

Abstract
Single particle cryo-electron microscopy is currently poised to produce high-resolution structures of many biological assemblies, but several pitfalls can trap the unwary. This critique highlights one problem that is particularly relevant when smaller structures are being studied. It is known as “Einstein from noise,” in which the experimenter honestly believes they have recorded images of their particles, whereas in reality, most if not all of their data consist of pure noise. Selection of particles using cross-correlation methods can then lead to 3D maps that resemble the model used in the initial selection and provide the illusion of progress. Suggestions are given about how to circumvent the problem.
Keywords: cryo-EM, SPEM, 3DEM, EMDB, validation
Single particle electron cryomicroscopy, abbreviated to cryo-EM or SPEM, has great potential as a method of determining biological structures. The associated technology has gradually improved since Dubochet and colleagues first developed their method for plunge-freezing suspensions of biological structures such as adenovirus (1) in thin films of amorphous ice in the 1980s. There are now better cold stages, better specimen environments with higher vacuum leading to less ice contamination, higher voltage electron guns with higher brightness and coherence, improved electron detectors, and improved computer programs running on much faster computers. All these improvements are bringing nearer the day when we can routinely determine atomic structures of biological assemblies from ice-embedded single particles and thus realize the theoretical potential of the method (2, 3).
At present, there are good examples of near-atomic resolution structures from a variety of well-behaved single particle specimens, such as icosahedral viruses (4), the ribosome (5), and a few others with sizes as small as the F420 enzyme complex at 1.2 MDa (6) and the 20S proteasome at 700 kDa (7). The size of the single particles is a critically important factor because, for 3D structure determination, the position and orientation of each molecule in an image must be accurately determined using a limited electron exposure. Beyond a certain electron dose, radiation damage prevents further useful information being accumulated in the image. In practice, the allowable dose (8) of 20–50 electrons/Å2 that can be used before the structure is degraded means there is not enough information in each image of a small structure to determine its orientation. In theory, atomic resolution structures for particles of 100 kDa should be obtainable from images of a few thousand single particles (2). The essential requirements for this to be possible include good specimen purification with conformational homogeneity, good ice embedding, absence of beam-induced specimen motion due to charging and radiation damage, high detective quantum efficiency (DQE) in the image recording, and careful attention to the treatment of the signal-to-noise ratio in the computer-based image processing. If any of these steps are deficient, then the minimum size and the highest resolution obtainable are rapidly compromised. For example, if the image contrast is reduced to 25% of that expected in a perfect image, then the minimum particle size that could be oriented would increase 16-fold (by a factor of 1/contrast2) and exceed 1 MDa (2).
Problems
In practice, there are many technical problems still to be overcome, and careful judgment is needed to differentiate theoretical potential from practical achievement. This situation has created a spectrum of different attitudes among researchers, ranging from those who are very careful and proceed step-by-step to ensure that progress is real to those who simply record images, follow an established (or sometimes a novel or inventive) protocol for 3D map calculation, and then boldly interpret and publish their map without any further checks or attempts to validate the result. Ten years ago, when the field was in its infancy, referees would simply have to accept the research results reported in manuscripts at face value. The researchers had recorded images, carried out iterative computer processing, and obtained a map that converged, but had no way of knowing whether it had converged to the true structure or some complete artifact. There were no validation tests, only an instinct about whether a particular map described in the publication looked right or wrong. Researchers could only cite consistency with a range of other data such as cross-linking when striving to be convinced whether their results were right. One of the most enlightening lessons was provided by the published maps of the 1.3-MDa inositol triphosphate (IP3) receptor, an interesting and important biological structure (9–13), and has become a classic and illustrative case study in the field. Three of these published 3D maps used cryo-EM, but they all showed different structures, which led to the obvious conclusion that most or possibly all of them must be wrong. Many years later, subsequent work with better microscopes, better specimens, and better computation has resulted in an IP3 receptor structure at 17-Å resolution that has been fully validated by recently introduced tests (14). The historically unsatisfactory situation, where the cryo-EM field had essentially no agreed standards, led to a meeting organized by the International Electron Microscopy Data Bank in September 2010, the EMDB Task Force, where the lack of validation tests and criteria were fully explored (15). Since then, a number of procedures have been developed that fill the need for criteria that might minimize the possibility of publication of either completely wrong cryo-EM structures, structures with overinflated claims of resolution achieved, or structures that are biased by an initial model either partially or completely.
Validation Tools
Among the best current validation tools are the tilt pair parameter plot that requires the recording of one or two additional pairs of single-particle cryo-EM images of the same area (16, 17), the use of gold standard Fourier shell correlation (FSC) plots (18, 19), where the initial single particle images are divided at the very start of an analysis into two independent halves (note they must be completely independent, because even the use of the same 3D mask can introduce false apparent correlations), and high-resolution noise (HR noise) substitution (20) to validate resolution. With these new tools, most cryo-EM practitioners have now become more confident in their ability to judge the correctness of their own results, the correctness of results that are described in manuscripts they are refereeing for possible publication, and the correctness of published papers. With these tools, it might even be useful to scrub clean the published literature over the last 10 y by encouraging authors of past papers to apply current validation techniques to papers published previously in good faith.
New Pitfall
Despite the promising development of rigorous validation tests and criteria, one must not underestimate the ingenuity of humans to invent new ways to deceive themselves. It was nevertheless still surprising to read a recently published paper in PNAS (21), which purports to show a 6-Å structure for the uncleaved HIV GP160 trimeric glycoprotein spike in its membrane state (i.e., in detergent). The GP160 trimer consists of three protein monomers each of molecular weight (MW) 100–107 kDa, together with 50–60 kDa of glycosylation per subunit, giving an overall MW for the trimer of 450–480 kDa. To provide some background to this work, I must mention that I was a referee for a virtually identical manuscript that had been submitted by the same authors to a different journal earlier in 2013 and that had been rejected. The authors appear to have committed one of the best-known and obvious mistakes that cryo-EM practitioners had been aware of for >20 y. They use a template consisting of projections of the structure they are hoping to observe to search many thousands of images. Their procedure results in the extraction of almost a million “single particle images.” Because the authors had prepared specimens by plunge freezing a solution of GP160, they naturally assumed that the field of view of their images consisted of many GP160 particles. Surprisingly, no particles can be seen. The computer algorithms that identify and then verify the particles are described in two sections of the Supporting Information (21) as “particle images were evaluated and verified comprehensively by unsupervised classification using multivariate data analysis and K-means clustering, as previously described” (22) and “image analysis was implemented in customized computational procedures and workflows, combining the functions of SPIDER and XMIPP.” As a referee, after examining the windowed images that I requested as part of the normal refereeing process and finding no evidence for particles, my conclusion was that “most if not all of Mao et al.’s windowed images and verified particles were nonparticles” and that their procedure was simply a more sophisticated 3D equivalent of the classical demonstration that the portrait of Einstein can be extracted from a few thousand images of pure random noise (Fig. 1). The authors had not shown that the images contained any GP160 particles. They had not carried out any of the validation tests that are currently expected (their 3D half-maps used the same mask as can be seen in Fig. 2 and thus were not independent). In my refereeing, I recommended a number of further steps of analysis and posed 9 or 10 questions that were designed not to remove any doubts about the validity of their work, because I personally had no doubt it was incorrect, but rather to raise the authors own awareness and encourage them to be more critical so that they themselves would realize the pitfall. My conclusion was that they had not determined the structure of GP160 at 6-Å resolution as the publication claims but that they had developed “customized computational procedure and workflows” so complicated that they were led to believe that the resulting map was real rather than simply the result of a sophisticated procedure for selecting noise biased toward the structure they wished to observe. I hope this critique will ensure that readers at least entertain a strong doubt about what they read in this paper from Mao et al. and do not trust that refereeing can weed out erroneous results. I was also an adjudicating referee for an earlier (rejected) manuscript from the same group, which at that stage about 2 y earlier made even more extreme claims for resolution from very similar images, again with no visible particles. This earlier manuscript eventually evolved into another paper that claimed a more modest 11-Å resolution (24), but, in light of the PNAS paper (21), must also be very likely to consist of template-selected and overfitted noise.
Fig. 1.

Illustration taken from a paper describing model bias (23). The image is copied from figure 2A in that paper. The familiar photograph of Einstein emerged from 1,000 images of pure white noise, after alignment to the model using a cross-correlation function. Reprinted from Journal of Structural Biology, Vol. 166, M. Shatsky et al., A method for the alignment of heterogeneous macromolecules from electron microscopy, pp. 67–78, Copyright 2009, with permission from Elsevier.
Fig. 2.

(A–F) Six individual windowed images from the stack of 423 that was supplied by the authors (21). (G) Average of 423 windowed images using the same gray scale as A–F. (H) Average of 423 windowed images with 15× increased contrast. The density in the central region of G and H shows the average of the many views used in particle picking and verification. The circle of dark density round the edge of the average, seen more clearly in H should not be present in the raw images so must arise from masked projections from the 3D map or model used to extract the particles. (I–L) Difference maps obtained by subtraction of sections from the two independent half maps [i.e., maps calculated using only half the data, normally even and odd particles in the stack (18)] supplied by the authors (21). The four panels represent sections at different heights along the spike, viewed from the apex. The differences are confined to a sharply defined region with no gradation into the flat background. This clearly visible and relatively sharp mask serves to constrain any density to the region inside the mask during iterative refinement. Use of masking plus the same initial reference suggests how the apparent resolution was extended from 11 to 6 Å. All images are on the same scale, with a window size of 190 Å.
This most recent Einstein from noise pitfall in cryo-EM, highlighted by the paper from Mao et al., arises because of the desire to exploit the growing potential of the method by trying to overcome the limitations of poor specimen and image quality by a brute force increase in numbers of single particle images. It is the combination of small (or invisible) particles, low (or zero) contrast, and the use of automatic particle (or nonparticle) picking procedures that is most dangerous.
It is perfectly reasonable for the experimenter, provided that clearly visible particles can be seen, to identify and pick them by hand, and then use established procedures to produce and validate a 3D map. This manual particle picking would be recommended for anyone carrying out an initial cryo-EM analysis of a new specimen with particles having a MW smaller than ∼500 kDa. This puts the user in direct contact with the images and allows him or her to decide whether they believe their images show the type of particles they expect. With a bit of practice, it is even possible to identify the different particle orientations by eye at this stage. Examples of images of this type can be seen in recent publications (20, 25). However, to avoid the tedious step of identifying and picking particles by hand, many people have started to use an automatic particle-picking procedure (26–28), possibly followed by an automatic particle verification procedure (22). Although this is acceptable once a firm foundation has been established through earlier work on a particular specimen, it is very dangerous to start out in this way, which is what Mao et al. have done. The genesis of their mistake appears to be a paper published in 2008 (22). This earlier paper presented an analysis of well-validated 2.6-MDa ribosomal single particle images. Ribosomes are composed of 60% RNA, which has 2× higher contrast and 2× lower radiation sensitivity than protein. Shaikh et al. (22) developed a procedure of particle verification to try to sort their single-particle images into those that represented genuine particles and those that were nonparticles, such as ice contamination, impurities, or even statistical fluctuations. Although their classification procedure was successful and introduced some useful ideas, they warned: “The underlying assumption in the use of classification for particle verification is that the real particles will be separated from the non-particles. The ribosome exceeds 2 MDa in molecular mass, has distinctive features, and has high contrast due to its RNA content, so the above assumption tends to hold…however—if the molecule has low molecular weight, smooth features, low contrast, or a small fraction of windowed images as real particles—that assumption may be invalidated.”
The HIV envelope glycoprotein trimer is 10 times smaller in terms of overall contrast than the 70S ribosome, possibly more than 10 times smaller if the polysaccharide component is poorly ordered. Therefore, at best, all their particles are small with low contrast but more likely are all nonparticles or simply pure noise. Others have also warned of the risks associated with particle-picking methods based on cross-correlation rather than local variance (29).
In Fig. 2, I show two panels, produced from data requested as part of my refereeing of the earlier, but almost identical, manuscript to that now published in PNAS (21). I thank the authors for permission to include this analysis of their data. Fig. 2H shows a circular shadow of the mask from the model or map used in particle picking. The density of this circular feature is almost as strong as that of the particles, which are averaged in the central white region. Even in the absence of real particles, noise from the images will be strongly correlated with the search motif and will lead to a 3D map that is similar to that used to pick the particles. Fig. 2 I–L shows that a tight mask was used to calculate the final map with improved resolution. The use of such a mask together with the same initial model invalidates the so-called gold standard approach (18, 19) in which all aspects of the structure determination of the two half sets of images must be independent and can explain how the apparent resolution increases from 11 to 6 Å, leading the authors to conclude that they have a 6-Å structure. The final map shows prominent radial density streaks, which are often due to overfitted noise.
Future Precautions
In the future, as the cryo-EM field moves toward higher-resolution structures of smaller molecular complexes and assemblies, there will be an increasing temptation to follow the same kind of procedure, so one might ask what advice should be given to those who would like to use the method. My own suggestions when starting a new project of this type would include the following:
i) The person carrying out the microscopy should make sure that their cryo-EM images show clearly the small particles they expect to find. They should consider the possibility that the macromolecular structures they expect to see might have attached to the carbon support film or the filter paper used to blot the specimen so that there are in fact no particles in the support film holes.
ii) The initial images should be recorded at large (e.g., 5 µm) defocus with relatively high-dose exposures, such as 80, 100, or even up to 140 el/Å2. An image of this type for the 450-kDa β-galactosidase structure is shown in Fig. 3. The particles stand out far above the noise level. Although these particles will then have more radiation damage than particles imaged at 25 el/Å2, the enormous increase in contrast ensures that the Einstein from noise pitfall will be avoided.
iii) In any initial analysis, the particles should always be identified and picked by hand. This ensures that the experimenter will interact with the data and consider carefully whether what they see corresponds with something related to what they expect.
iv) Ask whether the different views can be identified by eye or whether subsequent computer procedures will be relied on to do this?
v) If multivariate statistics or particle classification procedures are essential to obtain initial class sums and identify different views, this should not be done with particles that have been picked by a cross-correlation particle-picking program. If particle picking is carried out using a template, even random noise will result in the extraction of an image of the template and delude the experimenter into thinking they have performed a miracle.
Fig. 3.
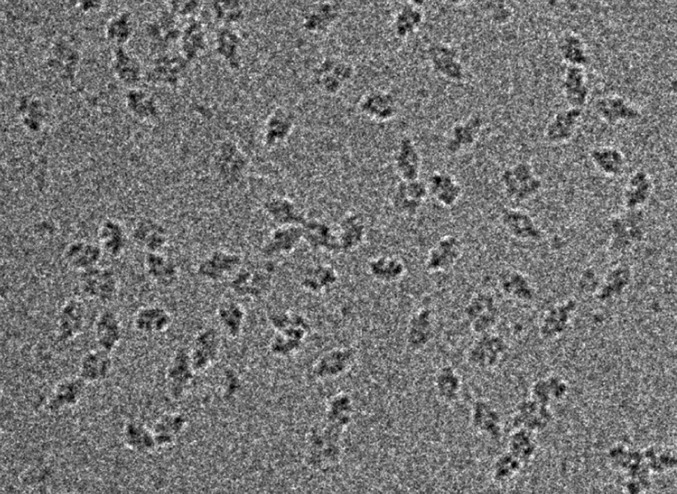
Cryo-EM image of a field of view of β-galactosidase single particles (molecular weight, 450 kDa). This image (number 04.51.13) was recorded on an FEI Falcon II detector at 300 keV and 80,000x magnification with a dose of 100 el/Å2 and a defocus of 3.5 μm. The particles are all clearly visible at a defocus value similar to that used to obtain the images presented in figure S1A of Mao et al. (21).
In the paper by Mao et al. (21), none of the above precautions were taken.
Once a Cryo-EM Project Reaches the Stage of Considering Publication
i) It is important to be self-critical. Ask whether the most rigorous validation tests have been avoided because of fear that they might show the structure is wrong or has a much lower resolution than hoped for. The tests should be carried out before submission and not afterward.
ii) All authors have the responsibility to ensure that the appropriate validation tests have been carried out, whether they are student, postdoc, or the senior author or group leader who has not personally carried out the research. In the absence of the necessary expertise, a suitable colleague should be asked to review the research critically before considering submission to a journal, especially if the journal has a broad scope: the more specialized journals tend to have more specialized referees where the likelihood of obtaining a knowledgeable and skillful referee is higher. The prearranged editor option in PNAS should not be encouraged for this type of research work.
iii) Any negative comments or criticisms from referees should be carefully considered. The paper should not simply be sent unchanged to a sequence of other journals until it gets published in the Nth journal where lucky statistics or an overworked editor results in the selection of referees who do not have the relevant experience or skill needed to spot any fundamental flaws.
Cryo-EM Referees
i) If there are any doubts about a paper, the authors should be asked to perform some or all of the validation tests mentioned above.
ii) If any response is still unconvincing or if the authors do not respond to requests for them to carry out appropriate validation tests, then the journal should be asked to request that the authors send (in confidence) to the referee all of the data that went into the paper; specifically, this should include the raw single particle boxed images plus the metadata that describe the images, especially magnification, defocus, and other microscope parameters used in the processing such as voltage, spherical aberration, and amplitude contrast. There is a 3DEM working group developing a metadata interchange format using an XML file. Hopefully this will be available soon and will make such interchanges and requests easier. This standard of data validation in which referees ask to see the raw data has been common practice in X-ray crystallography for many years. It is not compulsory, but many referees insist on it, in response to similar problems where completely wrong structures managed to evade rigorous refereeing in that field.
iii) Referees also have a responsibility to make sure they have enough time to referee the paper carefully. In the long run, it does not help the field if bad papers get published because of poor refereeing.
Note Added in Proof.
This perspective article is companion to two PNAS letters and a response. Subramaniam (30) and van Heel (31) provide independent critiques of Mao et al. (21), and Mao (32) provides a response.
Footnotes
The author declares no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Adrian M, Dubochet J, Lepault J, McDowall AW. Cryo-electron microscopy of viruses. Nature. 1984;308(5954):32–36. doi: 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- 2.Henderson R. The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q Rev Biophys. 1995;28(2):171–193. doi: 10.1017/s003358350000305x. [DOI] [PubMed] [Google Scholar]
- 3.Henderson R. Realizing the potential of electron cryo-microscopy. Q Rev Biophys. 2004;37(1):3–13. doi: 10.1017/s0033583504003920. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X, Jin L, Fang Q, Hui WH, Zhou ZH. 3.3 A cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell. 2010;141(3):472–482. doi: 10.1016/j.cell.2010.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai XC, Fernandez IS, McMullan G, Scheres SH. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. Elife. 2013;2:e00461. doi: 10.7554/eLife.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mills DJ, Vitt S, Strauss M, Shima S, Vonck J. De novo modeling of the F420-reducing [NiFe]-hydrogenase from a methanogenic archaeon by cryo-electron microscopy. Elife. 2013;2:e00218. doi: 10.7554/eLife.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li XM, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10(6):584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker LA, Smith EA, Bueler SA, Rubinstein JL. The resolution dependence of optimal exposures in liquid nitrogen temperature electron cryomicroscopy of catalase crystals. J Struct Biol. 2010;169(3):431–437. doi: 10.1016/j.jsb.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 9.Hamada K, Terauchi A, Mikoshiba K. Three-dimensional rearrangements within inositol 1,4,5-trisphosphate receptor by calcium. J Biol Chem. 2003;278(52):52881–52889. doi: 10.1074/jbc.M309743200. [DOI] [PubMed] [Google Scholar]
- 10.da Fonseca PCA, Morris SA, Nerou EP, Taylor CW, Morris EP. Domain organization of the type 1 inositol 1,4,5-trisphosphate receptor as revealed by single-particle analysis. Proc Natl Acad Sci USA. 2003;100(7):3936–3941. doi: 10.1073/pnas.0536251100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serysheva II, Hamilton SL, Chiu W, Ludtke SJ. Structure of Ca2+ release channel at 14 A resolution. J Mol Biol. 2005;345(3):427–431. doi: 10.1016/j.jmb.2004.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang QX, Thrower EC, Chester DW, Ehrlich BE, Sigworth FJ. Three-dimensional structure of the type 1 inositol 1,4,5-trisphosphate receptor at 24 A resolution. EMBO J. 2002;21(14):3575–3581. doi: 10.1093/emboj/cdf380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato C, et al. Inositol 1,4,5-trisphosphate receptor contains multiple cavities and L-shaped ligand-binding domains. J Mol Biol. 2004;336(1):155–164. doi: 10.1016/j.jmb.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 14.Murray SC, et al. Validation of cryo-EM structure of IP3R1 channel. Structure. 2013;21(6):900–909. doi: 10.1016/j.str.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henderson R, et al. Outcome of the first electron microscopy validation task force meeting. Structure. 2012;20(2):205–214. doi: 10.1016/j.str.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333(4):721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Henderson R, et al. Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy. J Mol Biol. 2011;413(5):1028–1046. doi: 10.1016/j.jmb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheres SHW, Chen SX. Prevention of overfitting in cryo-EM structure determination. Nat Methods. 2012;9(9):853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grigorieff N. Resolution measurement in structures derived from single particles. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 10):1270–1277. doi: 10.1107/s0907444900009549. [DOI] [PubMed] [Google Scholar]
- 20.Chen S, et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy. 2013;135C:24–35. doi: 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao Y, et al. Molecular architecture of the uncleaved HIV-1 envelope glycoprotein trimer. Proc Natl Acad Sci USA. 2013;110(30):12438–12443. doi: 10.1073/pnas.1307382110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaikh TR, Trujillo R, LeBarron JS, Baxter WT, Frank J. Particle-verification for single-particle, reference-based reconstruction using multivariate data analysis and classification. J Struct Biol. 2008;164(1):41–48. doi: 10.1016/j.jsb.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shatsky M, Hall RJ, Brenner SE, Glaeser RM. A method for the alignment of heterogeneous macromolecules from electron microscopy. J Struct Biol. 2009;166(1):67–78. doi: 10.1016/j.jsb.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mao YD, et al. Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat Struct Mol Biol. 2012;19(9):893–899. doi: 10.1038/nsmb.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harris A, et al. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc Natl Acad Sci USA. 2011;108(28):11440–11445. doi: 10.1073/pnas.1101414108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roseman AM. FindEM—a fast, efficient program for automatic selection of particles from electron micrographs. J Struct Biol. 2004;145(1-2):91–99. doi: 10.1016/j.jsb.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 27.Short JM. SLEUTH—a fast computer program for automatically detecting particles in electron microscope images. J Struct Biol. 2004;145(1-2):100–110. doi: 10.1016/j.jsb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 28.Tang G, et al. EMAN2: An extensible image processing suite for electron microscopy. J Struct Biol. 2007;157(1):38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 29. van Heel M, et al. (2012) Four-dimensional cryo-electron microscopy at quasi atomic resolution: IMAGIC 4D. International Tables for Crystallography Volume F: Crystallography of Biological Macromolecules, eds Arnold E, Himmel DM, Rossmann MG (Wiley, New York), 2nd Ed, pp 624–628.
- 30.Subramaniam S. 2013. Structure of trimeric HIV-1 envelope glycoproteins. Proc Natl Acad Sci USA 110:E4172–E4174.
- 31.van Heel M. 2013. Finding trimeric HIV-1 envelope glycoproteins in random noise. Proc Natl Acad Sci USA 110:E4175–E4177.
- 32.Mao Y, Castillo-Menendez LR, Sodroski JG. 2013. Reply to Subramaniam, van Heel, and Henderson: Validity of the cryo-electron microscopy structures of the HIV-1 envelope glycoprotein complex. Proc Natl Acad Sci USA 110:E4178–E4182.