

Significance
This manuscript links ApoE4-mediated signaling with Sirtuin function. Specifically, we show that ApoE4, but not ApoE3, reduces neuroprotective SirT1 levels. Our data support the hypothesis that neuronal connectivity, as reflected in the ratios of critical mediators such as sAPPα:Aβ, SirT1:SirT2, APP:p-APP, and Tau:p-Tau, is programmatically altered by ApoE4. Thus ApoE4, SirT1/2, p-Tau, and p-APP, all may be part of a signaling network that is affected in AD, providing a model for therapeutic candidate screening in AD drug discovery. These findings offer a unique insight into the mechanism by which ApoE4 confers risk for the development of Alzheimer’s disease.
Abstract
The canonical pathogenesis of Alzheimer’s disease links the expression of apolipoprotein E ε4 allele (ApoE) to amyloid precursor protein (APP) processing and Aβ peptide accumulation by a set of mechanisms that is incompletely defined. The development of a simple system that focuses not on a single variable but on multiple factors and pathways would be valuable both for dissecting the underlying mechanisms and for identifying candidate therapeutics. Here we show that, although both ApoE3 and ApoE4 associate with APP with nanomolar affinities, only ApoE4 significantly (i) reduces the ratio of soluble amyloid precursor protein alpha (sAPPα) to Aβ; (ii) reduces Sirtuin T1 (SirT1) expression, resulting in markedly differing ratios of neuroprotective SirT1 to neurotoxic SirT2; (iii) triggers Tau phosphorylation and APP phosphorylation; and (iv) induces programmed cell death. We describe a subset of drug candidates that interferes with the APP–ApoE interaction and returns the parameters noted above to normal. Our data support the hypothesis that neuronal connectivity, as reflected in the ratios of critical mediators such as sAPPα:Aβ, SirT1:SirT2, APP:phosphorylated (p)-APP, and Tau:p-Tau, is programmatically altered by ApoE4 and offer a simple system for the identification of program mediators and therapeutic candidates.
Alzheimer’s disease (AD) poses a health problem of pandemic proportions. It is estimated that by the year 2030 there will be greater than 60 million people worldwide with AD, and $375 billion will be spent annually in the United States to care for affected individuals. The tedious and expensive process of drug discovery in AD is complicated by the fact that the causes and underlying mechanisms of the disease are still incompletely defined, and presymptomatic diagnosis is not yet in routine clinical use (1).
The apolipoprotein E ε4 allele (ApoE; chromosomal locus 19q13) is the single most important genetic risk factor associated with AD. This allele confers increased risk for sporadic and familial AD (2). Individuals with two copies of the ApoE ε4 allele have an approximately eightfold increased risk of AD and have a significantly lower age of onset compared with AD patients not carrying this allele (2). Recent data indicate that the greater risk of AD associated with the ApoE4 isoform might relate to ApoE’s susceptibility to proteolysis and neurotoxicity or through its role in inhibiting Aβ clearance and/or stimulating Aβ deposition, leading to plaque formation (3, 4). Contrary to the previously reported work, one recent study provides evidence that ApoE and soluble Aβ have very minimal direct interaction, thus ApoE may influence soluble Aβ metabolism through its interactions with other receptors or transporters (5). Thus, despite knowing for over a decade that the ApoE ε4 allele is somehow contributory to the disease process, the precise molecular mechanisms underlying ApoE and APP interactions, direct or indirect, resulting in ApoE4-mediated toxicity, remain unclear.
The amyloid precursor protein, APP, has been shown to function as a molecular switch: cleavage at the β, γ, and caspase sites results in the production of four pro-AD-peptides—soluble amyloid precursor protein beta (sAPPβ) (from which N-APP is derived), Aβ, Jcasp, and C31—that mediate neurite retraction, synaptic reorganization, and ultimately programmed cell death. In contrast, cleavage at the α site produces the trophic peptide sAPPα and the inhibitor of APP γ-site cleavage, alpha-COOH-terminal fragment of APP (αCTF) (1, 6). The decision between these two proteolytic pathways is governed at least in part by ligand binding: interaction with the axon guidance and trophic factor netrin-1 increases α-site cleavage, whereas interaction with the antitrophin Aβ inhibits α-site cleavage and increases net production of the four neurite-retractive peptides (1, 6). According to a recent study, the level of cerebrospinal fluid (CSF) sAPPα is significantly lower in AD patients possessing one or two ApoE4 alleles than in those not possessing the ApoE4 allele (7). Therefore, it was of interest to determine whether ApoE isoforms impact this trophic–antitrophic peptide balance differentially, and, if so, by what mechanism.
Our data reveal differential effects of ApoE4 vs. ApoE3 on APP interaction, signaling, and processing and are compatible with the notion that the Alzheimer’s phenotype represents an imbalance between the trophic and antitrophic signaling of APP, reflected by the ratio of the four APP-derived neurite-retractive peptides to the two APP-derived trophic peptides. Our current results show that ApoE4 interacts with high affinity with APP, shifting the processing balance in the antitrophic direction, decreasing sAPPα secretion, and reducing sAPPα/Aβ and sAPPα/sAPPβ ratios in comparison with ApoE3. In addition, the presence of ApoE4 results in increased APP-Thr668 phosphorylation (p-APP) and Tau phosphorylation (p-Tau). Some of these ApoE4-mediated events were blocked by proteasomal inhibitors, and in a small pilot study, the proteasomal inhibitor disulfiram and a CDK inhibitor were shown to be effective in reversing some of the ApoE4-mediated effects. Furthermore, we identified initial therapeutic candidates—F03 and F05—that normalize some of the ApoE4 effects.
In addition to these effects on APP processing and signaling, ApoE4 expression was associated with a marked reduction in the ratio of SirT1 to SirT2, both in cultured neural cells and in the brains of patients with AD. Because SirT1 has been implicated in neuroprotection and SirT2 in neurodegeneration (8), this effect of ApoE4 may be important from both mechanistic and therapeutic development standpoints.
Our data support the view that ApoE4 modulates the connectivity balance, as reflected in the ratios of sAPPα:sAPPβ, sAPPα:Aβ, SirT1:SirT2, APP:p-APP, and Tau:p-Tau. The network of proteins that mediates this balance thus represents a set of candidate targets for the prevention and treatment of ApoE4-associated processes such as mild cognitive impairment and AD.
Results
ApoE Isoforms Associate with APP.
Although the lipidation status of ApoE affects its structure (9), it is not yet clear whether the risk associated with ApoE4 is related solely to lipidated ApoE4, to poorly lipidated ApoE4, to unlipidated ApoE4, or to a combination of these forms of ApoE4. Therefore, by transfecting and allowing cellular lipidation to occur naturally, we can assess both the unlipidated and the lipidated forms with respect to their effects on the AD phenotype. Hence our studies involved transfecting cells with ApoE4 vs. ApoE3 expression constructs.
All of the cells that we tested expressed one or more of the receptors or specific ER chaperone proteins involved in the uptake of ApoE, such as the low-density lipoprotein (LDL) receptor (∼110 kDa), LDL receptor-like protein (LRP; ∼85 kDa), or receptor-associated protein (RAP, ∼24 kDa) (Fig. S1). Coimmunoprecipitation (Co-IP) experiments suggested that ApoE3 and E4 associate with sAPPα (secreted in media) and APP (cell extracts) from several cell lines tested. Shown in Fig. 1 is the evidence of association between APP and ApoE isoforms in two different cell lines. Although A172 human glioblastoma cells (Fig. 1 A and B) were transfected with only the ApoE isoforms, the H4 human neuroglioma cells (Fig. 1 C and D) required transfection of both APP and ApoE due to low endogenous expression of APP in this cell line. Interestingly, in both paradigms, transfection of ApoE4 resulted in more than 50% reduction in the expression of sAPPα compared with ApoE3, suggesting that the presence of ApoE4 may either have blocked sAPPα formation or promoted its degradation. Immunoprecipitation (IP) of an unrelated receptor protein (TrkA) from A172 cell lysates obtained from transient cotransfections of human TrkA expression construct together with ApoE isoforms did not reveal any association with E3 or E4, suggesting specificity to the APP–ApoE interaction (Fig. S2).
Fig. 1.

ApoE and APP interaction. (A and B) Following transfection of A172 cells with ApoE isoforms alone or H4 cells with APP and ApoE isoforms (C and D), IP was performed with an anti–CT-15-APP antibody (A or C, extract) or N-terminal anti-APP antibody (B or D, media) followed by SDS/PAGE and WB to detect sAPPα, APP, or ApoE. The last panel in A or C represents endogenous GAPDH as a loading control before the pull-down. Band densities of sAPPα (B and D) measured by densitometric quantification of film autoradiograms are expressed as a percentage of sAPPα in control untransfected cells.
ApoE4 Binds to APP at Endocytic pH with a Significantly Higher Affinity than ApoE3.
We also confirmed the interaction of ApoE and APP by surface plasmon resonance (SPR) to measure the binding of recombinant ApoE isoforms with recombinant protein fragments of the ectodomain of APP, as well as the full ectodomain of APP695 (eAPP19–624) at pHs more characteristic of intracellular compartments. Analysis of the recombinant ApoE with a calibrated Superdex S-200 size-exclusion chromatography column gave the expected molecular weight of the ApoE tetramer for both ApoE3 and ApoE4, indicating that the amounts of lipid retained were small in comparison with the mass of the protein. The E2 domain of APP and the Aβ-cognate region (trx-eAPP290–624) gave an effective KD of 80 nM for ApoE4 and 300 nM for ApoE3 (Fig. 2 A and B, Table S1, and Fig. S3). Although both ApoE and APP self-associate (10), the amount of ApoE–APP complex formed at equilibrium approximated the isotherm expected for a single-binding equilibrium (Fig. 2 C and D), consistent with previous observations regarding the association of ApoE-containing VLDL particles at pH greater than 7 (11). At pH 6.5, we found a consistent trend that ApoE4 displayed tighter binding than ApoE3 independent of whether the ApoE or APP was attached to the flow cell surface. In addition, unlike previous experiments that suggested that the E1 domain of APP751 (residues 1–207) was the major binding site for ApoE-containing particles, our results suggest that at intracellular pH, the major binding site for ApoE in the APP695 ectodomain is within the E2 domain (residues 290–575). We did not observe significant binding to a fusion protein containing the Aβ 1–28 sequence (trx-eAPP575–624) (Table S1 and Fig. S3).
Fig. 2.

Surface plasmon resonance analysis of the binding of trx-eAPP290–624 to trx-ApoE4 (A), trx-ApoE3 (B), or thioredoxin (trx) (C). The sensograms are shown in gray, and the fits are shown in red. The arrows mark the direction of increasing concentration. The effective KD (KD, eff) was calculated with a single site binding model (D). Competition between disulfiram and ApoE4 for the ectodomain of APP was demonstrated by preincubating trx-ApoE4 with varying concentrations of disulfiram and then analyzing the binding of the mixture to a flow cell treated with biotinylated MBP-eAPP19–624 (E). At saturating concentrations of disulfiram (5 µM, ∼10 times the KD of disulfiram for APP), the binding of ApoE4 is reduced by 50% (Fig. S3).
ApoE4, but Not ApoE3, Significantly Reduces sAPPα Secretion, sAPPα/Aβ, and sAPPα/sAPPβ Ratios.
To understand the cellular consequences of ApoE–APP interactions further, we assessed the levels of sAPPα, sAPPβ, or Aβ in different cell lines. Addition of ApoE4, but not ApoE3, significantly decreased sAPPα secretion and reduced sAPPα/Aβ1–42 and sAPPα/sAPPβ ratios in A172 human glioblastoma cells (Fig. 3A) and H4 human neuroglioma cells (Fig. 3B), respectively. ApoE4Δ is the carboxyl-terminal truncated form of ApoE4 that triggers neurotoxicity and cell death (2). A significant ApoE4 or ApoE4Δ isoform-dependent inhibition of sAPPα secretion and reduction in the α/β ratio was observed in other neural cell lines as well (Fig. S4).
Fig. 3.

ApoE4 but not ApoE3 significantly reduces sAPPα and lowers α/β ratio. A172 cells (A) and H4 cells (B) were transfected with ApoE isoforms alone or APP and ApoE isoforms, respectively. sAPPα and sAPPβ secreted into the medium and Aβ1–42 in the cell extracts were assayed as mentioned in SI Materials and Methods. Data (mean ± SE) are from four experiments performed in triplicate, *P < 0.05.
Differential Effects of ApoE on Sirtuin Expression in Cells and AD Postmortem Tissue.
One of the mechanisms by which ApoE4 could trigger a reduction in sAPPα levels is by inhibiting the proteolysis of APP at the α-site. Recent studies have shown that SirT1, which belongs to the Sirtuin family of NAD-dependent protein deacetylases, suppresses AD-related biochemical events in cells, primary neurons, and mouse models by directly activating transcription of ADAM10, thus increasing the levels of the neuroprotective sAPPα (8, 12, 13).
To investigate what role ApoE has on SirT gene expression, we first performed quantitative real-time PCR in A172 cells transfected with ApoE3 or ApoE4. Our results indicated a significant down-regulation of SirT1 gene expression by ApoE4 compared with ApoE3 (Fig. 4A). Although SirT2 gene expression was unaffected by the ApoE isoforms, there was a slight decrease in SirT6 expression by ApoE4 alone (Fig. 4A). The data suggested that ApoE4 has a profound transcriptional effect on SirT1 expression.
Fig. 4.

ApoE’s effects on Sirtuin expression in cells and AD postmortem tissue. Following transfection of A172 cells with ApoE isoforms, cell pellets were collected and used for RNA isolation and PCR or for SDS/PAGE and WB. (A) The real-time PCR cycling was performed as described in SI Materials and Methods. Data (δCt values expressed as percentage of untransfected control) are from three experiments performed in triplicate, *P < 0.05. (B) Cell extracts were subjected to SDS/PAGE and WB to detect SirT1, T2, and T6. Band densities are expressed as a percentage of untransfected control. Data (mean ± SE) are from three independent experiments,*P < 0.05. (C) Representative immunoblots probed for SirT1, T2, and T6 from homogenates of the temporoparietal region of control subjects and AD patients. Band densities are expressed as a percentage of normal human brains. (D and E) Overexpression of SirT1 reverses ApoE4-mediated reduction in sAPPα. Following transfection of A172 cells with ApoE4 and SirT1 (1:1 and 1:2, respectively), sAPPα secreted into the medium (D) was assayed as mentioned in SI Materials and Methods. Cell extracts were subjected to IP with an N-terminal anti-APP antibody followed by SDS/PAGE and WB to detect sAPPα (E). The last three panels represent ApoE, SirT1, and GAPDH (loading control) before the pull-down. Band densities of sAPPα are expressed as a percentage of sAPPα in control untransfected cells.
To investigate the effect of ApoE isoforms on Sirtuin protein expression, we evaluated the levels of SirT1, SirT2, and SirT6 in cells and postmortem human brains. Cell extracts isolated from A172 cells transfected with ApoE3 or E4 were subjected to SDS/PAGE and Western blotting (WB) to detect the various Sirtuins. The presence of either ApoE3 or ApoE4 resulted in a significant reduction in SirT1 levels (Fig. 4B, Middle). In contrast, although the presence of ApoE3 resulted in a commensurate reduction in SirT2 levels, the presence of ApoE4 triggered an approximate twofold increase in SirT2 expression. The levels of SirT6 remained unchanged in ApoE-transfected cells. Thus, the ratio of SirT2:SirT1 was unaffected by ApoE3, but increased markedly—∼4.5-fold—by the expression of ApoE4 (Fig. 4B).
To extend these cellular observations, we performed a similar analysis on postmortem temporoparietal regions of normal subjects and individuals with advanced AD pathology. As in the cell culture results, the SirT2:SirT1 ratio was increased, although in these advanced cases, this effect was solely due to a reduction in SirT1 expression (Fig. 4C)—whether SirT2 expression was increased earlier in the pathogenetic process could not be determined from these samples. SirT6 expression was again unaltered.
Because the ApoE4-mediated reduction in SirT1 levels may in turn result in reduced sAPPα levels, it was important to determine whether the replacement of SirT1 in the presence of ApoE4 reverses the reduction in sAPPα. Following transfection of A172 cells with ApoE4 and SirT1 (1:1 and 1:2, respectively), cell culture media were collected, and the levels of sAPPα were assessed. Overexpression of SirT1 reversed the ApoE4-mediated reduction in sAPPα secretion and restored it to normal levels (Fig. 4D). Additionally, the cell culture media was subjected to IP with the N-terminal anti-APP antibody followed by SDS/PAGE and WB to detect sAPPα. Overexpression of SirT1 reversed the ApoE4-mediated decrease in sAPPα expression (Fig. 4E).
Pharmacological Reversal of ApoE4-Mediated Reduction in sAPPα Levels.
To determine whether the ApoE4-mediated reduction in sAPPα could be reversed by therapeutic candidates, A172 cells transfected with ApoE4 were incubated with various candidates including the pan-caspase inhibitor z-VAD, proteasomal inhibitors MG132 or epoxomicin, and disulfiram, a compound that we have previously shown to interact directly with the APP extracellular domain and increase its α-secretase cleavage (10). The addition of ApoE4 significantly decreased sAPPα secretion; however, this effect was reversed by the proteasomal inhibitors MG132 (5 μM) and epoxomicin (5 μM) and also by disulfiram (20 μM) but not by the pan-caspase inhibitor z-VAD or the γ-secretase inhibitor DAPT (Fig. 5 A and B).
Fig. 5.

ApoE4-mediated reduction in sAPPα levels is reversed by various inhibitors. A172 cells were transfected with ApoE4 in the presence of epoxomicin (5 μM), MG132 (5 μM), disulfiram (20 μM), z-VAD (40 μM), or DAPT (10 μM). (A and B) Cell culture media were collected and either assayed for sAPPα (A) as described in SI Materials and Methods or subjected to IP with an N-terminal anti-APP antibody followed by SDS/PAGE and WB to detect sAPPα (B). FO3 and FO5 (C and D) reverse ApoE4-mediated reduction in sAPPα levels. Twenty-four hours after transfecting A172 cells with ApoE4, culture medium was changed, and F03 (C) or F05 (D) was added. After an additional 24 h, cell culture media were collected and assayed for sAPPα. Data (mean ± SE) are expressed in arbitrary units of sAPPα released into the media (*P < 0.05).
Using small-angle X-ray scattering (SAXS) and intrinsic fluorescence studies, we previously reported that disulfiram binds to fragments of the extracellular domain of APP (10). We extended these observations here to show that disulfiram binds to APP575–624 with a calculated KD of 0.6 µM (Fig. S3); furthermore, incubation of saturating amounts of disulfiram (5 µM) with ApoE4 decreased the binding of ApoE4 to the ectodomain of APP (APP19–624) by 50% (Fig. 2E and Fig. S3).
Furthermore, in experiments involving screening of small molecule libraries, we also identified two therapeutic candidates—FO3 (tropisetron) and FO5 [1H-Indazole-3-carboxylicacid,N-(3S)-1-azabicyclo (2.2.2) oct-3-yL]—that reversed the ApoE4-mediated effects described above. In A172 cells transfected with ApoE4, the decrease in sAPPα was completely reversed by treatment with 1 μM FO3 (Fig. 5C). Similarly, FO5 (1 μM) also reversed the ApoE4-mediated reduction in sAPPα secretion and restored it to normal levels (Fig. 5D).
ApoE4 Triggers APP-Thr668 Phosphorylation and Tau Phosphorylation in Cells and AD Postmortem Tissue.
APP phosphorylation may be another possible mechanism by which the APP and ApoE interaction lead to decreased sAPPα secretion and α/β ratio (14, 15). Furthermore, the microtubule-associated protein Tau and its phosphorylation play a crucial role in the pathogenesis of AD (16). To investigate whether ApoE triggered APP and/or Tau phosphorylation, A172 cells were transfected with ApoE3 or E4. Twenty-four hours after transfection, cell extracts were subjected to IP with anti-APP antibody followed by SDS/PAGE and WB to detect p-APP and p-Tau. Increased phosphorylation of APP at Thr668 was observed in extracts isolated from cells transfected with the ApoE4 expression construct compared with ApoE3 (Fig. 6A). The IP experiments also suggested that APP associated with Tau both in the absence and presence of ApoE (Fig. 6B). Furthermore, increased p-Tau was observed only in those samples that were transfected with ApoE isoforms. ApoE3′s effect on p-Tau was considerably less than the effect of ApoE4 (Fig. 6B, top two panels).
Fig. 6.
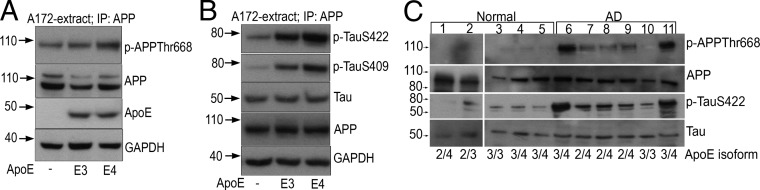
ApoE4 triggers APP-Thr668 phosphorylation and Tau phosphorylation. (A and B) Twenty-four hours after transfecting A172 cells with ApoE4, cell extracts were subjected to IP with anti-APP antibody followed by SDS/PAGE and WB to detect p-APP (A), p-TauSer422 (B, Top), p-TauSer409 (B, Middle), or Tau (B, Bottom). The last two panels in A represent ApoE and GAPDH (loading control) or APP and GAPDH (loading control) before the pull-down (B). (C) p-APP and p-Tau in AD. A total of 100 μg each of temporoparietal extracts isolated from normal and AD brains (Table S2) were examined by WB using APP, p-APPThr668, Tau, and p-TauSer422 antibodies. Isoform genotyping of ApoE was performed as described in SI Materials and Methods (Fig. S5).
The above-mentioned cellular results prompted us to assess the relationship between ApoE and phosphorylation of APP and Tau in human AD brains (Table S2). To do so, we initially performed ApoE genotype analysis on postmortem temporoparietal regions of normal subjects and individuals with advanced AD-like pathology (Fig. S5 and Table S2). With the exception of one control and one AD individual, all were heterozygotic for the three ApoE alleles. Although the expression of APP and Tau was similar in normal and AD individuals, increased phosphorylation of APP on Thr668 residue was observed only in the extracts isolated from AD brains (Fig. 6C). Similarly, immunoblotting with p-TauSer422 antibody revealed the presence of p-Tau in greater abundance in AD samples (Fig. 6C). Interestingly, increased APP and Tau phosphorylation was not observed in the AD patient who was homozygous for the ApoE3 allele.
ApoE4-triggered APP Thr668 phosphorylation and Tau phosphorylation have been reported to lead to disruption of the microtubule network, impairment of axonal transport, cellular damage, and cell death (16, 17). When A172 cells were transfected with ApoE isoforms and cell viability was measured 24 h later, cells transfected with ApoE4 were indeed found to be more susceptible to cell death compared with ApoE3 (Fig. S6).
Pharmacological Reversal of ApoE4-Mediated APP or Tau Phosphorylation.
To determine whether increased p-APP or p-Tau may be reversed pharmacologically, we tested ApoE4-mediated APP or Tau phosphorylation in the presence of specific kinase inhibitors. CHIR99021, a GSK-3β inhibitor, reversed ApoE4-mediated reduction in sAPPα secretion and restored it to normal levels (Fig. 7A) and also reversed ApoE4-mediated APP phosphorylation (Fig. 7B). Similarly, the cyclin-dependent kinase5 (CDK5) inhibitor PHA793887 also attenuated p-APP and p-Tau at all concentrations tested (Fig. 7C).
Fig. 7.
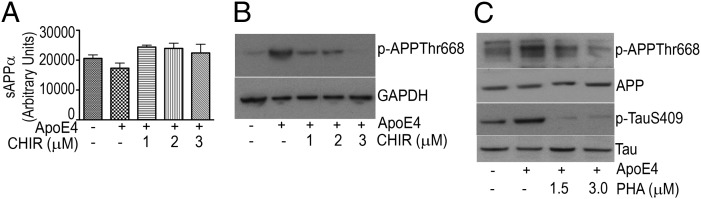
Inhibition of GSK-3β or CDK5 decreases p-APP or p-Tau. (A) Culture media from ApoE-transfected A172 cells exposed to the GSK-3β inhibitor CHIR9902 were collected and assayed for sAPPα as described in SI Materials and Methods. sAPPα values are depicted as a percentage of control. Data (mean ± SE) are from three independent experiments performed in triplicate (*P < 0.05). (B and C) Extracts were prepared from ApoE-transfected A172 cells exposed to the GSK-3β inhibitor CHIR9902 (B) or CDK5 kinase inhibitor PHA793887 (C) and subjected to SDS/PAGE and WB to detect p-APP (B) or APP, p-APP, Tau, and p-Tau (C).
Discussion
The amyloid precursor protein, APP, has been shown to function as a molecular switch: cleavage at the β, γ, and caspase sites results in the production of four pro-AD peptides—sAPPβ (from which N-APP is derived), Aβ, Jcasp, and C31—that mediate neurite retraction, synaptic reorganization, caspase activation, and ultimately programmed cell death. In contrast, cleavage at the α site produces the anti-AD trophic peptide sAPPα and the inhibitor of APP γ-site cleavage, αCTF (1, 6, 18). The decision between these two proteolytic pathways is governed at least in part by ligand binding (1, 6, 18). Therefore, it was of interest to determine whether ApoE isoforms impact this trophic–antitrophic peptide balance differentially, and, if so, by what mechanism(s).
Our data indicate that ApoE4 interacts with APP with nanomolar affinity, and whether via this interaction or others, ApoE4 decreases sAPPα secretion and reduces sAPPα/Aβ and sAPPα/sAPPβ ratios. ApoE4’s effect in reducing sAPPα varies from a modest but significant decrease to a near complete reduction in different cell lines. This may be due to varying levels of ApoE4 interactors that influence α-secretase activity and sAPPα levels (19). In our study, although the effect of E4 on Aβ varied in different cell lines, the ratio of sAPPα/Aβ decreased significantly in all cell lines that we tested, indicating that the ratio may serve as a more reliable indicator of the trophic–antitrophic peptide balance.
Our data are consistent with earlier reports demonstrating that the level of CSF sAPPα is significantly lower in AD patients possessing one or two ApoE4 alleles than in those not possessing the ApoE4 allele (7), treatment of human SH-SY5Y neuroblastoma cells with ApoE4 results in decreased secretion of sAPPα (20), and secretion of sAPPα is differentially affected by distinct ApoE isoforms as a functional consequence of the APP-ApoE interaction (21).
The mechanism by which ApoE4 decreases sAPPα secretion remains unclear. Recent studies point to SirT1, which belongs to the Sirtuin family of NAD-dependent protein deacetylases, as being a transcriptional activator of ADAM10. SirT1 suppresses AD in cells, primary neurons, and mouse models by activating transcription of ADAM10, thus increasing the levels of the neuroprotective sAPPα. In addition, SirT1 reduces production of β-amyloid (8, 12). Several studies point to opposing effects of SirT1 and SirT2: although SirT1 activation is associated with neuroprotection, SirT2 up-regulation is toxic to neuronal cells (22, 23). This led us to investigate the effect of ApoE isoforms on SirT levels. Although ApoE3 down-regulated SirT1 mRNA, there was a greater reduction in ApoE4-mediated SirT1 mRNA levels. Furthermore, the ApoE4-mediated reduction in SirT1 mRNA levels correlated strongly with a reduction in SirT1 protein levels, unlike for SirT2, whose protein expression did not correlate with the mRNA expression.
Although the effect of SirT1 in increasing sAPPα has been noted previously (8, 12), our work links ApoE4 to reduced sAPPα and a general shift of APP proteolysis away from the trophic peptides sAPPα and αCTF and toward the production of Aβ, via an effect on SirT1. The overexpression of SirT1 in the presence of ApoE4 prevented the phenotype observed with ApoE4, providing further support for our in vitro model.
Contrary to effects on SirT1 mRNA levels, neither isoform of ApoE affected SirT2 or SirT6 mRNA and protein levels (the decrease in SirT6 mRNA expression in ApoE4 transfected cells was not significant). The question arises as to how ApoE4-mediated SirT2 protein was increased posttranscriptionally. One possibility is that ApoE4 may be promoting an increased rate of SirT2 protein translation without affecting SirT2 transcription. Other explanations include the differential regulation of SirT2 mRNA expression, stability, and degradation compared with its protein expression, stability, and degradation, for example, via miRNA effects (24).
Furthermore, our results in cells, showing that ApoE4 expression is associated with a decrease in SirT1 and an increase in SirT2, support the notion that the set of alterations observed with ApoE4 expression may result at least in part from a reduction in SirT1 and an increase in SirT2. The results with the human AD brain—a decrease in SirT1—mirror our results in cells with ApoE4 and are in agreement with an earlier study (13). The lack of an associated increase in SirT2 was not observed in the AD brains, but this may have been due to the end-stage nature of the pathological samples. Our results suggest that ApoE4 causes a “Sirtuin inversion” such that SirT2 dominates SirT1, leading to a neurite-retractive and programmed cell death signal (22, 23). Reducing the SirT2:SirT1 expression ratio either by increasing the levels of SirT1 mRNA or protein or decreasing the levels of SirT2 protein may therefore serve as a feasible therapeutic approach. Because the SirT1 levels (mRNA and protein) were significantly reduced by ApoE4, another attractive therapeutic approach would be to screen for drugs that normalize the levels of SirT1 mRNA or protein. One question raised by these results is whether the evolutionary development of ApoE3 from the primordial ApoE4 in humans offered some advantage that is indeed associated with an alteration in the SirT1:SirT2 expression ratio (25).
Our studies on ApoE4-mediated APP and Tau phosphorylation are compatible with other studies that also suggest that ApoE4-mediated APP-Thr668 phosphorylation or Tau phosphorylation alters cellular APP processing, thereby affecting sAPPα or sAPPβ secretion and sAPPα/Aβ ratio (14–16). Increased p-APP and p-Tau were observed only in those samples that were transfected with ApoE4. The results with human AD brains—an increase in APP and Tau phosphorylation—mirror our results in cells. However, the increased APP and Tau phosphorylation pattern was not observed in one AD subject with an E3/E3 profile. With just n = 1, it is difficult to explain the lack of increased APP and Tau phosphorylation. If further samples support this observation, it may suggest that there may be events independent of APP or Tau phosphorylation that trigger the AD pathogenesis.
The mechanism by which ApoE4 triggers Tau phosphorylation is not clear. It has been reported that SIRT1 deficiency leads to hyperacetylation of Tau and accumulation of p-Tau and Aβ in mouse models of AD and in AD patients (26). Our results showing that ApoE4 expression is associated with a decrease in SirT1 mRNA and protein may partly explain the increase in p-Tau. Our results also demonstrated the interaction of APP with Tau and p-Tau. Thus, the trimeric complex of ApoE4-APP-Tau/p-Tau may represent one mediator of the pathological changes associated with AD (16, 17, 27).
Disulfiram, which we have reported to interact directly with APP (10), inhibited the ApoE4-mediated events. Interestingly, recent reports have shown that the previously observed antitumoral activity of disulfiram could be attributed to its potent inhibition of the ubiquitin-proteasome activity (28). Additionally, ApoE4-mediated reduction in sAPPα levels and increased p-APP and p-Tau were all reversed by the kinase inhibitors CHIR99021 and PHA793887. ApoE4 has been reported to activate GSK-3β more than other isoforms, and GSK-3β is believed to mediate phosphorylation of APP at Thr668 (20). In conjunction, CDK5 has also been proposed to be central to the phosphorylation of APP and Tau (29). Furthermore, in experiments involving the screening of chemical compound libraries, we identified two therapeutic candidates—FO3 and FO5—that prevent the ApoE4-mediated effects. Further secondary assay testing of these therapeutic candidates in neuronal cultures generated from platelet-derived growth factor B chain promoter-driven amyloid precursor protein (PDAPP) mice (huAPPSwe/Ind), together with a battery of in vivo tests in PDAPP mice, are in progress.
ApoE3′s effect on sAPPα was found to be less than the effect of ApoE4; furthermore, ApoE3 did not alter the SirT2/T1 ratio, nor did it have the same effect on p-Tau, p-APP, or cell death induction. Thus, it is clear that ApoE4 exerts isoform-specific signaling events that affect APP proteolysis and signaling, downstream kinase activity, and Sirtuin expression. These results are compatible with the notion that ApoE4-mediated signaling affects an endogenous program that mediates synaptic plasticity balance. Although the complete sequence of events initiated by ApoE4 leading finally to neurodegeneration remains to be defined, our studies demonstrate that ApoE4 triggers alternative cellular APP processing favoring the amyloidogenic route. The results also indicate that ApoE4, p-Tau, p-APP, and SirT1 all may be part of a signaling network that is affected in AD, providing a medium-throughput model for therapeutic candidate screening in AD drug discovery.
Materials and Methods
All experiments were performed in several different cell lines, some of which required transfection of ApoE isoforms alone and some that required transient transfections of APP and ApoE isoforms. Experimental details about the cell lines, cDNA constructs, transient transfection procedures, cellular extraction, immunoprecipitation, electrophoresis, western blotting and antibodies used for the experiments are provided in SI Materials and Methods. sAPPα or sAPPβ secreted into the cellular media were determined with the AlphaLISA sAPPα and sAPPβ immunoassay research kits as described in SI Materials and Methods. The Aβ1-42 or 1-40 was determined from media or cells using a sandwich ELISA kit as described in SI Materials and Methods. PCR primers, RNA isolation, real time PCR conditions, analysis of mRNA levels, postmortem human brain tissue extraction, ApoE isoform genotyping and SPR data are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Dr. Yadong Huang (Gladstone Institute of Neurological Disease, University of California, San Francisco) for providing the ApoE plasmids and for critical discussion of the results; Dr. Stephen DeArmond (Professor of Pathology, School of Medicine, University of California, San Francisco) for providing the human brains; members of the D.E.B. laboratory for helpful comments and discussions; Rowena Abulencia for administrative assistance; and Padma Rao for technical help. This work was supported in part by National Institutes of Health Grant AG034427-02 (to D.E.B.), and grants from the Joseph Drown Foundation (to D.E.B.) and the Stephen D. Bechtel, Jr. Foundation (to R.V.R.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1314145110/-/DCSupplemental.
References
- 1.Bredesen DE, John V. Next generation therapeutics for Alzheimer's disease. EMBO Mol Med. 2013;5(6):795–798. doi: 10.1002/emmm.201202307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103(15):5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris FM, et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci USA. 2003;100(19):10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ye S, et al. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: ApoE structure as a potential therapeutic target. Proc Natl Acad Sci USA. 2005;102(51):18700–18705. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verghese PB, et al. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci USA. 2013;110(19):E1807–E1816. doi: 10.1073/pnas.1220484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bredesen DE. Neurodegeneration in Alzheimer’s disease: Caspases and synaptic element interdependence. Mol Neurodegener. 2009;4:27. doi: 10.1186/1750-1326-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olsson A, et al. Measurement of alpha- and beta-secretase cleaved amyloid precursor protein in cerebrospinal fluid from Alzheimer patients. Exp Neurol. 2003;183(1):74–80. doi: 10.1016/s0014-4886(03)00027-x. [DOI] [PubMed] [Google Scholar]
- 8.Donmez G. Sirtuins as possible targets in neurodegenerative diseases. Curr Drug Targets. 2013;14(6):644–647. doi: 10.2174/1389450111314060004. [DOI] [PubMed] [Google Scholar]
- 9.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009;50(Suppl):S183–S188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Libeu CA, Descamps O, Zhang Q, John V, Bredesen DE. Altering APP proteolysis: Increasing sAPPalpha production by targeting dimerization of the APP ectodomain. PLoS ONE. 2012;7(6):e40027. doi: 10.1371/journal.pone.0040027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haas C, Cazorla P, Miguel CD, Valdivieso F, Vázquez J. Apolipoprotein E forms stable complexes with recombinant Alzheimer’s disease beta-amyloid precursor protein. Biochem J. 1997;325(Pt 1):169–175. doi: 10.1042/bj3250169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin W, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281(31):21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- 13.Julien C, et al. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68(1):48–58. doi: 10.1097/NEN.0b013e3181922348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee MS, et al. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163(1):83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu F, et al. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 2003;547(1-3):193–196. doi: 10.1016/s0014-5793(03)00714-2. [DOI] [PubMed] [Google Scholar]
- 16.Adalbert R, Gilley J, Coleman MP. Abeta, tau and ApoE4 in Alzheimer’s disease: The axonal connection. Trends Mol Med. 2007;13(4):135–142. doi: 10.1016/j.molmed.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Alonso AD, et al. Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. J Biol Chem. 2010;285(40):30851–30860. doi: 10.1074/jbc.M110.110957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bredesen DE, John V, Galvan V. Importance of the caspase cleavage site in amyloid-β protein precursor. J Alzheimers Dis. 2010;22(1):57–63. doi: 10.3233/JAD-2010-100537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Endres K, Fahrenholz F. Upregulation of the alpha-secretase ADAM10—risk or reason for hope? FEBS J. 2010;277(7):1585–1596. doi: 10.1111/j.1742-4658.2010.07566.x. [DOI] [PubMed] [Google Scholar]
- 20.Cedazo-Mínguez A, Wiehager B, Winblad B, Hüttinger M, Cowburn RF. Effects of apolipoprotein E (apoE) isoforms, beta-amyloid (Abeta) and apoE/Abeta complexes on protein kinase C-alpha (PKC-alpha) translocation and amyloid precursor protein (APP) processing in human SH-SY5Y neuroblastoma cells and fibroblasts. Neurochem Int. 2001;38(7):615–625. doi: 10.1016/s0197-0186(00)00128-5. [DOI] [PubMed] [Google Scholar]
- 21.Vincent B, Smith JD. Astrocytes down-regulate neuronal beta-amyloid precursor protein expression and modify its processing in an apolipoprotein E isoform-specific manner. Eur J Neurosci. 2001;14(2):256–266. doi: 10.1046/j.0953-816x.2001.01643.x. [DOI] [PubMed] [Google Scholar]
- 22.Korner S, et al. Differential sirtuin expression patterns in amyotrophic lateral sclerosis (ALS) postmortem tissue: Neuroprotective or neurotoxic properties of sirtuins in ALS? Neurodegener Dis. 2013;11(3):141–152. doi: 10.1159/000338048. [DOI] [PubMed] [Google Scholar]
- 23.Outeiro TF, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317(5837):516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 24.Yamakuchi M. MicroRNA Regulation of SIRT1. Front Physiol. 2012;3:68. doi: 10.3389/fphys.2012.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finch CE. Evolution in health and medicine Sackler colloquium: Evolution of the human lifespan and diseases of aging: Roles of infection, inflammation, and nutrition. Proc Natl Acad Sci USA. 2010;107(Suppl 1):1718–1724. doi: 10.1073/pnas.0909606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Min SW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67(6):953–966. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang KA, et al. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol Cell Biol. 2006;26(11):4327–4338. doi: 10.1128/MCB.02393-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cvek B, Dvorak Z. The value of proteasome inhibition in cancer. Can the old drug, disulfiram, have a bright new future as a novel proteasome inhibitor? Drug Discov Today. 2008;13(15-16):716–722. doi: 10.1016/j.drudis.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Wen Y, et al. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J Neurosci. 2008;28(10):2624–2632. doi: 10.1523/JNEUROSCI.5245-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.