

Significance
Many proteins solved by Structural Genomics have low sequence identity to other proteins and cannot be assigned functions. To address this problem, we present a computational approach that creates structural motifs of a few evolutionarily important residues, and these motifs probe local geometric and evolutionary similarities in other protein structures to detect functional similarities. This approach does not require prior knowledge of functional mechanisms and is highly accurate in computational benchmarks when annotations rely on homologs with low sequence identity. We further demonstrate the accuracy of this approach using biochemical and mutagenesis studies to validate two predictions of unannotated proteins.
Keywords: function annotation, evolutionary trace, structural motif, protein function
Abstract
Structural Genomics aims to elucidate protein structures to identify their functions. Unfortunately, the variation of just a few residues can be enough to alter activity or binding specificity and limit the functional resolution of annotations based on sequence and structure; in enzymes, substrates are especially difficult to predict. Here, large-scale controls and direct experiments show that the local similarity of five or six residues selected because they are evolutionarily important and on the protein surface can suffice to identify an enzyme activity and substrate. A motif of five residues predicted that a previously uncharacterized Silicibacter sp. protein was a carboxylesterase for short fatty acyl chains, similar to hormone-sensitive-lipase–like proteins that share less than 20% sequence identity. Assays and directed mutations confirmed this activity and showed that the motif was essential for catalysis and substrate specificity. We conclude that evolutionary and structural information may be combined on a Structural Genomics scale to create motifs of mixed catalytic and noncatalytic residues that identify enzyme activity and substrate specificity.
As the list of known genes grows exponentially, the elucidation of their function remains a major bottleneck and lags far behind the production of sequences (1–5). The best approach remains to search computationally for functionally characterized sequence homologs, ideally with greater than 50% sequence identity (6). Binding specificity, however, is sensitive to subtle amino acid differences, and the transfer of substrate between related enzymes is prone to errors when sequence identity is below 65–80% (7–9). These thresholds vary from case to case: Some orthologs will maintain identical functions down to 25% sequence identify (9), whereas paralogs can take on highly diverse activities (10). Other difficulties that plague annotation transfer between homologs are that individual small molecules may each bind to multiple and distinct molecular pockets (11), that different residues can support similar chemistries (12), and that activity can vary even when catalytic residues are conserved (13–18). To raise annotation accuracy, Structural Genomics (19) made structural information widely available and spurred the development of annotation methods dependent on local chemical and physical environments (20), sequence and structural comparisons (21), or 3D templates (22). In the case of the latter, these methods search between proteins for local structural similarities over a few signature residues that represent the telltale parts of a functional site, so-called “3D templates” (3, 14, 18, 22–24). The residue composition of 3D templates is critical, however, and derived from experiments (25) or from analyses of functional sites and determinants (14, 15, 26). The sensitivity and specificity of template-based annotations still needs to be established experimentally (27, 28), but retrospective controls suggest they often predict enzyme catalytic activity (14, 16, 17, 29, 30).
Here, to extend the functional resolution of 3D template annotations to substrates, we exploit Evolutionary Tracing (ET) (31, 32). ET ranks sequence positions by the tendency of their evolutionary variations to correlate with major or with minor divergences. Top-ranked ET sequence positions are the most evolutionarily and, presumably, functionally important, and indeed they map out functional sites and specificity determinants (33) accurately enough to efficiently design mutations that block or swap functions among homologs in vitro (34–36) or in vivo (37, 38).
Accordingly, given a query protein of unknown function, the ET Annotation pipeline (ETA) builds a 3D template from five or six top-ranked ET residues that also cluster together on surface regions of protein structures (31, 32). ETA then searches already annotated protein structures, the targets, for those that match the query 3D template (Fig. 1 and Movie S1). False positive matches are common but can be recognized because they typically (i) involve unimportant residues in the target (39), (ii) are not reciprocated back to the query (40), and (iii) point to multiple proteins that each bear unrelated functions. With appropriate specificity filters to eliminate these false positives, ETA identified enzyme activity down to the first three Enzyme Commission (EC) levels with 92% accuracy (40), as well as in nonenzymes (41) in large-scale Structural Genomics retrospective controls. The prediction of substrate specificity remains an open question and further requires accurate identification of the fourth and last EC level (42) presumably by adding a more discriminating use of 3D template residues than is sufficient to specify a general chemical process (43). Some sequence methods (29, 30) and other structure methods (14, 44) have aimed to predict all four EC levels, but to our knowledge they have not been directly tested on de novo predictions of substrate specificity.
Fig. 1.

ETA accurately determines substrate specificity. (A) The ET algorithm is applied to a protein from Sulfolobus tokadaii strain 7 (green, PDB ID code 2eer, chain A) to identify evolutionarily important residues. A cluster of 10 or more important residues is identified and a Template Picker algorithm further selects five or six residues to act as a template that is used to probe a target library of proteins with known functions. Paired-distance matching algorithm identifies regions in protein structures in the target library that are similar to the template. Found matches are next passed to the SVM, which identifies significant matches based on geometric and evolutionary similarities. ETA repeats all these steps reciprocally, generating templates from target structures and searching for matches in the query protein. Following this protocol, ETA suggests four matches: alcohol dehydrogenase from Saccharomyces cerevisae (blue left, PDB ID code 2hcy), alcohol dehydrogenase from S. solfataricus (blue middle, PDB ID code 1r37), human class II alcohol dehydrogenase (blue right, PDB ID code 3cos), and NADP(H)-dependent cinnamyl alcohol dehydrogenase from S. cerevisae (red, PDB ID code 1piw) to the query protein. (B) The most seen function among matches, alcohol dehydrogenase activity (EC 1.1.1.1), is identified with high confidence with a confidence value of 1.125 as calculated in the box. (C) Comparison of PPV versus confidence score binned at <1, =1, and >1 for both six-residue templates (Left) and five-residue templates (Right) when considering only matches of <30% sequence identity. For more detail, see Fig. S1. (D) Comparison of PPV when predictions are made using ETA or the closest structural match (TM-align). Horizontal axis shows the maximum sequence identity of matches for proteins depicted in corresponding bars; the vertical axis is the PPV for each bin range.
In this study, we improve the functional resolution of the ETA pipeline to identify relevant functional homology down to very low sequence identity and add substrate specificity to its large-scale predictions. We then experimentally validate the predictions and show that both catalytic and noncatalytic residues are essential for 3D templates to pinpoint activity and substrate specificity.
Results
Substrate-Level Predictions from 3D Templates.
To predict substrates, a new confidence score was created that empirically favored multiple mutually consistent matches between a query and other protein structures if they had identical fourth-level EC numbers (42) (Materials and Methods and Fig. 1B). In a retrospective control, 605 enzymes from all six major classes were stripped of their annotations and then matched to a set of 3,082 annotated target structures (Fig. S1A). Almost all of the ETA annotations that scored above 1 on this new substrate confidence score (Materials and Methods) were correct over all four EC levels (99%), and only two-thirds of those with scores below 1 were (63%, Fig. S1B). Strikingly, high scoring annotations remained nearly perfect even when the sequence identity between a query and its matches fell below 30% (Fig. 1 C and D). By contrast, the template-based matching method COFACTOR (45) performed worse than ETA (96% vs. 92%, Table S1) despite having a larger target set and matching to proteins with greater than 30% sequence identity (see Materials and Methods for further detail). Similarly, annotations based on overall structural matches became increasingly inaccurate below 45% sequence identity (Fig. 1D). These data show that when a plurality of ETA matches agree on all four EC levels, function predictions based on just a few evolutionarily important and structurally clustered residues yield accurate predictions of function and substrates that improve on current protein structure comparison methods.
A potential limitation of using plurality to determine confidence may occur when only a single structure of a functional class is solved that yields a maximum confidence score equal to 1. However, ETA remains accurate when the confidence score is equal to 1, with correct predictions in 92% of cases (Fig. S1A). Furthermore, in the 24 cases that were based on the only representative of a particular functional class, the ETA annotation was correct 100% of the time (n = 24). These data suggest that even in the absence of plurality ETA accurately identifies substrate specificity.
To probe the basis of ETA accuracy, we compared these ET-derived 3D template residues to those in the Mechanism, Annotation and Classification in Enzymes (MACiE) database of catalytic sites (Fig. 2A) (46). Histidine, aspartic acid, and arginine are preponderant in both, consistent with catalytic roles and hence with being structurally invariant in functionally similar enzymes (47). Noncatalytic residues, however, are mostly absent from MACiE but frequent in 3D templates. For example, glycines and prolines are in 69% and 27% of 3D templates, respectively. The low all-atom root-mean-square deviation (rmsd) between ETA templates and their cognate match sites, over a wide range of sequence identities (Fig. 2B), shows that just like catalytic residues these noncatalytic template residues are also structurally invariant among enzymes that catalyze the same reaction, and thus do not hinder recognition by ETA templates. This is distinct from the rest of the structure, as the rmsd between a query protein and its ETA matches, over the entire structures or between clusters of evolutionarily unimportant residues, increases as sequence identity decreases (Fig. 2B). This structural invariance of ETA templates does not compromise recognition of similarities among structures with and without ligand. In 248 enzymes for which both apo and holo conformation were available, ETA properly identified structurally invariant templates that led to reciprocal matches between these apo and holo structures in 89% of cases (Fig. S1C). These data show that ETA templates are robust, structurally invariant hybrids of catalytic and noncatalytic residues. They also suggest that noncatalytic 3D template residues, such as glycine and proline, take part in catalytic site stability and dynamics (48–51).
Fig. 2.

Noncatalytic residues are prevalent in structurally invariant ETA templates. (A) Comparison of log propensities of ETA six-residue templates and known catalytic residues from MACiE database 3.0. ETA templates use glycine and proline residues at higher propensity than they appear in catalytic sites (Pearson coefficient = 0.58 considering all residues; 0.91 when ignoring G and P). (B) The rmsd for structural alignments for ETA matches, binned according to sequence identity. Alignments were generated using ETA templates and the entire structures using all atoms (lovoalign) and only the alpha carbons (TM-Align) for all matches. Negative control templates were also made using clusters of evolutionarily unimportant residues and aligned (alpha carbon only).
In practice, the human zeta-crystallin [Protein Data Bank (PDB) ID code 1yb5, chain A] illustrates how both catalytic and noncatalytic residues contribute to predictions. Out of the 3,082 targets, a 3D template from this protein correctly matched a quinone oxidoreductase from Escherichia coli (PDB ID code1qor, chain B, EC 1.6.5.5) (52) (Fig. 3A). The matched residues had near identical geometry and evolutionary importance to those from the zeta-crystallin template, including four glycines previously described as contributing to stability due to their structural positioning (53, 54) (Fig. 3C). More generally, this template was made of the most important and most tightly clustered top-ranked ET residues from a larger group of mostly spatially invariant residues between the two proteins (Fig. 3B). By contrast, the best overall sequence identity match, at 32%, and the best overall structural match, at 1.99 Å rmsd, both pointed erroneously to an alcohol dehydrogenase from Sulfolobus solfataricus (PDB ID code 1r37, chain B, EC 1.1.1.1). Alcohol dehydrogenases have the same Rossmann fold as quinone reductases but, unlike them, require a zinc ion for proper function (53, 54). Therefore, in alcohol dehydrogenases, it is the residues near that zinc ion that are most evolutionarily important, and these are not matched by the zeta-crystallin query template (Fig. 3B). Indeed, such an erroneous match to alcohol dehydrogenases would include a residue as far as 4 Å off from its position in the template (Fig. 3D). These data show the extent to which both evolutionarily important catalytic and noncatalytic template residues capture local features that specifically determine function.
Fig. 3.

Using evolution as a guide, ETA identifies structural homologs that perform the same function. (A) The closest structural match to the human zeta-crystallin (blue, PDB ID code 1yb5, chain A) is an alcohol dehydrogenase from S. solfataricus (red, PDB: 1r37; chain B) with an rmsd of 1.99A, however they are functionally dissimilar. Conversely, ETA correctly matches the zeta-crystallin to E. coli quinone oxidoreductase (green, PDB ID code 1qor, chain B) despite a larger global rmsd value of 2.25 Å. (B) Comparison of the pretemplate residue clusters (alpha carbons) identified by ETA. The residue cluster for the E. coli quinone oxidoreductase more closely matches the cluster of human zeta-crystallin than S. solfataricus alcohol dehydrogenase. The S. solfataricus alcohol dehydrogenase coordinates zinc (orange), which leads to different substrate specificity despite the similarity in global topology. The cluster alignments are also shown in sequence form in A next to the target structures, where one dot signifies aligned residues and two dots signify an rmsd of less than 0.5 Å. (C and D) ETA alpha-carbon templates for zeta-crystallin (blue), quinone oxidoreductase (green), and alcohol dehydrogenase (red) represented as spheres. (E) Table of template residues with ET coverage values, where values closer to 0 are evolutionarily important and values closer to 100 are unimportant. The yellow highlights represent residues where the zeta-crystallin/alcohol dehyrodenase match site differ >5%.
Experimental Validation Studies.
We sought next to directly validate ETA with experimental controls. For this, we selected predictions for which there was no prior knowledge of activity and substrate and that were based on matches to proteins with less than 30% sequence identity, as accuracy is especially challenging in these distant homologs.
The first case is dhaf_2064 from Desilfutobacterium hafniense (PDB ID code 3db2, chain A; Fig. S2 and Fig. 4A), which is a member of the functionally diverse family of Rossmann fold proteins. In keeping with the prior discussion, the dhaf_2064 template, {G11, G13, E95, K96, P97, H184}, included two glycines and one proline that are noncatalytic (Fig. S2). This template matched three proteins that were evolutionarily distant with sequence identity of 21%, 17%, and 18%, namely Lactobacillus plantarum (PDB ID code 3cea), Corynebacterium glutamicum (PDB ID code 3euw), and Salmonella typhimurium (PDB ID code 3ec7), respectively. The mean rmsd of the matches was 0.8 Å, and the mean evolutionary importance rank of the matches was 1.5%. Strikingly, all three proteins carried the same oxidoreductase catalytic function, and thus dhaf_2064 was predicted to have that function as well: EC 1.1.1.18, namely, to convert myo-inositol into scyllo-inosose via the reduction of NAD+.
Fig. 4.

ETA template residues accurately identify substrate specificity and are necessary for function. (A) Validation of high-confidence predictions of enzymatic activity for two uncharacterized Structural Genomics proteins using crude lysate preps. (Left) dhaf_2064 has significantly more myo-inositol dehydrogenase activity than lysates lacking the protein (empty vector) and lysates containing the dhaf_2064 E95A template mutant (P = 0.005, n = 6). (Right) tm1040_2492 has significantly more carboxylesterase activity compared with control lysates lacking the protein (P = 0.0005, n = 6). (B) ETA matching of tm1040_2492 (PDB ID code 2pbl, chain C) to three carboxylesterases (EC 3.1.1.1): EstE2 (PDB ID code 2hm7, chain A), EstE1 (PDB ID code 2c7b, chain B), and AFEST (PDB ID code 1jji, chain D). ETA did not match tm1040_2492 to Lip1 (PDB ID code 1trh, chain A), a lipase (EC 3.1.1.3). (C) Structural alignment of tm1040_2492 and the three ETA matches; labels correspond to residue numbers in tm1040_2492. (D) Structural alignment of tm1040_2492 and to Lip1 shows that the Proline residue at position 104 does not have a reciprocal cognate residue in Lip1 (black arrow). (E) Dependence of catalytic activity on carbon chain length of substrate. tm1040_2492 only catalyzes hydrolysis when substrates have ≤10 carbon atoms in the fatty acid chain. (F) Specific activity of WT tm1040_2492 and template and control mutants toward 4-nitrophenyl acetate (C2 in D). All template mutants have significantly less carboxylesterase activity compared with wild-type and control mutants (all P values ≤ 0.005, n = 6). Additionally, the W73F mutation had significantly more activity than the W73A mutation (P values < 0.005, n = 6). All error bars represent SD.
To confirm this prediction, we expressed dhaf_2064 in E. coli and tested crude extracts in vitro for myo-inositol activity. Extracts containing dhaf_2064 had significantly more activity toward the predicted substrates (4 U/mg) than negative control extracts that lacked the protein (0 U/mg) (Fig. 4A). As a further control, and because template residue E95 is thought to mediate a key binding event to cofactor NAD+ (55), we showed that an extract from an E95A mutant abolished activity (0 U/mg). These data confirm three aspects of this ETA prediction: the enzyme activity, the substrate specificity, and the critical role of at least one template residue.
A second case focused on tm1040_2492 from Silicibacter sp. (PDB ID code 2pbl, chain C), hereafter protein x for short, selected because it belongs to the highly populated α/β hydrolase fold and may therefore match a wide spectrum of possible functions. ETA matched protein x to three proteins: EstE1 (PDB ID code 2c7b, chain B; 18% identity) from the metagenome of thermophilic organisms, Est2 (PDB ID code 2hm7, chain A; 18% identity) from Alicyclobacillus acidocaldarius, and AFEST (PDB ID code 1jji, chain D; 16% identity; Fig. 4B) from the archaeon Archaeoglobus fulgidus. Strikingly, all were carboxylesterases from the hormone-sensitive lipase (HSL)–like family (Fig. 4B, EC 3.1.1.1), suggesting that, like them, protein x catalyzed the hydrolysis of an ester bond into an alcohol and a carboxylic acid.
To test this prediction in vitro, we monitored the degradation of a carboxylesterase substrate, 4-nitrophenyl acetate, by a crude extract of E. coli containing recombinant protein x. This extract had significantly more activity (13 U/mg) than a negative control extract that lacked protein x (1.5 U/mg) (Fig. 4A). Although these data show that as predicted protein x hydrolyzes some ester bonds, it is important to recognize that 4-nitrophenyl acetate is not representative of the entire spectrum of substrates with ester bonds. HSL-like carboxylesterases (EC 3.1.1.1) target ester bonds from short fatty acid chains, and their activity falls dramatically in chains longer than 8 or 10 carbons (56, 57). By contrast, lipases (EC 3.1.1.3) hydrolyze ester bonds from fatty acid carbon chains with more than 10 carbons (58, 59).
To confirm the substrate selectivity of protein x, we purified it (Fig. S3) and tested its activity in vitro against fatty acid chains of increasing length: acetate (2C), butyrate (4C), octanoate (8C), decanoate (10C), and palmitate (16C, lipase substrate) (Fig. 4E). Protein x activity decreased from 100% against 4-nitrophenyl acetate (2C) to 15%, 3%, 2.5%, and 0.5% activity toward 4-nitrophenyl butyrate, octanoate, decanoate, and palmitate, respectively. These data show that like other HSL-like carboxylesterases, protein x has extremely low activity toward fatty acid carbon chain lengths of 8 or 10, and no activity toward the lipase substrate containing a fatty acid chain length of 16 carbons (Fig. 4E). Consequently, we can conclude that protein x is a true member of the HSL-like family of carboxylesterase, rather than a lipase.
Because both HSL-like carboxylesterases and lipases share the same Ser–His–Glu catalytic triad of residues, their respective substrate bias toward shorter or longer chain lengths must depend on other residues. To probe the functional role of the 3D template residues, {G71, W73, P104, S136, H241}, we structurally aligned protein x with the HSL-like carboxylesterases. The mean rmsd of these residues with their targets is 0.6 Å, so they are structurally invariant in HSL-like family members (Fig. 4C). The template contains two of the three residues from the Ser–His–Glu catalytic triad, serine 136, and histidine 241. The glutamic acid at position 214 was not included in the template as it is relatively buried and ETA preferentially picks surface-exposed residues. The template also contains glycine 71, which forms the critical oxyanion hole in HSL-like carboxylesterases (60, 61). The role of the tryptophan at position 73, and of the proline at position 104, remains unknown, but their spatial invariance suggests they may stabilize and promote hydrophobic interactions in the active site cleft consistent with a role in substrate specificity and reaction mechanisms (60).
This last possibility is further supported by a second structural alignment of protein x with the lipase from Candida rugosa (Fig. 4D). In that case, the catalytic serine and histidine are structurally invariant between the two proteins, as is the glycine in the oxyanion hole and the tryptophan that lines the active site. The proline, however, cannot be aligned with any residue in the C. rugosa lipase. Lipases, unlike the HSL-like carboxylesterases, undergo conformational changes to accommodate large lipid substrates in their active sites, and in this region lipases are rich with flexible random coils. This suggests that, unlike HSL-like carboxylesterases, lipases might not tolerate a rigid proline residue at the cognate location. These data show that a single noncatalytic template residue, proline 104, is critical to distinguish substrate specificity, whereas the catalytic residues cannot do so by themselves, as these are common to both the lipases and the HSL-like carboxylesterases enzyme families.
To confirm that the template residues were essential to function, we individually mutated each one to alanine, purified the mutants, and tested for activity toward 2-nitrophenyl acetate, the most reactive substrate (Fig. 4F). Mutation of any of the template residues, whether catalytic or not, always abolishes enzymatic activity. In contrast, mutations of nontemplate residues near the catalytic triad do not necessarily inactivate the protein. For instance, the control mutation, E105A, preserved activity even though this residue is within 2 Å of the active site. Likewise, Y12A, which is within 10 Å of the active site, retains significantly greater activity than any of the template mutations (Fig. 4F). These control mutations were picked based on their ET ranks of evolutionary importance, which put E105 in the bottom 5% of importance and Y12 in the bottom 20%. These data show that the template residues are necessary for activity, unlike nearby residues with poor ET rank.
ETA exploits an important feature of top-ranked ET positions—namely, that although they are important, they are often not invariant. Instead the residues at these positions vary, in direct correlation with evolutionary branches. This fact is reflected by allowing 3D template residues to “wobble,” meaning that they can match to alternative side chains if they appear recurrently in the multiple sequence alignment. This wobble is therefore different from case to case, and in practice, it increases sensitivity from 62% to 70% in our benchmarks (Fig. S4). Interestingly, two of the three proteins that matched protein x had such wobbles. Whereas the native protein x template contains W73, the matched cognate structural position was a phenylalanine in both EstE1 and AFEST. Because all of these proteins have similar substrate specificity, we hypothesize that regardless of this tryptophan to phenylalanine wobble, these positions fulfill identical mechanistic roles.
To confirm that the wobble does not fundamentally alter function, we introduced a W73F mutation into protein x. The specific activity of the mutant was reduced (229 U/mg), but still remained larger than the activity of the Y12A control mutation (198 U/mg), and it was much more active than a control W73A mutation (1.9 U/mg) (Fig. 4F). These data show that position 73 is critical to function and yet tolerates a phenylalanine substitution remarkably well despite dramatic sequence differences between protein x, EstE1, and AFEST.
Discussion
The key findings of this work are that ETA can now reliably predict substrate-level specificity and the experimental demonstration of this fact on two previously unannotated proteins. The approach relies on narrowly identifying molecular similarity between proteins in the immediate vicinity of their most evolutionarily important site, which can potentially include both catalytic and noncatalytic residues. The role of catalytic residues is well understood and has motivated several other template-based annotations methods (16, 62, 63) and spurred many approaches to annotate structural genomics structures based on local structural similarities (14–16, 23, 24, 62–67). However, because the fundamental catalytic mechanism may be invariant between proteins that operate on varied substrates (68), the challenge has been to add noncatalytic residues that constrain the catalytic mechanism to a more specific substrate (24, 69). The selection of which noncatalytic residues in the vicinity of an active site to include in a template is not trivial, however because not all residues near a catalytic site are themselves important.
To solve this problem, ETA relies on evolution and on structure to pick the most evolutionarily important residues that also cluster tightly at or near the protein surface. It is has been previously shown that evolutionarily important residues are necessary for function (31–33, 70, 71), map functional sites (71, 72), guide a variety of targeted mutational experiments that efficiently block function (73, 74), separate functions in multifunctional proteins (38, 75), or recode function by swapping amino acids between functionally distinct homologs (34, 36). For these reasons, these residues are good candidates from which to pick templates.
As a result, the ETA annotation pipeline reliably predicted both the activity and the substrate of an enzyme structure. In retrospective controls, ETA accuracy was consistently high (96%) even when the annotations were based on matches to homologs that were deep in the twilight zone, with homology below 30% sequence identity. These results were equivalent and in some cases better than previously defined template (14, 15, 23, 65) sequence- (29, 30) or topology- (67, 76) based annotation methods. We then extended our testing to annotate previously uncharacterized proteins (14, 25, 26, 46) to validate unique ETA predictions of function. Additionally, mutagenesis studies verified the essential role played by the noncatalytic residues that were selected to be in the 3D template. Thus, all template residues, catalytic or noncatalytic, are necessary for complete enzyme activity toward its substrate. Our data also show that evolutionary wobble substitutions enhance computational specificity.
Taken together, this work suggests that ETA 3D templates capture the essential local elements of the enzyme structure, dynamics, and chemical activity that combine to define substrate-specific mechanisms. A central finding is the essential role that noncatalytic 3D template residues play in specifying substrates. As a result, this study validates a high-throughput method that combines evolution and structure to identify the activity and substrate specificity of unique enzymes, with applications to enzyme annotations on a structural genomics scale.
An ETA webserver is available that makes 4 E.C. digit predictions using five or six residue templates: http://mammoth.bcm.tmc.edu/AminErdinetalPNAS/eta. This webserver also integrates the newly described confidence scoring. We are actively maintaining this site, and several updates to enhance prediction power and protein visualization are currently in production.
Materials and Methods
Evolutionary Trace Annotation Pipeline.
ETA is made of five modules that separately operate to suggest a predicted function for a given query protein. In the first module, ET algorithm (31, 32) assigns evolutionary importance ranks to the query protein’s residues based on correlation of branches in the phylogenetic tree, with the variations in the multiple sequence alignment generated by ClustalX (77) for homologs identified for the sequence of query protein by BLAST (78). The second module is the Template Picker algorithm (26), which selects five or six best-ranked residue positions near the center of a cluster of at least 11 evolutionarily important residues on the protein surface where solvent accessibility of the residues is greater than 2 Å2, as calculated by DSSP algorithm (79). Each position is geometrically represented by the 3D Cartesian coordinates of the selected residue’s alpha carbon atoms. ETA templates use both residue labels in the query structure (native templates) and a combination of variations that were observed at least twice in the multiple sequence alignment (variations) for the identified positions (40). Further, the paired-distance algorithm (40) searches the query template against a “target” library of proteins with known functions for geometric similarity and, in doing so, identifies geometric matches in which residue labels are alike with those in the query template and subject to the criterion that each pair of residues in the template and the matched region are within a distance of 2.5 Å. In the fourth module, found matches are passed to the support vector machine (SVM), which identifies significant matches based on geometric and evolutionary similarities. ETA uses a support SVM trained for a set of 53 enzymes based on six-residue templates (40) using SPIDER package (www.kyb.tuebingen.mpg.de/bs/people/spider) for MATLAB with a radial basis function kernel and σ = 0.5. SVM feature space is seven-dimensional, with one dimension representing rmsd between the query template residues and found matched site in the target structure, whereas six other dimensions are for ET percentile rank differences of the query six-template residues and the residues they are linked with in the target structures. In the case of the five-residue ETA, we use the same six-residue SVM by constructing a virtual sixth residue as the average of the other five positions for the five-residue data. ETA repeats all these steps reciprocally, generating templates from target structures and searching for significant matches on the “query” protein. In the last module, ETA suggests the most seen function as a prediction among a set of identified reciprocal significant matches.
Confidence Score.
In developing a confidence score, we considered the following two criteria: First, confidence should be high if the number of matches that have the same EC number is large, and second, confidence should be low if the number of matches with differing EC numbers is large. A simple model that holds these criteria is defined to be 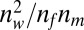 , where
, where 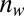 is the number of matches with winning function,
is the number of matches with winning function,  is the number of identified distinct function, and
is the number of identified distinct function, and  is the number of matches.
is the number of matches.
Negative Templates.
To generate negative templates, we started with the surface residue (solvent accessible area greater than 2 Å2 as calculated by the DSSP algorithm) (79) with the poorest ET percentile rank. Next we selected the neighbor surface residue with the poorest ET percentile rank that lies between 70% and 100% ET percentile to the first selected one within a distance of 8 Å. We further selected the surface residues iteratively with the poorest percentile rank that lies between 70% and 100% ET percentile within the distance of 8 Å with the center of mass of chosen residues in the previous iteration.
Comparison of Template, Negative Template, and Global Structure Alignments.
Alignment data are retrieved from 1,157 pairs of matches in which query and target protein functions are in full agreement for 451 proteins. We used TM-align (80) and lovoalign (81) structural alignment methods for alpha-carbon rmsd and all-atom rmsd, respectively. To calculate template alignment rmsd, we aligned query templates with their corresponding match sites that are identified by ETA through TM-align and lovoalign. Again, TM-align and lovoalign were used to align the whole structures of query and target proteins in reciprocal match pairs. We applied the negative templates generated as described above from the query proteins to the previously found reciprocal match partners. Among those, ETA’s paired-distance matching algorithm identified a matched site for 445 proteins, with 1,127 matches in total. In generation of Fig. 2B, these pairs of matches were used for comparison of alignments at levels of templates, clusters, and global structures.
ETA and TM-Align Comparison for Annotation Performance.
We compared performance of ETA and TM-align (80) on a set of 430 protein structures where ETA made correct or incorrect predictions with six-residue templates. TM-align annotation was made by identifying the lowest rmsd hit upon aligning 430 proteins with the protein structures in the target set. Next, we grouped ETA and TM-align predictions according to the maximum sequence identity of reciprocal matches found by ETA for 430 proteins.
ETA and COFACTOR Comparison for Annotation Performance.
We specifically selected test proteins that were matched with high (>1) or medium (=1) confidence to proteins with less than 30% sequence identity for the comparison (n = 137). All proteins were submitted to the COFACTOR webserver (http://zhanglab.ccmb.med.umich.edu/COFACTOR/) between 8/5/13 and 8/14/13. Due to the fact that the COFACTOR webserver has a larger target enzyme set than the one used in this publication, many of the query proteins were matched to homologs of >30% sequence identity. Therefore, both the top hit (highest CscoreEC) and the top hit with less than 30% sequence identity (defined as IDEN*Cov. < 30%) were recorded. For the library size comparison, only the four-digit PDB codes were used; chains were not considered. For example, 1jjiD and 1jjiC would be considered the same protein.
Propensity Calculation.
Propensity of a particular residue is defined to be the ratio of its frequency among all template residues for the protein structures in the experiment to its frequency within all residues for the protein structures (82). To get better insight, we used log propensity, which is defined to be the logarithm of the calculated ratio. Log propensity becomes positive for the values with a ratio greater than 1, whereas it becomes negative when the ratio is less than 1. MACiE’s log propensities were retrieved from MACiE database version 3.0 (www.ebi.ac.uk/thornton-srv/databases/MACiE/) (46):
 |
where  is the total number of measurements,
is the total number of measurements,  is the ith measurement, and
is the ith measurement, and 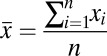 .
.
Performance Measures.
Positive predictive value (PPV) = TP/(TP + FP), where TP and FP stand for the numbers of True Positives and False Positives, respectively. True Positive is defined for cases where the prediction fully agrees with the known function at the four-digit EC level. False Positive is the case in which the predicted EC number for a protein does not agree fully with its known EC number. Sensitivity = TP/(TP + FN), where FN denotes False Negatives, which is defined for the cases for which ETA does not make predictions.
Datasets.
In selecting proteins for the benchmark test set, we started with 837 Structural Genomics enzymes with full EC annotations in the SwissProt database (83). The structures were retrieved from PDB (84) as of October 2010, and any pair of enzymes has at most 90% sequence identity with one another. One distinct feature of those proteins was that they had a varying truncation ratio, which was defined to be the ratio of number of amino acids in the protein structure to the actual sequence length. This ratio quantifies how much the structure represents the whole protein sequence. Therefore, a ratio of less than 1 means that the whole sequence is not represented in the structure. The cases with such ratios might pose a potential problem in automated computational annotation efforts, as these structures might lack the regions essential for function. This motivated us to assess how ETA performed on those cases. To this end, we calculated the prediction coverage of query proteins in a given bin of truncation ratios (Fig. S1). The prediction coverage reaches its maximum values (>71%) when the ratio is >0.9, whereas the PPV does not show a clear pattern as the prediction coverage below this threshold. Therefore, we used 605 protein structures with a truncation ratio greater than 0.9 as a benchmark test set.
The target set contains 3,082 protein structures with a truncation ratio greater than 0.95 and full EC annotation from 2008PDB90, among which any pair has at most 90% sequence identity with one another. EC annotations of these proteins are retrieved from PDB (317), SwissProt (2454), and Trembl (311) databases, where the numbers inside the parentheses denote the number of protein structures with the associated annotation source.
Apo–holo pairs were taken from ref. 85. In our analysis, we used only those that were enzymes, which were identified based on annotation in the PDBSprotEC (86). From these, we further restrict to apo–holo pairs with precalculated templates from the ETA server and, if the apo and holo structures have different truncation lengths, with a ratio of the number of amino acids in the shorter to that of the longer structure of at least 0.9.
The log propensity dataset contains 413 SG enzymes that are predicted correctly with ETA’s six-residue templates.
The modified template control set contains 476 protein structures whose native six-residue ETA templates have at least one glycine or proline residue.
The benchmark test set and target set are available at http://mammoth.bcm.tmc.edu/AminErdinetalPNAS/Sup/.
Cloning and Expression.
Each uncharacterized gene was PCR amplified from vectors received from a structural genomics center using the primers identified in Table S1. The resulting PCR products contained Not1 and Sal1 restriction sites, which were used to insert the genes into the pet28a vector, resulting in an N-terminal His tag. The vectors were electroporated into BL21D cells for protein expression. Protein expression was carried out as previously described (87).
Initial enzymatic assays were carried out using crude lysate. Crude lysates were produced by spinning down 15 mL of induced culture, freezing the pellets overnight at –20 °C, and resuspending the pellets in 1 mL of Bugbuster Mastermix (Novagen). Lysates were kept shaking at room temperature for 20 min, followed by centrifugation to remove the insoluble debris. Protein concentration of the supernatant was then determined using the MicroBCA kit (Thermo).
Determination of Enzymatic Activity.
Activity of myo-inositol dehydrogenase was determined by adding crude lysate to a mixture of 25 mM myo-inositol, 10 mM sodium phyrophosphate (pH 9.0), and 0.5 mM β-nicotinamide adenine dinucleotide and monitoring absorbance at 340 nm (88). Activity of the putative aspartate aminotransferase was determined by adding crude lysate to a mixture of 50 mM l-Aspartate, 5 mM alpha-ketoglutarate, 100 mM Tris⋅HCl (pH 8.0), 0.1 mM NADH, and two units of malate dehydrogenase (Sigma) and monitoring absorbance at 340 nm. All reactions were initiated following the addition of the substrate (myo-inositol or l-aspartate) and mixed vigorously. For both reactions, one unit of activity is defined as the conversion of 1.0 µmole of NAD/NADH per minute using 6.22 as the millimolar extinction coefficient (89). P values were obtained using Student t test.
Carboxylesterase Purification, Activity Measurement, and Mutation.
Purification and measurement of activity was conducted as previously defined (87). All 4-nitrophenyl substrates were dissolved in 100% DMSO at a stock solution of 100 mM. The final reaction volumes consisted of 50 mM 2-(N-Morpholino)ethanesulfonic acid, pH 6.0, 3% (vol/vol) DMSO, and 1 mM 4-nitrophenyl substrate and enzyme, either as crude lysate or purified. The reaction was initiated upon addition of the substrate and monitored at 405 nm using an Amersham Ultrospec 3100pro spectrophotometer. All mutations were carried out using the Quickchange II Site-Directed Mutagenesis Kit (Agilent) using primers found in Table S1.
Supplementary Material
Acknowledgments
We thank the following University of Saint Thomas students for bioinformatic support: Anthony Pham, Anthony Tran, Brian Bui, and Medina Baitemirova. We gratefully acknowledge grant support from the National Institutes of Health (GM079656, GM066099, and K12 GM084897) and from the National Science Foundation (CCF 0905536 and DBI 1062455).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305162110/-/DCSupplemental.
References
- 1.Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol. 2007;8(12):995–1005. doi: 10.1038/nrm2281. [DOI] [PubMed] [Google Scholar]
- 2.Loewenstein Y, et al. Protein function annotation by homology-based inference. Genome Biol. 2009;10(2):207. doi: 10.1186/gb-2009-10-2-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erdin S, Lisewski AM, Lichtarge O. Protein function prediction: Towards integration of similarity metrics. Curr Opin Struct Biol. 2011;21(2):180–188. doi: 10.1016/j.sbi.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rentzsch R, Orengo CA. Protein function prediction—The power of multiplicity. Trends Biotechnol. 2009;27(4):210–219. doi: 10.1016/j.tibtech.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Wilkins AD, Bachman BJ, Erdin S, Lichtarge O. The use of evolutionary patterns in protein annotation. Curr Opin Struct Biol. 2012;22(3):316–325. doi: 10.1016/j.sbi.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devos D, Valencia A. Practical limits of function prediction. Proteins. 2000;41(1):98–107. [PubMed] [Google Scholar]
- 7.Rost B. Enzyme function less conserved than anticipated. J Mol Biol. 2002;318(2):595–608. doi: 10.1016/S0022-2836(02)00016-5. [DOI] [PubMed] [Google Scholar]
- 8.Tian W, Skolnick J. How well is enzyme function conserved as a function of pairwise sequence identity? J Mol Biol. 2003;333(4):863–882. doi: 10.1016/j.jmb.2003.08.057. [DOI] [PubMed] [Google Scholar]
- 9.Addou S, Rentzsch R, Lee D, Orengo CA. Domain-based and family-specific sequence identity thresholds increase the levels of reliable protein function transfer. J Mol Biol. 2009;387(2):416–430. doi: 10.1016/j.jmb.2008.12.045. [DOI] [PubMed] [Google Scholar]
- 10.Nagano N, Orengo CA, Thornton JM. One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J Mol Biol. 2002;321(5):741–765. doi: 10.1016/s0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- 11.Kahraman A, Morris RJ, Laskowski RA, Favia AD, Thornton JM. On the diversity of physicochemical environments experienced by identical ligands in binding pockets of unrelated proteins. Proteins. 2010;78(5):1120–1136. doi: 10.1002/prot.22633. [DOI] [PubMed] [Google Scholar]
- 12.Gerlt JA, Babbitt PC, Rayment I. Divergent evolution in the enolase superfamily: The interplay of mechanism and specificity. Arch Biochem Biophys. 2005;433(1):59–70. doi: 10.1016/j.abb.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 13.Goyal K, Mohanty D, Mande SC. PAR-3D: A server to predict protein active site residues. Nucleic Acids Res. 2007;35(Web Server issue):W503–W505. doi: 10.1093/nar/gkm252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tseng YY, Dundas J, Liang J. Predicting protein function and binding profile via matching of local evolutionary and geometric surface patterns. J Mol Biol. 2009;387(2):451–464. doi: 10.1016/j.jmb.2008.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Redfern OC, Dessailly BH, Dallman TJ, Sillitoe I, Orengo CA. FLORA: A novel method to predict protein function from structure in diverse superfamilies. PLOS Comput Biol. 2009;5(8):e1000485. doi: 10.1371/journal.pcbi.1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li GH, Huang JF. CMASA: An accurate algorithm for detecting local protein structural similarity and its application to enzyme catalytic site annotation. BMC Bioinformatics. 2010;11:439. doi: 10.1186/1471-2105-11-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pugalenthi G, et al. SMpred: A support vector machine approach to identify structural motifs in protein structure without using evolutionary information. J Biomol Struct Dyn. 2010;28(3):405–414. doi: 10.1080/07391102.2010.10507369. [DOI] [PubMed] [Google Scholar]
- 18.Tseng YY, Li WH. PSC: Protein surface classification. Nucleic Acids Res. 2012;40(Web Server issue):W435–W439. doi: 10.1093/nar/gks495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chance MR, et al. Structural genomics: A pipeline for providing structures for the biologist. Protein Sci. 2002;11(4):723–738. doi: 10.1110/ps.4570102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu T, Altman RB. Using multiple microenvironments to find similar ligand-binding sites: Application to kinase inhibitor binding. PLOS Comput Biol. 2011;7(12):e1002326. doi: 10.1371/journal.pcbi.1002326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie L, Bourne PE. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments. Proc Natl Acad Sci USA. 2008;105(14):5441–5446. doi: 10.1073/pnas.0704422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng EC, Polacco BJ, Babbitt PC, Rigden DJ. 3D motifs. In: Rigden DJ, editor. From Protein Structure to Function with Bioinformatics. The Netherlands: Springer; 2009. pp. 187–216. [Google Scholar]
- 23.Laskowski RA, Watson JD, Thornton JM. Protein function prediction using local 3D templates. J Mol Biol. 2005;351(3):614–626. doi: 10.1016/j.jmb.2005.05.067. [DOI] [PubMed] [Google Scholar]
- 24.Polacco BJ, Babbitt PC. Automated discovery of 3D motifs for protein function annotation. Bioinformatics. 2006;22(6):723–730. doi: 10.1093/bioinformatics/btk038. [DOI] [PubMed] [Google Scholar]
- 25.Porter CT, Bartlett GJ, Thornton JM. The Catalytic Site Atlas: A resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004;32(Database issue):D129–D133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kristensen DM, et al. Prediction of enzyme function based on 3D templates of evolutionarily important amino acids. BMC Bioinformatics. 2008;9:17. doi: 10.1186/1471-2105-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hermann JC, et al. Structure-based activity prediction for an enzyme of unknown function. Nature. 2007;448(7155):775–779. doi: 10.1038/nature05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalyanaraman C, et al. Discovery of a dipeptide epimerase enzymatic function guided by homology modeling and virtual screening. Structure. 2008;16(11):1668–1677. doi: 10.1016/j.str.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tian W, Arakaki AK, Skolnick J. EFICAz: A comprehensive approach for accurate genome-scale enzyme function inference. Nucleic Acids Res. 2004;32(21):6226–6239. doi: 10.1093/nar/gkh956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu C, Zavaljevski N, Desai V, Reifman J. Genome-wide enzyme annotation with precision control: Catalytic families (CatFam) databases. Proteins. 2009;74(2):449–460. doi: 10.1002/prot.22167. [DOI] [PubMed] [Google Scholar]
- 31.Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257(2):342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 32.Mihalek I, Res I, Lichtarge O. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J Mol Biol. 2004;336(5):1265–1282. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 33.Lichtarge O, Yamamoto KR, Cohen FE. Identification of functional surfaces of the zinc binding domains of intracellular receptors. J Mol Biol. 1997;274(3):325–337. doi: 10.1006/jmbi.1997.1395. [DOI] [PubMed] [Google Scholar]
- 34.Sowa ME, et al. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat Struct Biol. 2001;8(3):234–237. doi: 10.1038/84974. [DOI] [PubMed] [Google Scholar]
- 35.Raviscioni M, Gu P, Sattar M, Cooney AJ, Lichtarge O. Correlated evolutionary pressure at interacting transcription factors and DNA response elements can guide the rational engineering of DNA binding specificity. J Mol Biol. 2005;350(3):402–415. doi: 10.1016/j.jmb.2005.04.054. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez GJ, Yao R, Lichtarge O, Wensel TG. Evolution-guided discovery and recoding of allosteric pathway specificity determinants in psychoactive bioamine receptors. Proc Natl Acad Sci USA. 2010;107(17):7787–7792. doi: 10.1073/pnas.0914877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quan XJ, et al. Evolution of neural precursor selection: Functional divergence of proneural proteins. Development. 2004;131(8):1679–1689. doi: 10.1242/dev.01055. [DOI] [PubMed] [Google Scholar]
- 38.Adikesavan AK, et al. Separation of recombination and SOS response in Escherichia coli RecA suggests LexA interaction sites. PLoS Genet. 2011;7(9):e1002244. doi: 10.1371/journal.pgen.1002244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kristensen DM, et al. Recurrent use of evolutionary importance for functional annotation of proteins based on local structural similarity. Protein Sci. 2006;15(6):1530–1536. doi: 10.1110/ps.062152706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ward RM, et al. De-orphaning the structural proteome through reciprocal comparison of evolutionarily important structural features. PLoS ONE. 2008;3(5):e2136. doi: 10.1371/journal.pone.0002136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Erdin S, Ward RM, Venner E, Lichtarge O. Evolutionary trace annotation of protein function in the structural proteome. J Mol Biol. 2010;396(5):1451–1473. doi: 10.1016/j.jmb.2009.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. International Union of Biochemistry and Molecular Biology; Nomenclature Committee; Webb EC (1992) Enzyme Nomenclature 1992: Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes (Academic, San Diego, CA), pp xiii, 862.
- 43.Perona JJ, Craik CS. Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J Biol Chem. 1997;272(48):29987–29990. doi: 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]
- 44.Brylinski M, Skolnick J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc Natl Acad Sci USA. 2008;105(1):129–134. doi: 10.1073/pnas.0707684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roy A, Yang J, Zhang Y. COFACTOR: An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012;40(Web Server issue):W471–W477. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holliday GL, et al. MACiE (Mechanism, Annotation and Classification in Enzymes): Novel tools for searching catalytic mechanisms. Nucleic Acids Res. 2007;35(Database issue):D515–D520. doi: 10.1093/nar/gkl774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torrance JW, Bartlett GJ, Porter CT, Thornton JM. Using a library of structural templates to recognise catalytic sites and explore their evolution in homologous families. J Mol Biol. 2005;347(3):565–581. doi: 10.1016/j.jmb.2005.01.044. [DOI] [PubMed] [Google Scholar]
- 48.Betts MJ, Russell RB. Amino acid properties and consequences of substitutions. In: Barnes MR, Gray IC, editors. Bioinformatics for Geneticists. Chichester, UK: Wiley; 2003. [Google Scholar]
- 49.Peters GH, Bywater RP. Computational analysis of chain flexibility and fluctuations in Rhizomucor miehei lipase. Protein Eng. 1999;12(9):747–754. doi: 10.1093/protein/12.9.747. [DOI] [PubMed] [Google Scholar]
- 50.Krieger F, Möglich A, Kiefhaber T. Effect of proline and glycine residues on dynamics and barriers of loop formation in polypeptide chains. J Am Chem Soc. 2005;127(10):3346–3352. doi: 10.1021/ja042798i. [DOI] [PubMed] [Google Scholar]
- 51.Jacob J, Duclohier H, Cafiso DS. The role of proline and glycine in determining the backbone flexibility of a channel-forming peptide. Biophys J. 1999;76(3):1367–1376. doi: 10.1016/S0006-3495(99)77298-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thorn JM, Barton JD, Dixon NE, Ollis DL, Edwards KJ. Crystal structure of Escherichia coli QOR quinone oxidoreductase complexed with NADPH. J Mol Biol. 1995;249(4):785–799. doi: 10.1006/jmbi.1995.0337. [DOI] [PubMed] [Google Scholar]
- 53.Borrás T, Persson B, Jörnvall H. Eye lens zeta-crystallin relationships to the family of “long-chain” alcohol/polyol dehydrogenases. Protein trimming and conservation of stable parts. Biochemistry. 1989;28(15):6133–6139. doi: 10.1021/bi00441a001. [DOI] [PubMed] [Google Scholar]
- 54.Edwards KJ, et al. Structural and sequence comparisons of quinone oxidoreductase, zeta-crystallin, and glucose and alcohol dehydrogenases. Arch Biochem Biophys. 1996;328(1):173–183. doi: 10.1006/abbi.1996.0158. [DOI] [PubMed] [Google Scholar]
- 55.van Straaten KE, Zheng H, Palmer DR, Sanders DA. Structural investigation of myo-inositol dehydrogenase from Bacillus subtilis: Implications for catalytic mechanism and inositol dehydrogenase subfamily classification. Biochem J. 2010;432(2):237–247. doi: 10.1042/BJ20101079. [DOI] [PubMed] [Google Scholar]
- 56.Manco G, et al. Overexpression and properties of a new thermophilic and thermostable esterase from Bacillus acidocaldarius with sequence similarity to hormone-sensitive lipase subfamily. Biochem J. 1998;332(Pt 1):203–212. doi: 10.1042/bj3320203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim SB, Lee W, Ryu YW. Cloning and characterization of thermostable esterase from Archaeoglobus fulgidus. J Microbiol. 2008;46(1):100–107. doi: 10.1007/s12275-007-0185-5. [DOI] [PubMed] [Google Scholar]
- 58.Zhang J, Deng Y, Fang J, McKay G. Enantioselective analysis of ritalinic acids in biological samples by using a protein-based chiral stationary phase. Pharm Res. 2003;20(11):1881–1884. doi: 10.1023/b:pham.0000003389.77585.be. [DOI] [PubMed] [Google Scholar]
- 59.Rhee JK, Ahn DG, Kim YG, Oh JW. New thermophilic and thermostable esterase with sequence similarity to the hormone-sensitive lipase family, cloned from a metagenomic library. Appl Environ Microbiol. 2005;71(2):817–825. doi: 10.1128/AEM.71.2.817-825.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Simone G, et al. The crystal structure of a hyper-thermophilic carboxylesterase from the archaeon Archaeoglobus fulgidus. J Mol Biol. 2001;314(3):507–518. doi: 10.1006/jmbi.2001.5152. [DOI] [PubMed] [Google Scholar]
- 61.Mandrich L, et al. Functional and structural features of the oxyanion hole in a thermophilic esterase from Alicyclobacillus acidocaldarius. Proteins. 2008;71(4):1721–1731. doi: 10.1002/prot.21877. [DOI] [PubMed] [Google Scholar]
- 62.Wallace AC, Laskowski RA, Thornton JM. Derivation of 3D coordinate templates for searching structural databases: Application to Ser-His-Asp catalytic triads in the serine proteinases and lipases. Protein Sci. 1996;5(6):1001–1013. doi: 10.1002/pro.5560050603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fetrow JS, Siew N, Skolnick J. Structure-based functional motif identifies a potential disulfide oxidoreductase active site in the serine/threonine protein phosphatase-1 subfamily. FASEB J. 1999;13(13):1866–1874. doi: 10.1096/fasebj.13.13.1866. [DOI] [PubMed] [Google Scholar]
- 64.Wallace AC, Borkakoti N, Thornton JM. TESS: A geometric hashing algorithm for deriving 3D coordinate templates for searching structural databases. Application to enzyme active sites. Protein Sci. 1997;6(11):2308–2323. doi: 10.1002/pro.5560061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang MP, Brutlag DL, Altman RB. Automated construction of structural motifs for predicting functional sites on protein structures. Pac Symp Biocomput. 2003;8:204–215. doi: 10.1142/9789812776303_0020. [DOI] [PubMed] [Google Scholar]
- 66.Brylinski M, Skolnick J. FINDSITE: A threading-based approach to ligand homology modeling. PLOS Comput Biol. 2009;5(6):e1000405. doi: 10.1371/journal.pcbi.1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petrey D, Fischer M, Honig B. Structural relationships among proteins with different global topologies and their implications for function annotation strategies. Proc Natl Acad Sci USA. 2009;106(41):17377–17382. doi: 10.1073/pnas.0907971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bashton M, Thornton JM. Domain-ligand mapping for enzymes. J Mol Recognit. 2010;23(2):194–208. doi: 10.1002/jmr.992. [DOI] [PubMed] [Google Scholar]
- 69.Zhang S, Barr BK, Wilson DB. Effects of noncatalytic residue mutations on substrate specificity and ligand binding of Thermobifida fusca endocellulase cel6A. Eur J Biochem. 2000;267(1):244–252. doi: 10.1046/j.1432-1327.2000.00988.x. [DOI] [PubMed] [Google Scholar]
- 70.Lichtarge O, Bourne HR, Cohen FE. Evolutionarily conserved Galphabetagamma binding surfaces support a model of the G protein-receptor complex. Proc Natl Acad Sci USA. 1996;93(15):7507–7511. doi: 10.1073/pnas.93.15.7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yao H, et al. An accurate, sensitive, and scalable method to identify functional sites in protein structures. J Mol Biol. 2003;326(1):255–261. doi: 10.1016/s0022-2836(02)01336-0. [DOI] [PubMed] [Google Scholar]
- 72.Sowa ME, He W, Wensel TG, Lichtarge O. A regulator of G protein signaling interaction surface linked to effector specificity. Proc Natl Acad Sci USA. 2000;97(4):1483–1488. doi: 10.1073/pnas.030409597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cushman I, et al. Computational and biochemical identification of a nuclear pore complex binding site on the nuclear transport carrier NTF2. J Mol Biol. 2004;344(2):303–310. doi: 10.1016/j.jmb.2004.09.043. [DOI] [PubMed] [Google Scholar]
- 74.Bonde MM, et al. An angiotensin II type 1 receptor activation switch patch revealed through evolutionary trace analysis. Biochem Pharmacol. 2010;80(1):86–94. doi: 10.1016/j.bcp.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shenoy SK, et al. Beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281(2):1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 76.Laskowski RA, Luscombe NM, Swindells MB, Thornton JM. Protein clefts in molecular recognition and function. Protein Sci. 1996;5(12):2438–2452. doi: 10.1002/pro.5560051206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 78.Altschul SF, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y, Skolnick J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33(7):2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martínez L, Andreani R, Martínez JM. Convergent algorithms for protein structural alignment. BMC Bioinformatics. 2007;8:306. doi: 10.1186/1471-2105-8-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Holliday GL, Mitchell JB, Thornton JM. Understanding the functional roles of amino acid residues in enzyme catalysis. J Mol Biol. 2009;390(3):560–577. doi: 10.1016/j.jmb.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 83. Anonymous; UniProt Consortium (2010) The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res 38(Database issue):D142–D148. [DOI] [PMC free article] [PubMed]
- 84.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brylinski M, Skolnick J. What is the relationship between the global structures of apo and holo proteins? Proteins. 2008;70(2):363–377. doi: 10.1002/prot.21510. [DOI] [PubMed] [Google Scholar]
- 86.Martin AC. PDBSprotEC: A web-accessible database linking PDB chains to EC numbers via SwissProt. Bioinformatics. 2004;20(6):986–988. doi: 10.1093/bioinformatics/bth048. [DOI] [PubMed] [Google Scholar]
- 87.Venner E, et al. Accurate protein structure annotation through competitive diffusion of enzymatic functions over a network of local evolutionary similarities. PLoS ONE. 2010;5(12):e14286. doi: 10.1371/journal.pone.0014286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Berman T, Magasanik B. The pathway of myo-inositol degradation in Aerobacter aerogenes. Dehydrogenation and dehydration. J Biol Chem. 1966;241(4):800–806. [PubMed] [Google Scholar]
- 89.Wilkie SE, Warren MJ. Recombinant expression, purification, and characterization of three isoenzymes of aspartate aminotransferase from Arabidopsis thaliana. Protein Expr Purif. 1998;12(3):381–389. doi: 10.1006/prep.1997.0845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.