replication activity was greatly enhanced in the presence of an ApG dinucleotide primer that is complementary to the 3′ end of the vRNA template (Fig. 4A). vRNPs were also able to synthesize 5′ capped mRNA when β-globin mRNA was provided as a cap donor. In the presence of the cap donor, vRNPs also synthesized de novo initiated cRNA, as reported previously (19), although we cannot exclude entirely that some of these products represent de-capped viral mRNAs. In contrast, cRNPs were unable to synthesize detectable amounts of vRNA de novo or in the presence of an ApG dinucleotide that is complementary to the 3′ end of the cRNA template (Fig. 4A).
Fig. 4.

A trans-activating RdRp is required for cRNP activity. In vitro activity assays of purified NA cRNPs (c) and vRNPs (v) in the absence of rNTPs as a negative control to indicate template RNA (Template) or (A) in the presence of rNTPs but absence of primer (–Primer), presence of ApG dinucleotide primer (ApG), or capped RNA (β-globin mRNA), (B) in the presence of rNTPs and ApG dinucleotide primer without (–Pol) or with (+Pol) added recombinant influenza virus RdRp purified from insect cells. Templates and transcription products were analyzed by primer extension using primers specific for positive sense (mRNA and cRNA) or negative sense (vRNA) NA-specific viral RNAs as described for Fig 2A. c* denotes primer extension analysis in the absence of vRNA-specific primer. Small arrows indicate products of vRNP activity, and large arrows indicate products of cRNP activity that form a doublet. Note that the weak vRNA-specific signals observed in the Template and –Pol cRNP lanes represent small amounts of vRNA that copurify with cRNPs.
A Trans-Activating RdRp Is Required for cRNP Activity.
Earlier work has proposed a model whereby vRNA synthesis relies on a trans-acting RdRp for replication (20). We therefore hypothesized that the inability of the cRNP to synthesize vRNA is due to the absence of an RdRp acting in trans. To test this hypothesis, we included recombinant RdRp purified from Spodoptera frugiperda cells (Fig. S4A) in the cRNP activity assays. Only in the presence of additional RdRp did we observe the synthesis of vRNA from the cRNA template. Two primer extension products with slightly different sizes were observed (Fig. 4B), the shorter product likely representing vRNA initiated internally at position 4 on the cRNA promoter, consistent with the previously proposed internal initiation model for the cRNA promoter (21). In the absence of a vRNA-specific radiolabeled primer the products of cRNP activity were not detected (Fig. 4B). A 4.1-fold increase in vRNA signal was observed in the presence of added recombinant RdRp, compared with no recombinant RdRp being present.
The Trans-Activating RdRp Acts Cooperatively with the Resident RdRp, Which Carries Out the vRNA Synthesis.
Does the recombinant RdRp act in trans in agreement with previously proposed models (3, 20), or does it instead have an uncharacterized function for supporting the cRNP-associated RdRp to act in cis? To address this question, recombinant wild-type RdRp or PB1 active-site mutant (PB1a) RdRp was purified from mammalian cells (Fig. S4 B and C) and included in the in vitro assays. Unexpectedly, both wild-type and active-site mutant RdRp were able to promote cRNP activity, resulting in the synthesis of vRNA 3.1-fold and 2.8-fold over background for the wild-type and the active-site mutant RdRp, respectively (Fig. 5A). These results suggest that the recombinant RdRp does not act as a replicase in trans, but instead, it acts cooperatively with the resident RdRp, which acts as a replicase in cis.
Fig. 5.
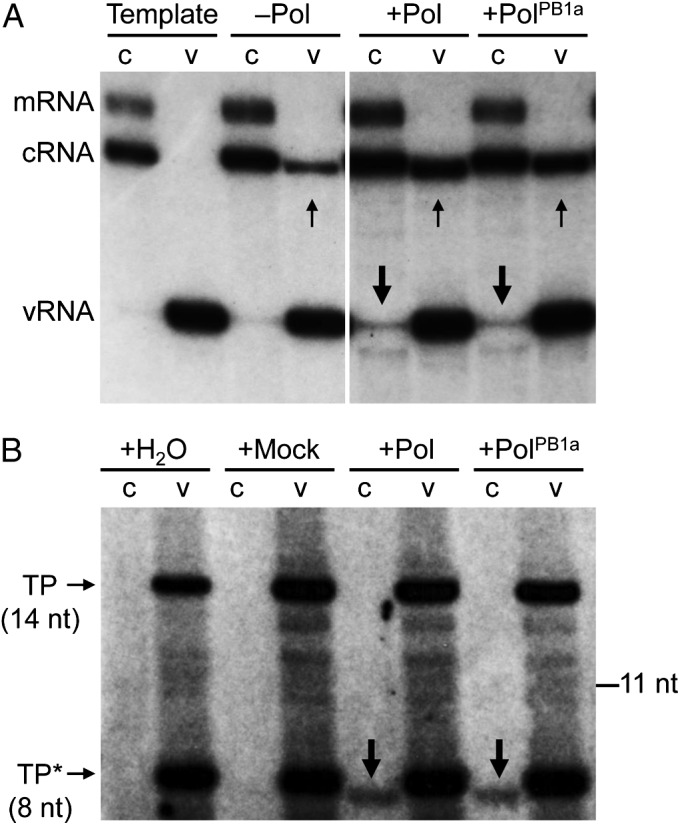
Trans-activating RdRp acts cooperatively with the resident RdRp, which carries out vRNA synthesis. (A) In vitro activity assays of purified NA cRNPs (c) and vRNPs (v) in the absence of rNTPs as a negative control to indicate template RNA (Template) or in the presence of rNTPs without (–Pol) or with added recombinant wild-type (+Pol) or active-site mutant (+PolPB1a) influenza virus RdRp purified from transfected HEK 293T cells. Templates and transcription products were analyzed by primer extension using primers specific for positive sense (mRNA and cRNA) or negative sense (vRNA) NA-specific viral RNAs. Small arrows indicate products of vRNP activity, and large arrows indicate products of cRNP activity that form a doublet. Note that small amounts of vRNA copurifying with cRNP were detected in the Template and –Pol cRNP lanes. (B) In vitro activity assays of purified NA cRNPs (c) and vRNPs (v) using [α-32P]NTP incorporation in the presence of ApG primer and water (+H2O), mock polymerase (+Mock), wild-type (+Pol), or active-site mutant (+PolPB1a) RdRp purified from transfected HEK 293T cells. TP (14 nt) denotes the transcription product derived from the vRNA template, and TP* (8 nt) denotes a premature termination product due to limiting CTP concentrations. The 8-nt-long transcription product derived from the cRNA template is denoted by large arrows.
To investigate this further, we used an alternative in vitro transcription assay that involves the incorporation of radiolabeled nucleoside triphosphates. To analyze vRNP activity we set up a reaction leaving out UTP, expecting a 14-nt-long transcription product as the first ATP occurs at position 15 in the vRNA template. Analogously, to analyze cRNP activity we left out CTP, expecting an 8-nt-long transcription product. Consistent with previous results (Fig. 5A), cRNPs showed activity in presence of wild-type RdRp as well as in the presence of the PB1a active-site mutant RdRp (Fig. 5B). Taken together, these data show that the cRNP-associated RdRp requires a trans-activating RdRp with a nonenzymatic role.
Discussion
The principal advancement of this study is the isolation of a replicative intermediate of a negative-strand virus, permitting visualization and functional characterization of such complexes. Our data revealed that the replicative intermediate of influenza virus exists as an individual structured particle that comprises cRNA, NP, and RdRp. We observed that the cRNP particle contains three major domains, the central filament and two distinct termini, one terminus harboring a loop and the other a structure that we interpret as the RdRp, on the basis of analogy to previously published structures of vRNPs (2, 3). The central filament exhibited a helical arrangement, similar to the reported vRNP filament structure (2, 3) in which the helical arrangement of the filament is facilitated by NP–NP interactions. We observed a varying diameter of the loop at the terminus of the cRNP, with three to seven NP molecules being present within the loop. This observation is consistent with previous reports that the loop end of the vRNP comprises five to eight NP molecules in one study (3) and three NP molecules in another study (2). In contrast to previous studies, which analyzed a mixture of vRNPs corresponding to RNA segments of various lengths, here we analyze one particular cRNP with an RNA segment of a defined length. The fact that these cRNPs exhibit loops with varying numbers of NP molecules implies either slightly different degrees of helical winding in the central filament or, perhaps more likely, different degrees of unwinding at the loop-end of the filament. The opposite terminus contained the RdRp exhibiting a small and a large structural feature. Previous EM studies of vRNP-associated RdRp also found that the RdRp shows two structural features, the larger feature possibly corresponding to PB1, PB2, and the N terminus of PA, whereas the smaller feature corresponds to the C-terminal domain of PA (3). Further studies using higher-resolution approaches (e.g. cryo-EM) are required to answer whether a similar or different arrangement of subunits exists within the cRNP-associated RdRp.
The specific purification of PP7 RNA-tagged RNPs relies on the accessibility of a surface exposed RNA tag that is not bound by NP. It remains unclear whether the formation of the PP7 RNA secondary structure is sufficient to prevent encapsidation by NP or the binding of PP7CP is required to stabilize the RNA structure and prevent the melting by NP. The presence of an RNA tag that is accessible within the vRNP and cRNP structures of influenza virus for the binding of PP7CP provides experimental evidence that sufficient flexibility exists within viral ribonucleoproteins for secondary RNA structures to form and that such structures do not prevent read-through by the RdRp. Local secondary RNA structures are also believed to play a role in the packaging process of the eight vRNP complexes into progeny virions. Interactions between such RNA secondary structures may mediate the interaction between the eight vRNPs, leading to the observed 7+1 arrangement of vRNPs within budding virions as visualized by EM (22–25).
Interestingly, viral mRNA was purified with a significantly lower efficiency than cRNA or vRNA (Fig. 2B). This finding suggests that the PP7 RNA tag is less accessible for PP7CP binding in the NA mRNPs, possibly reflecting the association of viral mRNAs with numerous cellular factors involved in their processing, nuclear export, and translation (26). The potential for secondary RNA structures to form in the ribonucleoproteins of other negative-strand RNA viruses is not known. In contrast to influenza virus RNPs, the RNA associated with the majority of negative-strand RNA virus RNPs is more resistant to nuclease digestion, suggesting a tighter packaging of RNA within the RNP structure (27). The described method using an RNA tag might help to address the question of whether RNA secondary structures can form within these RNPs.
The isolation of cRNPs presented in this study allowed us to advance the model of genome replication for influenza viruses by performing in vitro functional assays. Previously proposed models of influenza virus RNP function suggest that replication is performed by a trans-acting RdRp (3, 20). It has been observed, by using a genetic trans-complementation approach, that the replication activity of a replication-defective RNP could be rescued in the presence of a replication-competent trans-acting RdRp (20). Consistent with this observation, EM studies of RNPs purified from transfected cells revealed short RNPs branching from longer RNPs with an RdRp being present at both the branch point and termini (3).
In contrast to this model, our model on cRNP function suggests that replication (cRNA→vRNA) is performed by the RdRp that is resident in the cRNP complex and that a trans-activating polymerase is required for activation of vRNA synthesis. This model is supported by two findings from our in vitro experiments. First, cRNP showed no detectable activity in vRNA synthesis assays, but activity could be gained by the addition of purified RdRp in trans. Second, the result that the addition of an RNA synthesis inactive mutant of the RdRp had the same effect as the wild-type RdRp demonstrates that replication is performed by the resident RdRp in cis. The trans-activating RdRp, however, is required for activation of the resident RdRp. It could be argued that there is an exchange at the cRNA promoter between the active and inactive RdRps such that the active RdRp could then act in trans on the template. Although this possibility cannot be excluded, previous data showed that binding of RdRp to the 5′ end of the promoter cannot be outcompeted by a preexpressed RdRp (19, 20), suggesting that an exchange is unlikely. We speculate that the trans-activating RdRp either provides structural support or induces a conformational change of the resident RdRp. This may occur through the formation of a quaternary structure by an interaction between the two RdRps. Indeed, it has been proposed that the influenza virus RdRp can form oligomers, possibly through interactions between PB1 and/or PB2 subunits (28, 29).
The cooperation and oligomerization between RdRps has been reported to be necessary for viral RNA synthesis in several RNA viruses, including poliovirus (30), hepatitis C virus (31), and Sendai virus (32). Additionally, the HIV-1 reverse transcriptase functions as a dimeric complex of the p66 subunit and a processed product p51 (33). Together with our study, these findings suggest that multimerization-driven stimulation of RNA polymerase activity is a common theme in RNA viruses. Whether the arrangement of RdRps plays an architectural role, required for the reorganization of RNA structural motifs or the rearrangement of the active site of the cRNP-associated replicating RdRp, remains unknown.
To conclude, the presented approach for isolating the replicative intermediates of negative-strand RNA viruses represents a powerful method for understanding how vRNA replication works in influenza virus infection. The approach should be applicable to the study of any other negative-strand RNA virus. This will undoubtedly yield new mechanistic insights into the workings of the RNA synthesis machines shared by a broad group of important human, animal, and plant pathogens.
Materials and Methods
Cells.
Human embryonic kidney 293T (HEK 293T) cells and Madin-Darby bovine kidney epithelial (MDBK) cells were maintained in DMEM (PAA) or modified Eagle medium with Earle’s salts (MEM; PAA), respectively, supplemented with 2 mM l-glutamine (PAA) and 10% FCS (PAA). An HEK 293T-PP7CP stable cell line was produced under the selection of hygromycin-B (Roche). For this purpose, 30 µg of pcDNA-PP7CP-His was transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions into ∼4 × 106 HEK 293T cells in 10-cm dishes. After 48 h, the cells were replated into 15-cm dishes containing DMEM supplemented with 10% FCS and hygromycin-B (500 µg/mL). Stable clone selection occurred in the presence of hygromycin-B (500 µg/mL) for ∼30 d. Stable colonies resistant to hygromycin-B were picked and recultured, and protein expression was confirmed by Western blotting.
Viruses.
Influenza A/WSN/33 virus (WSN) was cultured on MDBK cells in MEM supplemented with 2 mM l-glutamine and 0.5% FCS. Influenza A/WSN/33 NA cRNA-PP7 and vRNA-PP7 influenza viruses were generated using the 12 plasmid-based reverse genetics rescue system (11). An XbaI site was introduced into the pPOLI-NA-RT plasmid by site-directed mutagenesis [(5′-GGCAGCATTACCTCTAGAGTTGTTGC-3′) and (5′-GCAACAACTCTAGAGGTAATGCTGCC-3′)]. The PP7 RNA tag was inserted by annealing two complementary oligonucleotides with XbaI restriction sites at the 5′ and 3′ ends [(5′-CTAGTAAGGAGTTTATATGGAAACCCTTAT-3′) and (5′-CTAGATAAGGGTTTCCATATAAACTCCTTA-3′)] for NA cRNA-PP7 and [(5′-CTAGTAAGGGTTTCCATATAAACTCCTTAT-3′) and (5′-CTAGATAAGGAGTTTATATGGAAACCCTTA-3′)] for NA vRNA-PP7 and ligating into the XbaI-digested pPOLI-NA-XbaI plasmid. Underlined nucleotides correspond to the PP7 RNA tag recognized by PP7CP. Insertion of the PP7 RNA tag resulted in the replacement of amino acids Y56 and K57 in the NA stalk with SSKEFIWKPLSR in NA cRNA-PP7 and SSKGFHINSLSR in NA vRNA-PP7.
Plasmids.
The pcDNA-PP7CP-His and pcDNA-PP7CP-Strep expression plasmids were constructed by PCR amplifying the PP7 gene from pETZZ-tev-PP7CP (kind gift from J. Robert Hogg, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda) (10) with NotI or KpnI at the 5′ end and a 6-His tag or One-Strep and a XbaI or NotI restriction site at the 3′ end, respectively. The PCR product and pcDNA3A (34) were double-digested with the relevant restriction endonucleases, and the PCR amplified gene was ligated into pcDNA3A.
Growth Curve Analysis.
Growth curve analysis was performed by infecting 80% confluent MDBK cells in 3.5-cm dishes at a multiplicity of infection (MOI) of 0.001 with wild-type or PP7-tagged influenza/A/WSN/33 viruses and harvesting the supernatant every 12 h after infection for 72 h. Viral titers were determined by plaque assay.
Isolation of RNPs by RNA-Based Affinity Purification.
Approximately 3 × 107 HEK 293T-PP7CP-His cells were seeded into 15-cm dishes and cultured for 48 h in DMEM supplemented with 10% FCS and hygromycin-B (500 µg/mL) at 37 °C, 5% CO2. Alternatively, ∼3 × 107 HEK 293T cells were transfected with pcDNA-PP7CP-Strep using Lipofectamine 2000 according to the manufacturer’s instructions and incubated for 48 h in DMEM supplemented with 10% FCS at 37 °C, 5% CO2. Cells were rinsed with PBS and infected at an MOI of 5 for 1 h at room temperature with wild-type or PP7-tagged influenza A/WSN/33 viruses and incubated for a further 6–9 h in DMEM (0.5% FCS) at 37 °C, 5% CO2. Six to nine hours after infection, cells were collected by centrifugation at 200 × g for 5 min at 4 °C and washed with ice-cold PBS. Cells were collected by centrifugation at 200 × g for 5 min at 4 °C, flash-frozen, and stored at −80 °C.
vRNPs were purified either by Ni-NTA or Strep-tag affinity purification. For Ni-NTA affinity purification of RNPs, cells were thawed on ice and lysed in 4 mL of Tris lysis buffer [50 mM Tris·HCl (pH 8.0), 200 mM NaCl, 33% (vol/vol) glycerol, 20 mM imidazole, 0.5% (vol/vol) Igepal CA-630 (Sigma), 1 mM DTT, and 1× complete EDTA-free protease inhibitor mixture (Roche)] by rotation for 1 h at 4 °C, and then the soluble and insoluble fractions were separated by centrifugation at 17,000 × g for 5 min at 4 °C. The soluble fraction was diluted 1:5 with Ni-NTA binding buffer [20 mM Tris·HCl (pH 8.0), 200 mM NaCl, 20 mM imidazole, 1 mM MgCl2, and 1× complete EDTA-free protease inhibitor mixture] and incubated with 4 mL Ni-NTA agarose (Qiagen) for 2 h by rotation at 4 °C. Ni-NTA agarose was washed extensively with Ni-NTA wash buffer [20 mM Tris·HCl (pH 8.0), 200 mM NaCl, 40 mM imidazole, 1 mM MgCl2, 0.1% (vol/vol) Igepal CA-630, 10% (vol/vol) glycerol, and 1× phenylmethanesulfonylfluoride], and PP7CP-bound RNPs were eluted in 5 mL of Ni-NTA elution buffer [20 mM Tris·HCl (pH 8.0), 200 mM NaCl, 250 mM imidazole, 1 mM MgCl2, 0.1% (vol/vol) Igepal CA-630, 10% (vol/vol) glycerol, and 1× complete EDTA-free protease inhibitor mixture] for 2 h at 4 °C by rotation. Eluates were subsequently analyzed for proteins by SDS/PAGE and silver staining, and RNA was extracted and analyzed for by primer extension using NA-specific primers as previously described (18). Quantitation of primer extension products was performed by phosphorimage analysis using Aida software (Raytest). Eluates were concentrated to 1 mL using Amicon Ultra-4 (100K MWCO) centrifugation devices (Millipore) before density gradient centrifugation.
For Strep-tag affinity-purification of RNPs, cells were thawed on ice and lysed in 4 mL of Tris lysis buffer [50 mM Tris·HCl (pH 8.0), 200 mM NaCl, 33% (vol/vol) glycerol, 0.5% (vol/vol) Igepal CA-630, 1 mM DTT, and 1× complete EDTA-free protease inhibitor mixture] by rotation for 1 h at 4 °C, and then the soluble and insoluble fractions were separated by centrifugation at 17,000 × g for 5 min at 4 °C. The soluble fraction was diluted 1:5 with Strep-Tactin binding buffer [20 mM Tris·HCl (pH 8.0), 200 mM NaCl, 1 mM MgCl2, and 1× complete EDTA-free protease inhibitor mixture] and incubated overnight at 4 °C with 0.5 mL of 50% suspension Strep-Tactin Superflow high-capacity resin (IBA GmbH). Strep-Tactin resin was washed extensively with Strep-Tactin wash buffer [20 mM Tris·HCl (pH 8.0), 200 mM NaCl, 1 mM MgCl2, 0.1% (vol/vol) Igepal CA-630, 10% (vol/vol) glycerol, and 1× phenylmethanesulfonylfluoride], and PP7CP-bound RNPs were eluted in 5 mL of Strep-Tactin elution buffer [20 mM Tris·HCl (pH 8.0), 200 mM NaCl, 1 mM MgCl2, 0.1% (vol/vol) Igepal CA-630, 10% (vol/vol) glycerol, 2 mM d-Desthiobiotin (IBA GmbH), and 1× complete EDTA-free protease inhibitor mixture] for 2 h at 4 °C by rotation. Eluates were subsequently analyzed for proteins by SDS/PAGE and silver staining, and RNA was extracted and analyzed for by primer extension using NA-specific primers. Eluates were concentrated to 1 mL using Amicon Ultra-4 (100K MWCO) centrifugation devices (Millipore) before density gradient centrifugation.
RNPs were isolated by ultracentrifugation on a discontinuous glycerol gradient [1 mL each of 70%, 50%, 40%, and 33% (vol/vol)] in 50 mM Tris·HCl (pH 8.0), 200 mM NaCl, and 1 mM MgCl2. The gradients were centrifuged at 192,000 × g in a Beckman SW55Ti rotor for 4 h at 4 °C. Fractions of ∼300 μL were collected dropwise from the bottom of the tube. Fractions were analyzed for RNA by primer extension using NA-specific primers. Proteins were analyzed by SDS/PAGE and silver staining and Western blotting using polyclonal antibodies directed against PB2 and NP. Fractions enriched with RNPs were pooled and used for EM and in vitro activity assays.
EM and Image Processing.
A 5-μL aliquot of purified cRNP was applied to a glow-discharged 200 mesh carbon-formvar copper grid, and grids were stained with 2% uranyl acetate. Images were taken on CCD (UltraScan 4000SP; Gatan) with a transmission electron microscope (Tecnai F30; FEI) operated at 200 kV and at 39,000× nominal magnification. Contrast transfer function estimation and phase flipping was carried out using XMIPP (http://xmipp.cnb.csic.es/) (35). Images were down-sampled by a factor of two, resulting in a pixel size of 6.08 Å/pixels. In total, 529 images were taken, resulting in a dataset of 2,456 cRNP particles. Extracted particles (150 × 150 pixels) were subjected to simultaneous alignment and classification in Relion (http://www2.mrc-lmb.cam.ac.uk/relion/) (36). The approximate coordinates of the termini were defined in the selected class averages, and 2,456 corresponding subimages (80 × 80 pixels) were extracted from the original micrographs. Subimages were aligned and classified using the in-plane rotation angle of the central filament (psi) as prior information. To derive final class-average images of the RdRp and NP-RNA loop ends, particles assigned to either group were subjected to further 2D classification.
Recombinant Influenza Virus RdRp Purification.
Recombinant influenza A/NT/60/68 virus RdRp with a protein-A tag on the PB2 subunit was expressed using the Multibac system in Sf9 insect cells (37, 38). Sf9 cell suspension cultures at 2.0 ×106 cells/mL were infected with recombinant baculovirus at an MOI of 0.5–1.0 and incubated for 72 h at 27 °C. Cells were harvested by centrifugation and lysed by sonication in lysis buffer [50 mM Hepes·NaOH (pH 7.5), 500 mM NaCl, 10% (vol/vol) glycerol, 0.05% (wt/vol) octylthioglucoside, 1× complete EDTA-free protease inhibitor mixture, and 100 μg/mL RNase A]. The lysate was clarified by centrifugation, and the supernatant was incubated with washed IgG Sepharose (GE Healthcare; ∼2 mL bead slurry per liter of original culture) for 3–4 h at 4 °C. After binding, the beads were washed with 50 mM Hepes·NaOH (pH 7.5), 500 mM NaCl, 10% (vol/vol) glycerol, and 0.05% (wt/vol) octylthioglucoside. The polymerase was released using AcTEV protease (150 U per mL of IgG Sepharose) at 4 °C overnight in the above wash buffer supplemented with 2.5 mM reduced glutathione. The final purification step was size exclusion chromatography through a HiLoadTM 16/60 Superdex S200 column (GE Healthcare) at 4 °C, using 500 mM NaCl, 50 mM Hepes·NaOH (pH 7.5), 10% (vol/vol) glycerol, 1 mM MgCl2, and 0.1 mM MnCl2.
Recombinant influenza A/WSN/33 virus RdRp expressed in mammalian cells was prepared by tandem affinity purification (37) by transiently transfecting ∼2 × 107 HEK 293T cells with 60 µg of each of the RdRp expression plasmids (pcDNA-PA, pcDNA-PB2-TAP, and pcDNA-PB1 for wild-type RdRp or pcDNA-PB1a for active-site mutant RdRp) (7, 34, 39) using Lipofectamine 2000 according to the manufacturer’s instructions. Cells were incubated for 48 h in DMEM supplemented with 10% FCS at 37 °C, 5% CO2 before collection by centrifugation at 200 × g for 5 min at 4 °C. Cells were then washed with ice-cold PBS and pelleted by centrifugation at 200 × g for 5 min at 4 °C and then lysed in 2 mL of Tris lysis buffer [50 mM Tris·HCl (pH 8.0), 200 mM NaCl, 33% (vol/vol) glycerol, 0.5% (vol/vol) Igepal CA-630, 1 mM DTT, and 1× complete EDTA-free protease inhibitor mixture] by rotation for 1 h at 4 °C. The soluble and insoluble fractions were separated by centrifugation at 17,000 × g for 5 min at 4 °C, and the soluble fraction was diluted 1:5 with binding buffer [20 mM Tris·HCl (pH 8.0), 150 mM NaCl, and 1× complete EDTA-free protease inhibitor mixture] and incubated with 400 µL of IgG Sepharose (GE Healthcare) for 2 h by rotation at 4 °C. IgG Sepharose was washed extensively with wash buffer [20 mM Tris·HCl (pH 8.0), 150 mM NaCl, 0.1% (vol/vol) Igepal CA-630, 10% (vol/vol) glycerol, and 1× phenylmethanesulfonylfluoride], and the RdRp was released by cleavage overnight at 4 °C with 20 U of AcTEV (Invitrogen) in 500 µL of cleavage buffer [10 mM Tris·HCl (pH 8.0), 150 mM NaCl, 0.1% (vol/vol) Igepal CA-630, 10% (vol/vol) glycerol, 1 mM DTT, and 1× complete EDTA-free protease inhibitor mixture]. AcTEV was removed by binding the eluate to 100 µL Ni-NTA agarose for 30 min at 4 °C by rotation and centrifuging at 17,000 × g for 5 min at 4 °C. The supernatant was concentrated to 50 µL using Amicon Ultra-4 (100K MWCO) centrifugation devices (Millipore) and stored at 4 °C. Mock samples as negative controls were prepared by processing HEK 293T cells transfected with empty vector (pcDNA3A) as described above.
In Vitro Activity Assays of RNPs.
RNP activity was assessed in a 20-µl reaction mixture containing 10 µL of purified RNPs, incubated for 16 h at 30 °C with 1 mM ATP, 0.5 mM of each CTP, GTP, and UTP (Amersham), 5 mM MgCl2, 2 mM DTT, and 5 U RNasin (Promega), with 360 ng β-globin mRNA (Sigma) or 0.5 mM ApG (Sigma) as indicated. Where indicated, 500 ng of purified recombinant influenza virus RdRp expressed in Sf9 cells or 40 ng of wild-type or PB1a mutant RdRp expressed in mammalian HEK 293T cells was included. Transcription products were extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions and precipitated with isopropanol in the presence of 10 µg of tRNA from Escherichia coli MRE 600 (Roche) before analysis by NA gene-specific primer extension and 6% PAGE in 7 M urea as described previously (7). Quantitation of primer extension products was performed by phosphorimage analysis using Aida software (Raytest).
Alternatively, RNP activity was assessed in a 10-µL reaction mixture containing 5 µl of purified RNPs incubated for 2 h at 30 °C with 1 mM ATP, 0.5 mM GTP, and 0.16 µM of [α-32P]UTP (3,000 Ci/mmole; Perkin-Elmer) for cRNPs or 0.16 µM of [α-32P]CTP (3,000 Ci/mmole; Perkin-Elmer) for vRNPs, 0.5 mM ApG, 5 mM MgCl2, 2 mM DTT, and 5 U RNasin, with 10 ng of wild-type RdRp, PB1a active-site mutant RdRp, or mock sample. RNA was extracted by adding an equal volume of phenol/chloroform 1:1 (vol/vol) (pH 4.3) and vortexing for 5 min at room temperature before centrifuging at 17,000 × g for 20 min at 4 °C. RNA was precipitated from the aqueous fraction by adding 5 volumes of acetone/ethanol 3:1 (vol/vol) in the presence of 10 µg of tRNA from E. coli MRE 600 and incubating for 30 min at −20 °C before centrifuging at 17,000 × g for 20 min at 4 °C. The RNA pellet was washed in ice-cold 70% ethanol before dissolving in 10 µL formamide [90% (vol/vol) formamide (Sigma), 10 mM EDTA, 0.001% (wt/vol) bromophenol blue, and 0.001% (wt/vol) xylene cyanol] and heating to 95 °C for 3 min and cooling directly on ice before being analyzed by 20% PAGE containing 7 M urea.
In Vitro Activity Assay of Recombinant RdRp.
Activity of purified recombinant RdRp polymerase was assessed using an [α-32P]GTP incorporation assay as previously described (40). Briefly, 1.5 µL of purified polymerase and 15 pmol of each of 5′ end [15 nt (5′-AGUAGAAACAAGGCC-3′)] and 3′ end [14 nt (5′- GGCCUGCUUUUGCU-3′)] promoter templates were incubated in a 3-µL reaction mixture for 2 h at 30 °C with 1 mM ATP, 0.5 mM CTP, 0.5 mM UTP, 0.1 µM [α-32P]GTP (3,000 Ci/mmole; Perkin-Elmer), 0.5 mM ApG, 5 mM MgCl2, 2 mM DTT, and 5 U RNasin. After 2 h, 10 µL formamide was added [90% (vol/vol) formamide (Sigma), 10 mM EDTA, 0.001% (wt/vol) bromophenol blue, and 0.001% (wt/vol) xylene cyanol] and heated to 95 °C for 3 min and cooling directly on ice before being analyzed by 20% PAGE containing 7 M urea.
Supplementary Material
Acknowledgments
We thank J. Robert Hogg for the pET-ZZ-tev-PP7CP plasmid, Otto Haller for the NP antibody, Imre Berger for providing the baculovirus technology, Zoe Gage for assistance with protein expression optimization, and Errin Johnson and Alistair Siebert for assistance with EM. This work was supported by Medical Research Council (MRC) Grants G0700848 and MR/K000241/1 (to E.F.) and G1100138 (to F.T.V.), an MRC studentship (to A.Y.), by Wellcome Trust Studentship 092931/Z/10/Z (to N.H.), and by Academy of Finland Grants 130750 and 218080 (to J.T.H.). The Wellcome Trust Centre for Human Genetics is supported by Wellcome Trust Core Award 090532/Z/09/Z. The Oxford Particle Imaging Centre EM facility was founded by Wellcome Trust Joint Infrastructure Fund Award 060208/Z/00/Z and is supported by Wellcome Trust Equipment Grant 093305/Z/10/Z.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315068110/-/DCSupplemental.
References
- 1.Morin B, Kranzusch PJ, Rahmeh AA, Whelan SP. The polymerase of negative-stranded RNA viruses. Curr Opin Virol. 2013;3(2):103–110. doi: 10.1016/j.coviro.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arranz R, et al. The structure of native influenza virion ribonucleoproteins. Science. 2012;338(6114):1634–1637. doi: 10.1126/science.1228172. [DOI] [PubMed] [Google Scholar]
- 3.Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. Organization of the influenza virus replication machinery. Science. 2012;338(6114):1631–1634. doi: 10.1126/science.1227270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fodor E. The RNA polymerase of influenza a virus: Mechanisms of viral transcription and replication. Acta Virol. 2013;57(2):113–122. doi: 10.4149/av_2013_02_113. [DOI] [PubMed] [Google Scholar]
- 5.Krug RM, Fodor E. The virus genome and its replication. In: Webster RG, Monto AS, Braciale TJ, Lamb RA, editors. Textbook of Influenza. 2nd Ed. New York: John Wiley & Sons; 2013. pp. 57–66. [Google Scholar]
- 6.Palese P, Shaw ML. In: Orthomyxoviridae. Fields Virology. 6th Ed. Knipe DM, Howley PM, editors. Philadelphia: Lippincott Williams and Wilkins; 2013. pp. 1151–1185. [Google Scholar]
- 7.Vreede FT, Jung TE, Brownlee GG. Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J Virol. 2004;78(17):9568–9572. doi: 10.1128/JVI.78.17.9568-9572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatada E, Hasegawa M, Mukaigawa J, Shimizu K, Fukuda R. Control of influenza virus gene expression: Quantitative analysis of each viral RNA species in infected cells. J Biochem. 1989;105(4):537–546. doi: 10.1093/oxfordjournals.jbchem.a122702. [DOI] [PubMed] [Google Scholar]
- 9.Lim F, Peabody DS. RNA recognition site of PP7 coat protein. Nucleic Acids Res. 2002;30(19):4138–4144. doi: 10.1093/nar/gkf552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogg JR, Collins K. RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA. 2007;13(6):868–880. doi: 10.1261/rna.565207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fodor E, et al. Rescue of influenza A virus from recombinant DNA. J Virol. 1999;73(11):9679–9682. doi: 10.1128/jvi.73.11.9679-9682.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Air GM. Influenza neuraminidase. Influenza Other Respi Viruses. 2012;6(4):245–256. doi: 10.1111/j.1750-2659.2011.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo G, Chung J, Palese P. Alterations of the stalk of the influenza virus neuraminidase: Deletions and insertions. Virus Res. 1993;29(2):141–153. doi: 10.1016/0168-1702(93)90055-r. [DOI] [PubMed] [Google Scholar]
- 14.Castrucci MR, Kawaoka Y. Biologic importance of neuraminidase stalk length in influenza A virus. J Virol. 1993;67(2):759–764. doi: 10.1128/jvi.67.2.759-764.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorrell EM, Song H, Pena L, Perez DR. A 27-amino-acid deletion in the neuraminidase stalk supports replication of an avian H2N2 influenza A virus in the respiratory tract of chickens. J Virol. 2010;84(22):11831–11840. doi: 10.1128/JVI.01460-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munier S, et al. A genetically engineered waterfowl influenza virus with a deletion in the stalk of the neuraminidase has increased virulence for chickens. J Virol. 2010;84(2):940–952. doi: 10.1128/JVI.01581-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Compans RW, Content J, Duesberg PH. Structure of the ribonucleoprotein of influenza virus. J Virol. 1972;10(4):795–800. doi: 10.1128/jvi.10.4.795-800.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robb NC, Smith M, Vreede FT, Fodor E. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J Gen Virol. 2009;90(Pt 6):1398–1407. doi: 10.1099/vir.0.009639-0. [DOI] [PubMed] [Google Scholar]
- 19.Vreede FT, Brownlee GG. Influenza virion-derived viral ribonucleoproteins synthesize both mRNA and cRNA in vitro. J Virol. 2007;81(5):2196–2204. doi: 10.1128/JVI.02187-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jorba N, Coloma R, Ortín J. Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog. 2009;5(5):e1000462. doi: 10.1371/journal.ppat.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng T, Vreede FT, Brownlee GG. Different de novo initiation strategies are used by influenza virus RNA polymerase on its cRNA and viral RNA promoters during viral RNA replication. J Virol. 2006;80(5):2337–2348. doi: 10.1128/JVI.80.5.2337-2348.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noda T, et al. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature. 2006;439(7075):490–492. doi: 10.1038/nature04378. [DOI] [PubMed] [Google Scholar]
- 23.Hutchinson EC, von Kirchbach JC, Gog JR, Digard P. Genome packaging in influenza A virus. J Gen Virol. 2010;91(Pt 2):313–328. doi: 10.1099/vir.0.017608-0. [DOI] [PubMed] [Google Scholar]
- 24.Fournier E, et al. A supramolecular assembly formed by influenza A virus genomic RNA segments. Nucleic Acids Res. 2012;40(5):2197–2209. doi: 10.1093/nar/gkr985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noda T, et al. Three-dimensional analysis of ribonucleoprotein complexes in influenza A virus. Nat Commun. 2012;3:639. doi: 10.1038/ncomms1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.York A, Fodor E. Biogenesis, assembly and export of viral messenger ribonucleoproteins in the influenza A virus infected cell. RNA Biol. 2013;10(8):1274–1282. doi: 10.4161/rna.25356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruigrok RW, Crépin T, Kolakofsky D. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr Opin Microbiol. 2011;14(4):504–510. doi: 10.1016/j.mib.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 28.Jorba N, Area E, Ortín J. Oligomerization of the influenza virus polymerase complex in vivo. J Gen Virol. 2008;89(Pt 2):520–524. doi: 10.1099/vir.0.83387-0. [DOI] [PubMed] [Google Scholar]
- 29.Digard P, Blok VC, Inglis SC. Complex formation between influenza virus polymerase proteins expressed in Xenopus oocytes. Virology. 1989;171(1):162–169. doi: 10.1016/0042-6822(89)90523-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyle JM, Bullitt E, Bienz K, Kirkegaard K. Visualization and functional analysis of RNA-dependent RNA polymerase lattices. Science. 2002;296(5576):2218–2222. doi: 10.1126/science.1070585. [DOI] [PubMed] [Google Scholar]
- 31.Wang QM, et al. Oligomerization and cooperative RNA synthesis activity of hepatitis C virus RNA-dependent RNA polymerase. J Virol. 2002;76(8):3865–3872. doi: 10.1128/JVI.76.8.3865-3872.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smallwood S, Cevik B, Moyer SA. Intragenic complementation and oligomerization of the L subunit of the sendai virus RNA polymerase. Virology. 2002;304(2):235–245. doi: 10.1006/viro.2002.1720. [DOI] [PubMed] [Google Scholar]
- 33.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256(5065):1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 34.Fodor E, et al. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol. 2002;76(18):8989–9001. doi: 10.1128/JVI.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheres SHW, Núñez-Ramírez R, Sorzano COS, Carazo JM, Marabini R. Image processing for electron microscopy single-particle analysis using XMIPP. Nat Protoc. 2008;3(6):977–990. doi: 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheres SHW. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180(3):519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puig O, et al. The tandem affinity purification (TAP) method: A general procedure of protein complex purification. Methods. 2001;24(3):218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 38.Bieniossek C, Richmond TJ, Berger I. MultiBac: Multigene baculovirus-based eukaryotic protein complex production. Curr Protoc Protein Sci. 2008;Chapter 5:Unit 5.20. doi: 10.1002/0471140864.ps0520s51. [DOI] [PubMed] [Google Scholar]
- 39.Engelhardt OG, Smith M, Fodor E. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol. 2005;79(9):5812–5818. doi: 10.1128/JVI.79.9.5812-5818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brownlee GG, Sharps JL. The RNA polymerase of influenza A virus is stabilized by interaction with its viral RNA promoter. J Virol. 2002;76(14):7103–7113. doi: 10.1128/JVI.76.14.7103-7113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.