

Abstract
Glaucoma is a neurodegenerative disease affecting 70 million people worldwide. For some time, analysis of human glaucoma and animal models suggested that RGC axonal injury in the optic nerve head (where RGC axons exit the eye) is an important early event in glaucomatous neurodegeneration. During the last decade advances in molecular biology and genome manipulation have allowed this hypothesis to be tested more critically, at least in animal models. Data indicate that RGC axon degeneration precedes soma death. Preventing soma death using mouse models that are mutant for BAX, a proapoptotic gene, is not sufficient to prevent the degeneration of RGC axons. This indicates that different degeneration processes occur in different compartments of the RGC during glaucoma. Furthermore, the Wallerian degeneration slow allele (Wlds) slows or prevents RGC axon degeneration in rodent models of glaucoma. These experiments and many others, now strongly support the hypothesis that axon degeneration is a critical pathological event in glaucomatous neurodegeneration. However, the events that lead from a glaucomatous insult (e.g. elevated intraocular pressure) to axon damage in glaucoma are not well defined. For developing new therapies, it will be necessary to clearly define and order the molecular events that lead from glaucomatous insults to axon degeneration.
Introduction
Glaucoma is a leading neurodegenerative cause of blindness and is reported to be the second leading cause of blindness worldwide (Quigley & Broman 2006). It is a heterogeneous group of diseases characterized by the dysfunction and death of retinal ganglion cells (RGCs). Approximately 70 million people are affected by glaucoma (Quigley 1996). Major risk factors include increasing age and intraocular pressure (IOP) elevation. However, high IOP by itself is not sufficient to cause glaucoma, as many individuals with high IOP do not develop glaucoma (Ritch et al 1996). Treatments currently center on lowering IOP levels, but do not prevent the development or progression of visual abnormalities in all patients (Gordon et al 2002; Kass et al 2002). To develop better treatments, it is necessary that we better understand pressure-dependent and pressure-independent mechanisms that contribute to RGC loss. Given the limitations of working with humans, animal models are an essential component for understanding mechanisms of glaucoma. No single animal model captures all of the nuances of human glaucoma, nor could one since glaucoma differs between patients. However, models used to study glaucoma aim to model the key event in glaucomatous neurodegeneration, early damage to axons in the optic nerve head, and this has been clearly shown for some models. In this review, we do not extensively review animal models of glaucoma as this has been done recently elsewhere (Howell et al 2008; Lasker/IRRF 2010; McKinnon et al 2009; Morrison et al 2005; Pang & Clark 2007; Ruiz-Ederra et al 2005). Instead, we focus on mechanisms contributing to RGC axon degeneration and discuss the early changes that occur in axons prior to axon degeneration. We highlight studies that have directly manipulated biological pathways to test the importance of different pathways/processes that are involved. More extensive discussion on axon degeneration pathways is provided in other reviews in this issue and elsewhere, and many of these pathways need to be tested in glaucoma (reviewed in (Nickells et al submitted)).
Compartmentalized axon degeneration
Neurons have distinct functional compartments and can execute specific self-destruct pathways that are spatially compartmentalized (Whitmore et al 2005). In glaucoma, work using DBA/2J mice (a widely used mouse model of glaucoma) showed that the mechanisms by which RGC somata die differ from those involved in the degeneration of their axons. Genetic ablation of BAX, a proapoptotic molecule, prevented the death of essentially all RGC somata in DBA/2J mice but the axons of these mice still degenerated (Libby et al 2005b). Therefore, for glaucoma, somal death is a BAX-dependant apoptotic process, whereas BAX is not necessary for axonal degeneration. Interestingly, axon degeneration may not be completely independent of BAX as BAX-deficiency did slow axon degeneration in DBA/2J mice (Libby et al 2005b). This possibility is supported by recent work showing that BAX participates in the axon degeneration pathway, independently of somal degeneration (Nikolaev et al 2009).
Further evidence of an axonal injury in glaucoma came from testing the effects of the Wallerian degeneration slow (Wlds) allele in two different animal models of glaucoma (Figure 1) (Beirowski et al 2008; Howell et al 2007). The Wlds gene creates a fusion protein containing 70 N-terminal amino acids of ubiquitination factor Ube4b linked to full-length nicotinamide mononucleotide adenylyltranserase 1 (Nmnat1) (Mack et al 2001). This chimeric protein protects axons from degeneration induced by axonal trauma (Lunn et al 1989; Mack et al 2001; Perry et al 1991; Perry et al 1990; Ribchester et al 1995). Wlds is proposed to directly protect axons but not somata (Adalbert et al 2005; Deckwerth & Johnson 1994; Glass et al 1993; Ikegami & Koike 2003), and so the allele can be used to test the importance of axon degeneration in disease. In DBA/2J mice, the Wlds allele protects from axon degeneration (Howell et al 2007). Wlds more than doubled the number of eyes with no detectable glaucoma compared to standard DBA/2J mice and preserved RGC function (as determined by the pattern electroretinogram). Wlds had a strong protective effect on the survival of optic nerve axons for at least a few months following IOP elevation. In addition, RGC somata survived in DBA/2J.Wlds eyes whose axons were spared. Although the somata were spared, they were not completely protected as on average somal diameter had shrunk by ~10%. RGC somata were largely absent in DBA/2J.Wlds eyes with severe axon loss, indicating that Wlds cannot protect the somata from death if the axon degenerates.
Figure 1. Wallerian degeneration slow (Wlds) allele protects or delays axon degeneration in two models of glaucoma.

(A–F) In DBA/2J mice, the Wlds allele protected both RGC axons (A–C) and soma (D–F) at two key time points (Libby et al 2005a). A and B show representative images of optic nerve cross sections stained with paraphenylene diamine from a wild type DBA/2J eye with severe glaucoma (A) and a DBA/2J.Wlds eye with no glaucoma (B). (C) Assessment of over 100 eyes showed that Wlds more than doubled the number of eyes with no glaucoma (green, hashed bars) compared to wild type DBA/2J mice. (D–F) Wlds also protected RGC somata. In eyes with no optic nerve damage, RGC somata were also preserved. (G–O) In a model where IOP elevation was induced experimentally in rats, Wlds delayed axon degeneration (G–H) but had no effect on somal survival (J–O). (G–H) At 2 weeks following IOP elevation, proximal axons form Wlds rats (H) had significantly less axon damage compared to wild type rats (G). Interestingly, in the wild type rats, greater axon damage was observed in proximal axons (I, squares) compared to distal axons (I, triangles) two weeks after IOP elevation. This data suggests dying back is not the major mechanism of axon degeneration in this model. (J–O) Wlds does not protect the soma. Higher magnification confocal stacks showing loss of dendritic arborization and cell body shrinkage in glaucomatous RGCs from both wild-type (K, M) and Wlds rats (L, N) as compared to control RGCs from untreated retinas (J). The majority of control RGCs showed more than one immunopositive process (arrow in J depicts example RGC with six processes) while, in particular, 4 weeks of glaucoma triggered massive loss of arborization that resulted in RGCs with only one (K and L, arrows) or no processes (M and N, arrows).
A–F reproduced from Howell et. al., 2007 (J. Cell Biology). G–O reproduced from Beirowski et. al., 2008 (Eur. J. Neuro.). Permission requested.
In the second study, the ability of the Wlds protein to protect from RGC loss was assessed in an inducible rat model of glaucoma (Beirowski et al 2008). Transgenic rats were generated carrying the Wlds gene driven by the β-actin promoter. (This is in contrast to the mouse version of Wlds where the Ube4b promoter controls expression of the Wlds protein.) IOP was artificially elevated by translimbal laser photocoagulation of the trabecular meshwork. Due to the different promoter use between mouse and rat, the authors first confirmed that the transgenic rat RGCs expressed the Wlds protein and that RGC axons underwent delayed axon degeneration following optic nerve transection. They then tested whether Wlds affects RGC axon degeneration and somal loss in rats with high IOP. Survival of RGC axons and cell bodies was assessed two and four weeks after induction of ocular hypertension. In wild-type rats, substantial loss of axons was observed and correlated well with cumulative IOP exposure. The Wlds gene delayed axonal degeneration, with a duration similar to that seen in optic nerve transection (approximately 2 weeks). In contrast to the study in DBA2J, Wlds had little or no effect on somal death.
The two studies report two clear differences. First, axonal protection by Wlds was longer lasting in DBA/2J mice than in the rats. IOP elevation is detected in some DBA/2J eyes by 6 months and in many eyes by 9 months of age (Libby et al 2005a). Wlds protected from optic nerve damage to at least 12 months of age, indicating that eyes were protected for at least a few months. This compares to a two-week protection that was observed in the rat study. Second, Wlds genotype saved somata (when axons were spared) in DBA/2J mice but had little or no effect on somal survival in the rat study. These contrasting results are likely due to the different glaucoma models used. DBA/2J is an inherited, chronic model of glaucoma where IOP elevation and subsequent RGC loss occurs progressively over time. In contrast, the rat model is an inducible, acute model where IOP levels are artificially increased and RGC loss occurs over a shorter window. However, species, genetic background and/or Wlds expression differences could also explain the contrasting results between the two models.
Irrespective of the differences, both studies indicate that protecting RGCs from axonal degeneration should be explored further as a treatment for human glaucoma (possibly as part of a combinatorial treatment regimen). The mechanisms by which Wlds protects neurons are not fully understood (Araki et al 2004; Laser et al 2006) and are discussed in more detail elsewhere in this special issue. However, these studies suggest that neuroprotective strategies that involve NAD biosynthesis or additional functions of Nmnat, possibly in combination with BAX inhibitors, should be tested for glaucoma.
Mechanisms of distal axon degeneration in glaucoma
Both dying back and Wallerian degeneration have been proposed as mechanisms of distal axon degeneration in glaucoma. Dying back is the process that involves the slow and progressive (weeks or months) degeneration of a stressed neuron from its terminals and distal axon towards the cell body (Whitmore et al 2005). Wallerian degeneration occurs in response to severe focal damage such as transection (Whitmore et al 2005). The distal axon, separated from the cell body by the lesion, rapidly disassembles (days) in a characteristic way with the accumulation of dense bodies and neuroaxonal spheroids. The exact molecular processes are not clear but Wallerian degeneration and dying back likely share similar mechanisms, as the Wlds allele slows both forms of degeneration. Some studies support a role for dying back as the major mechanism of distal axon degeneration in glaucoma (Figure 2). Disruption of axonal transport, as observed in many glaucoma models, may limit transport and communication between distal axons and cell bodies leading to the degeneration of the axons by dying back. In DBA/2J mice, axon degeneration is evident at distal locations of the optic pathway and coincides with a lack of axonal transport at the level of the superior colliculus (Figure 2 and (Crish et al 2010; Schlamp et al 2006).
Figure 2. Dying back as a mechanism for axon degeneration in glaucoma.
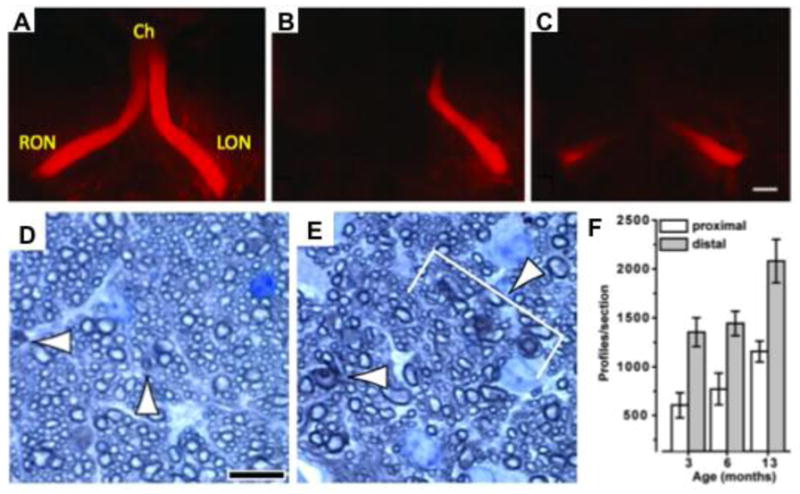
(A–C) Schlamp et al used the diI labeling technique in DBA/2J mice to show that axon degeneration followed a die-back pattern from the distal end of the optic nerve to the proximal end. The images shown are dorsal views of a healthy young mouse (A), and two old mice with degenerating nerves that typically exhibit reduced label distally (in relation to cell body) (B, C). Ch = Chiasm, RON = right optic nerve, LON = left optic nerve. (D–F) Crish et al stained optic nerve cross sections stained with Toluidine blue to identify and quantify the number of degenerating axons (or profiles, arrowed). Sections were assessed from the proximal (D) and the distal (E) portions of the optic nerve. (F) A greater number of degenerating profiles were observed in distal optic nerve sections compared to proximal optic nerve sections. This supports dying back as the major mechanism of axon degeneration in DBA/2J glaucoma.
A–C reproduced from Schlamp et. al., 2006 (BMC Neuro). D–F reproduced from Crish et. al., 2010 (PNAS).
However, data also support a more Wallerian degeneration-like mechanism for distal axon degeneration in glaucoma. In DBA/2J mice, swollen axon regions with disorganization of axonal contents and accumulation of organelles and neurofilaments, reminiscent of the neuroaxonal swellings of Wallerian degeneration, are observed from the optic nerve head all the way back to the superior colliculus (Howell et al 2007). However, in the same eyes, milder and highly focal swelling of individual axons was the far more common form of damage and was restricted to axon segments in the ONH. We speculate that these focally swollen and damaged axon segments represent regions of slowed or disrupted axonal transport, which may lead to the degeneration of the damaged axon by dying back. The presence of two types of damage in DBA/2J mice suggests that distal degeneration of axons occurs by both Wallerian degeneration and dying back, depending on the severity of damage to individual axons. Both of these degeneration processes are likely to occur in human glaucoma. It is possible that the relative contribution of each process to axon degeneration varies between different glaucoma patients (e.g. a greater contribution of Wallerian degeneration in severe angle-closure glaucoma patients) and even between different axons in the same eye.
Axon degeneration is caused by an insult in the ONH
Early studies in humans and primates suggested that the ONH was an important site in glaucoma (Anderson & Hendrickson 1974; Quigley & Addicks 1980a; b; 1981; Quigley et al 1981; Quigley & Anderson 1977; Quigley et al 1979; Quigley et al 1983). RGC axons exit the eye through a specialized region of the ONH known either as the lamina cribrosa (LC) or the glial lamina. In humans and species with larger optic nerves the LC is comprised of plates of extracellular matrix (ECM) that provide support for the bundles of axons as they pass through the posterior wall of the eye. The ECM plates are covered by astrocytes that provide support to the neurons (Anderson DR & HA 1992). The laminar region in the rodent ONH has been termed the glial lamina (Howell et al 2007). It has striking similarities to the LC in primates, primarily because of the presence of a network of astrocytes but the ECM plates are absent (Howell et al 2007; May & Lütjen-Drecoll 2002; Schlamp et al 2006). Despite these differences, the earliest RGC axon damage appears to occur in the laminar region in species with either an LC or a glial lamina. Also, the characteristic pattern of RGC dysfunction/loss observed in both human glaucoma (Shields 1992) and animal models (Howell et al 2007; Jakobs et al 2005; Schlamp et al 2006) is best explained by crucial damage to axon bundles in the laminar region (reviewed in (Nickells et al submitted)).
Various studies have shown that the earliest damage to axons is observed in the ONH in glaucoma (Anderson & Hendrickson 1974; Anderson & Hendrickson 1977; Howell et al 2007; Quigley & Anderson 1976; Quigley & Addicks 1980c; Quigley et al 1981; Quigley et al 1983; Schlamp et al 2006). However, demonstrating that the first signs of axon damage occur within the lamina is not proof that axons are insulted within the lamina. In the general case, it is well established that the first site of neuronal degeneration may be remote from the site of insult (reviewed in (Conforti et al 2007)). For example, in transected motor axons, neuromuscular junctions that are many centimeters from the lesion degenerate first. The axons immediately adjacent to the lesion remain intact for two to three times longer than the distant terminals (Beirowski et al 2005). To test if an axon injury occurs in the glial lamina in glaucoma, a study was performed that was based on the well-established pattern of axon degeneration and survival that occurs in response to axon injury in the peripheral nervous system (reviewed in (Whitmore et al 2005)). Direct and focal axon injury in the peripheral nervous system results in degeneration of the distal portion of the axon that is isolated from the cell body by the lesion. However the proximal portion of the axon attached to the surviving cell body can survive up to the proximity of the axon insult. It was reasoned that in BAX-deficient DBA/2J mice, because all the RGC somata survive after insult, the proximal axon should survive up to the site of injury. This reasoning was first tested and confirmed using optic nerve crush in BAX-deficient DBA/2J mice. Following crush, the proximal axon segment survived up to the crush site for at least 30 days. Next, survival of the proximal axon was used to locate a site of insult in glaucoma. Importantly, in eyes of DBA/2J mice with severe glaucomatous damage (e.g. distal axons degenerating), RGC axons survived from the RGC bodies to the anterior edge of the glial lamina. No axons survived to the middle or posterior of the lamina. This provided experimental evidence for a major axon insult occurring within the lamina of the optic nerve or very close to its anterior edge.
Early axonal changes that occur prior to axon degeneration
The processes that damage RGC axons in the ONH are not clear. However, observations from several different animal models of glaucoma show that disruption of axonal transport at the ONH is consistently seen early in the disease process. In monkeys with experimental glaucoma, anterograde and retrograde transport is affected in the ONH (Anderson & Hendrickson 1974; Dandona et al 1991; Radius & Anderson 1981). Electron microscopy studies showed that mitochondria and dense bodies accumulate anterior and posterior to the lamina cribrosa after IOP elevation (Gaasterland et al 1978) (Quigley & Anderson 1976). This accumulation of organelles is observed as early as one hour after induction of high IOP (Quigley & Anderson 1976). Under conditions of transient IOP elevation, this axonal blockade is reversed when IOP is normalized (Levy 1974; Minckler et al 1977; Quigley & Anderson 1976). Similarly, acute elevation of IOP in the rat causes disruption of axonal transport and cytoskeletal changes that presumably sustain axonal transport blockade (Chidlow et al 2011; Pease et al 2000; Quigley et al 2000; Salinas-Navarro et al 2010). Finally, in DBA/2J mice, anterograde axonal transport is disrupted from the retina to the superior colliculus and in regional patterns that are similar to the regional degeneration of axons in glaucoma (Crish et al 2010). Compromised axonal transport likely results in a reduction in the flow of neurotrophic factors in RGCs (reviewed in (Baltmr et al 2010)). It has not yet been shown that a loss of neurotrophic factors directly leads to axon degeneration. However, delivery of neurotrophic factors improves RGC survival in animal models of glaucoma (Martin et al 2003; Pease et al 2009) and may be a therapeutic target for human glaucoma (Baltmr et al 2010; Saragovi et al 2009). Overall though, many early changes occur to RGC axons at the optic nerve head that could trigger axon degeneration, and all these early changes may be considered as avenues for neuroprotective treatments.
While it is clear that damage to axons in the ONH is an important early event in glaucoma, the molecular signaling pathways that trigger axon degeneration are not well defined. Elevation of intra-axonal calcium levels, and its downstream affects on mitochondrial dysfunction, are often proposed to be important in axonal self destruction in glaucoma (reviewed in (Kong et al 2009; Whitmore et al 2005)). Increased release of calcium from the ER causes the uptake and accumulation of calcium in the mitochondrial matrix. This accumulation of calcium promotes the opening of the permeability transition pore (mPTP) that results in the swelling, membrane rupture, and depolarization of mitochondria leading to an energetic failure (Whitmore et al 2005). Recent data have demonstrated that opening of the mPTP induces axonal degeneration, and that inhibition of this complex protects axons (Barrientos et al 2011). Opening of the mPTP, mitochondrial swelling and mitochondrial dysfunction were all downstream of important axonal changes that follow injury, such as intra-axonal calcium elevation and activation of signaling pathways that are known to be important in axonal degeneration (Barrientos et al 2011).
Recently, the importance of axonal mitochondria in RGC death in glaucoma has gained traction (Osborne 2008; 2010). The role of mitochondrial dysfunction in axonal degeneration is highly relevant in glaucoma, since higher concentrations of mitochondria are found in the prelaminar and laminar regions of RGC axons (Barron et al 2004; Minckler et al 1977; Morgan 2004). Axons in the prelamina and lamina regions are unmyelinated resulting in higher energy demands and the need for more mitochondria. Consequently, disturbances in mitochondrial function can lead to metabolic stress and dysfunction of RGC axons with possible degeneration (Yu-Wai-Man et al 2011). In fact, disruption of axonal transport at the lamina region in glaucoma could be indicative of mitochondrial dysfunction and energy failure (Anderson 1999; Anderson & Hendrickson 1974). Changes in mitochondrial dynamics/morphology in mice (Abe & Cavalli 2008), and mitochondrial dysfunction in glaucoma patients (Abu-Amero et al 2006) have been reported. In addition to lower energy production, mitochondrial abnormalities could result in increased oxidative stress. Mice with a mutation that impairs mitochondrial polymerase proofreading function, accumulate mitochondrial mutations, have accelerated, age-related loss of retinal function, and their RGCs are more vulnerable to high IOP (Kong et al 2011). Thus, increases in mitochondrial dysfunction with increasing age, as well as loss of function, are likely to be important components of the age-related increase in glaucoma risk.
Regardless of the cause of axonal injury in glaucoma, an active molecular process is likely to occur both proximally (axon injury signaling to cell body) and distally (axon degeneration) to the site of injury. To optimally prevent RGC death in glaucoma, it will be important to understand the signaling pathways that control these distinct molecular processes. c-Jun N terminal kinase (JNK) signaling could potentially be a key mediator of both processes. JNK signaling is known to contribute to both axon injury signaling and axon degeneration in other systems (Abe & Cavalli 2008; Cavalli et al 2005; Lindwall & Kanje 2005; Miller et al 2009; Sengupta Ghosh et al 2011). JNK signaling appears to be involved in RGC death in various animal models of glaucoma (Kwong & Caprioli 2006; Tezel et al 2004; Yang et al 2008) and was shown to be active in human glaucoma (Tezel et al 2004). In a recent study, we found that activated JNK was present at the site of injury soon after mechanical injury to RGC axons (Libby, unpublished observations). Genetic ablation of JNK 2 and 3 protected the RGC somas from degeneration following the axonal injury, supporting the hypothesis that a JNK pathway signals axonal injury to the soma (Libby, unpublished observations). In contrast, a recent study proposed that targeting JNK3 alone did not prevent RGC loss or axon degeneration in an inducible model of glaucoma (Quigley et al 2011). This may agree with our study where JNK2 disruption alone was not protective, and/or may reflect difficulties of identifying a protection given the little damage that occurred in the inducible model. Further experiments are needed to fully determine the effectiveness of inhibiting JNK signaling as a potential treatment for glaucoma.
The role of glial cells in axon injury and degeneration
Glaucoma is very complex and axon degeneration may be initiated, modulated or propagated by different cell types including glia. Axon injury not only has a profound effect on the neuron, but also triggers changes in glial cell types such as astrocytes, microglia and Muller glia (reviewed in (Bringmann et al 2006; Perry et al 2010; Sofroniew 2009)). In addition to responding to injury, it is possible that glial cells may contribute to the initiation or propagation of axon injury. This is true for retinal and optic nerve glia, which may independently affect RGC health. These other cell types may also mediate protective responses. The protective and damaging responses of different cell types will need to be distinguished when prioritizing potential neuroprotective therapies for glaucoma.
Astrocytes are a major component of the neurovascular unit providing nutrients and support for neurons (Kimelberg & Nedergaard 2010). Microglia are the resident macrophages of the central nervous system sensing and responding to changes in the environment (Perry et al 2010). For glaucoma, a number of studies have used gene expression profiling to identify glial responses using a variety of animal models and human tissue (e.g. (Howell et al 2011b; Johnson et al 2011; Johnson et al 2007; Kompass et al 2008; Nikolskaya et al 2009; Panagis et al 2009; Steele et al 2006; Yang et al 2007). Two profiling studies propose that glial changes occur at the ONH prior to any significant RGC axon damage. In DBA/2J mice, upregulation of extracellular matrix (ECM) proteins by astrocytes and activation of both the endothelin and complement systems by non-neuronal cells occurred very early in the ONH and retina (Howell et al 2011a). In a rat model of glaucoma, astrocyte proliferation was reported to be a very early process (Johnson et al 2011).
For glaucoma, the changes described in glia are likely to consist of both protective and damaging responses (Johnson & Morrison 2009; Morgan 2000). In addition, these changes may be primary (in response to intraocular pressure changes) and/or secondary (in response to early RGC stress and/or damage). Early changes in extracellular matrix proteins may be a direct result of increased mechanical stress and strain due to IOP elevation. These changes occur in primates and rodents and may reflect an astrocyte-mediated response to protect axons from strain-induced injury (Burgoyne 2010; Del Zoppo et al 2006; Nickells et al submitted; Roberts et al 2009). Recently, fortified astrocytes (with dense, cytoskeletal filaments) in the ONH have been described in a rat model of glaucoma. In response to pressure, dorsal processes of the fortified astrocytes are torn away from the sheath and it is proposed that the damage to axons is not mechanical but as a consequence of regional loss of metabolic support from the astrocytes (Dai et al 2011).
Astrocytes in the ONH are phagocytic (Nguyen et al 2011), a process that may be increased as a result of injured or stressed axons expressing ‘eat me’ signals (Grimsley & Ravichandran 2003). Phagocytosis of cellular debris can be beneficial (to minimize inflammatory damage), but if the astrocytes directly phagocytose parts of the RGC axon in glaucoma it may turn out to be detrimental.
Non-neuronal cells have also been shown to express potentially damaging molecules including complement components and endothelins (Chauhan 2008; Rosen & Stevens 2010). The complement cascade has important roles in innate immunity and in the pruning of neuronal synapses. Microglia synthesize and secrete key molecules of the complement cascade and may phagocyotse synapses (reviewed in (Perry et al 2010; Rosen & Stevens 2010)). Increased complement expression is reported in the optic nerve and retina for various glaucoma models and human glaucoma (reviewed in (Nickells et al submitted)). DBA/2J mice mutant for complement component C1qa mice have a significant reduction in RGC loss and axon degeneration compared to wild-type DBA/2J mice (Howell et al 2011a). Endothelins are reported to be expressed in multiple cell types in glaucoma including microglia (Howell et al 2011a) and astrocytes (Murphy et al 2010; Prasanna et al 2005). Endothelins can cause vasoconstriction, glial cell activation, inhibition of axonal transport, and induce RGC death (Howell et al 2011a; Sasaoka et al 2006; Taniguchi et al 2006). Inhibition of the endothelin system by administration of the endothelin receptor antagonist Bosentan reduced RGC loss in DBA/2J mice (Howell et al 2011a).
Conclusion
It is clear that an early site of injury to RGCs in glaucoma is the axon segment in the ONH, but fundamental questions remain. While it is now understood that many different cell types respond in glaucoma, it is unclear if the earliest pathological events are intrinsic or extrinsic to RGC axons. Furthermore, it is not known which pathways control distal axon degeneration and proximal axon signaling after injury. Although early changes are known to occur in glial cells, the contribution of these early changes to glaucoma needs further investigation. Some of these changes will be protective while others will be damaging. Targeting early, damaging events has the potential to provide robust neuroprotection for glaucoma patients and perhaps for patients suffering from other axonopathies.
Acknowledgments
The authors thank Robert Nickells for helpful discussions, and K. Saidas Nair, Krish Kizhatil and Mimi de Vries for critical comments. This work was supported by EY011721 (SWMJ), EY018606 (RTL), The Glaucoma Foundation (RTL, GRH), American Health Assistance Foundation (GRH), Glaucoma Research Foundations (GRH), and Research to Prevent Blindness unrestricted grant to the Department of Ophthalmology at the University of Rochester. SWMJ is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe N, Cavalli V. Nerve injury signaling. Curr Opin Neurobiol. 2008;18:276–83. doi: 10.1016/j.conb.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Amero KK, Morales J, Bosley TM. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2006;47:2533–41. doi: 10.1167/iovs.05-1639. [DOI] [PubMed] [Google Scholar]
- Adalbert R, Gillingwater TH, Haley JE, Bridge K, Beirowski B, et al. A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur J Neurosci. 2005;21:271–7. doi: 10.1111/j.1460-9568.2004.03833.x. [DOI] [PubMed] [Google Scholar]
- Anderson DR. Introductory comments on blood flow autoregulation in the optic nerve head and vascular risk factors in glaucoma. Surv Ophthalmol. 1999;43(Suppl 1):S5–9. doi: 10.1016/s0039-6257(99)00046-6. [DOI] [PubMed] [Google Scholar]
- Anderson DR, HAQ . The Optic nerve. In: WMHJ, editor. Adler’s Physiology of the Eye. St Louis: Mosby Year Book; 1992. pp. 616–40. [Google Scholar]
- Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest Ophthalmol. 1974;13:771–83. [PubMed] [Google Scholar]
- Anderson DR, Hendrickson AE. Failure of increased intracranial pressure to affect rapid axonal transport at the optic nerve head. Invest Ophthalmol Vis Sci. 1977;16:423–6. [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–3. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Baltmr A, Duggan J, Nizari S, Salt TE, Cordeiro MF. Neuroprotection in glaucoma - Is there a future role? Exp Eye Res. 2010;91:554–66. doi: 10.1016/j.exer.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Barrientos SA, Martinez NW, Yoo S, Jara JS, Zamorano S, et al. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci. 2011;31:966–78. doi: 10.1523/JNEUROSCI.4065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron MJ, Griffiths P, Turnbull DM, Bates D, Nichols P. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br J Ophthalmol. 2004;88:286–90. doi: 10.1136/bjo.2003.027664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Adalbert R, Wagner D, Grumme DS, Addicks K, et al. The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci. 2005;6:6. doi: 10.1186/1471-2202-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Coleman MP, Martin KR. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur J Neuro. 2008;28:1166–79. doi: 10.1111/j.1460-9568.2008.06426.x. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, et al. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Burgoyne CF. A biomechanical paradigm for axonal insult within the optic nerve head in aging and glaucoma. Exp Eye Res. 2010 doi: 10.1016/j.exer.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli V, Kujala P, Klumperman J, Goldstein LS. Sunday Driver links axonal transport to damage signaling. J Cell Biol. 2005;168:775–87. doi: 10.1083/jcb.200410136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan BC. Endothelin and its potential role in glaucoma. Can J Ophthalmol. 2008;43:356–60. doi: 10.3129/i08-060. [DOI] [PubMed] [Google Scholar]
- Chidlow G, Ebneter A, Wood JP, Casson RJ. The optic nerve heas is the site of axonal transport disruption, axonal cutoskeleton damage and putative axonal regeneration failure in a rat model of glaucoma. Acta Neuropathol. 2011;121:737–51. doi: 10.1007/s00401-011-0807-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Adalbert R, Coleman MP. Neuronal death: where does the end begin? Trends Neurosci. 2007 doi: 10.1016/j.tins.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci U S A. 2010;107:5196–201. doi: 10.1073/pnas.0913141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Khaw PT, Yin ZQ, Li D, Raisman G, Li Y. Structural basis of glaucoma: The fortified astrocytes of the optic nerve head are the target of raised intraocular pressure. Glia. 2011 doi: 10.1002/glia.21242. [DOI] [PubMed] [Google Scholar]
- Dandona L, Hendrickson A, Quigley HA. Selective effects of experimental glaucoma on axonal transport by retinal ganglion cells to the dorsal lateral geniculate nucleus. Invest Ophthalmol Vis Sci. 1991;32:484–91. [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM., Jr Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis) Dev Biol. 1994;165:63–72. doi: 10.1006/dbio.1994.1234. [DOI] [PubMed] [Google Scholar]
- Del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Koziol JA. Vascular matrix adhesion and the blood-brain barrier. Biochem Soc Trans. 2006;34:1261–6. doi: 10.1042/BST0341261. [DOI] [PubMed] [Google Scholar]
- Gaasterland D, Tanishima T, Kuwabara T. Axoplasmic flow during chronic experimental glaucoma I. Light and electron microscopic studies of the monkey optic nerve head during development of glaucomatous cupping. Invest Ophthalmol Vis Sci. 1978;17:838–46. [PubMed] [Google Scholar]
- Glass JD, Brushart TM, George EB, Griffin JW. Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon. J Neurocytol. 1993;22:311–21. doi: 10.1007/BF01195555. [DOI] [PubMed] [Google Scholar]
- Gordon MO, Beiser JA, Brandt JD, Heuer DK, Higginbotham EJ, et al. The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120:714–20. doi: 10.1001/archopht.120.6.714. discussion 829–30. [DOI] [PubMed] [Google Scholar]
- Grimsley C, Ravichandran KS. Cues for apoptotic cell engulfment: eat-me, don’t eat-me and come-get-me signals. Trends Cell Biol. 2003;13:648–56. doi: 10.1016/j.tcb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–37. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Libby RT, John SW. Mouse genetic models: an ideal system for understanding glaucomatous neurodegeneration and neuroprotection. Prog Brain Res. 2008;173:303–21. doi: 10.1016/S0079-6123(08)01122-9. [DOI] [PubMed] [Google Scholar]
- Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest. 2011a;121:1429–44. doi: 10.1172/JCI44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Walton DO, King BL, Libby RT, John SWM. Datgan, a reusable software system for facile interrogation and visualization of complex transcription profiling data. BMC Genomics. 2011b;12 doi: 10.1186/1471-2164-12-429. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami K, Koike T. Non-apoptotic neurite degeneration in apoptotic neuronal death: pivotal role of mitochondrial function in neurites. Neuroscience. 2003;122:617–26. doi: 10.1016/j.neuroscience.2003.08.057. [DOI] [PubMed] [Google Scholar]
- Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol. 2005;171:313–25. doi: 10.1083/jcb.200506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelsma TN, Aguayo AJ. Trophic factors. Curr Opin Neurobiol. 1994;4:717–25. doi: 10.1016/0959-4388(94)90015-9. [DOI] [PubMed] [Google Scholar]
- Johnson EC, Doser TA, Cepurna WA, Dyck JA, Jia L, et al. Cell proliferation and interleukin-6-type cytokine signaling are implicated by gene expression responses in early optic nerve head injury in rat glaucoma. Invest Ophthalmol Vis Sci. 2011;52:504–18. doi: 10.1167/iovs.10-5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EC, Jia L, Cepurna WA, Doser TA, Morrison JC. Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2007;48:3161–77. doi: 10.1167/iovs.06-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EC, Morrison JC. Friend or foe? Resolving the impact of glial responses in glaucoma. J Glaucoma. 2009;18:341–53. doi: 10.1097/IJG.0b013e31818c6ef6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass MA, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, et al. The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120:701–13. doi: 10.1001/archopht.120.6.701. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Nedergaard M. Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics. 2010;7:338–53. doi: 10.1016/j.nurt.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kompass KS, Agapova OA, Li W, Kaufman PL, Rasmussen CA, Hernandez MR. Bioinformatic and statistical analysis of the optic nerve head in a primate model of ocular hypertension. BMC Neurosci. 2008;9:93. doi: 10.1186/1471-2202-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong GY, Van Bergen NJ, Trounce IA, Crowston JG. Mitochondrial dysfunction and glaucoma. J Glaucoma. 2009;18:93–100. doi: 10.1097/IJG.0b013e318181284f. [DOI] [PubMed] [Google Scholar]
- Kong YX, Van Bergen N, Trounce IA, Bui BV, Chrysostomou V, et al. Increase in mitochondrial DNA mutations impairs retinal function and renders the retina vulnerable to injury. Aging Cell. 2011;10:572–83. doi: 10.1111/j.1474-9726.2011.00690.x. [DOI] [PubMed] [Google Scholar]
- Kwong JM, Caprioli J. Expression of phosphorylated c-Jun N-terminal protein kinase (JNK) in experimental glaucoma in rats. Exp Eye Res. 2006;82:576–82. doi: 10.1016/j.exer.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Laser H, Conforti L, Morreale G, Mack TG, Heyer M, et al. The slow Wallerian degeneration protein, WldS, binds directly to VCP/p97 and partially redistributes it within the nucleus. Mol Biol Cell. 2006;17:1075–84. doi: 10.1091/mbc.E05-04-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker/IRRF. Astrocytes and Glaucomatous Neurodegeneration. 2010. [DOI] [PubMed] [Google Scholar]
- Levy NS. The effects of elevated intraocular pressure on slow axonal protein flow. Invest Ophthalmol Vis Sci. 1974;13:691–40. [PubMed] [Google Scholar]
- Libby RT, Anderson MG, Pang IH, Robinson ZH, Savinova OV, et al. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005a;22:637–48. doi: 10.1017/S0952523805225130. [DOI] [PubMed] [Google Scholar]
- Libby RT, Li Y, Savinova OV, Barter J, Smith RS, et al. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005b;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindwall C, Kanje M. Retrograde axonal transport of JNK signaling molecules influence injury induced nuclear changes in p-c-Jun and ATF3 in adult rat sensory neurons. Mol Cell Neurosci. 2005;29:269–82. doi: 10.1016/j.mcn.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Martin KR, Quigley HA, Zack DJ, Levkovitch-Verbin H, Kielczewski J, et al. Gene therapy with brain-derived neurotrophic factor as a protection: retinal ganglion cells in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2003;44:4357–65. doi: 10.1167/iovs.02-1332. [DOI] [PubMed] [Google Scholar]
- May CA, Lütjen-Drecoll E. Morphology of the murine optic nerve. Invest Ophthalmol Vis Sci. 2002;43:2206–12. [PubMed] [Google Scholar]
- McKinnon SJ, Schlamp CL, Nickells RW. Mouse models of retinal ganglion cell death and glaucoma. Exp Eye Res. 2009;88:816–24. doi: 10.1016/j.exer.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–9. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minckler DS, Bunt AH, Johanson GW. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Invest Ophthalmol Vis Sci. 1977;16:426–41. [PubMed] [Google Scholar]
- Morgan JE. Optic nerve head structure in glaucoma: astrocytes as mediators of axonal damage. Eye. 2000;14 ( Pt 3B):437–44. doi: 10.1038/eye.2000.128. [DOI] [PubMed] [Google Scholar]
- Morgan JE. Circulation and axonal transport in the optic nerve. Eye (Lond) 2004;18:1089–95. doi: 10.1038/sj.eye.6701574. [DOI] [PubMed] [Google Scholar]
- Morrison JC, Johnson EC, Cepurna WA, Jia L. Understanding mechanisms of pressure-induced optic nerve damage. Prog Retin Eye Res. 2005;24:217–40. doi: 10.1016/j.preteyeres.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Murphy JA, Archibald ML, Chauhan BC. The role of endothelin-1 and its receptors in optic nerve head astrocyte proliferation. Br J Ophthalmol. 2010;94:1233–8. doi: 10.1136/bjo.2009.172098. [DOI] [PubMed] [Google Scholar]
- Nguyen JV, Soto I, Kim KY, Bushong EA, Oglesby E, et al. Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci U S A. 2011;108:1176–81. doi: 10.1073/pnas.1013965108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickells RW, Howell GR, Soto I, John SW. Under pressure: cellular and molecular responses in glaucoma. Ann Rev Neuroscience. doi: 10.1146/annurev.neuro.051508.135728. submitted. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–9. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nikolskaya T, Nikolsky Y, Serebryiskaya T, Zvereva S, Sviridov E, et al. Network analysis of human glaucomatous optic nerve head astrocytes. BMC Med Genomics. 2009;2:24. doi: 10.1186/1755-8794-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne NN. Pathogenesis of ganglion “cell death” in glaucoma and neuroprotection: focus on ganglion cell axonal mitochondria. Prog Brain Res. 2008;173:339–52. doi: 10.1016/S0079-6123(08)01124-2. [DOI] [PubMed] [Google Scholar]
- Osborne NN. Mitochondria: Their role in ganglion cell death and survival in primary open angle glaucoma. Exp Eye Res. 2010;90:750–7. doi: 10.1016/j.exer.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Panagis L, Zhao X, Ge Y, Ren L, Mittag TW, Danias J. Gene Expression Changes in Areas of Focal Loss of Retinal Ganglion Cells (RGC) in the Retina of DBA/2J Mice. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.09-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang IH, Clark AF. Rodent models for glaucoma retinopathy and optic neuropathy. J Glaucoma. 2007;16:483–505. doi: 10.1097/IJG.0b013e3181405d4f. [DOI] [PubMed] [Google Scholar]
- Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000;41:764–74. [PubMed] [Google Scholar]
- Pease ME, Zack DJ, Berlinicke C, Bloom K, Cone F, et al. Effect of CNTF on retinal ganglion cell survival in experimental glaucoma. Invest Ophthalmol Vis Sci. 2009;50:2194–200. doi: 10.1167/iovs.08-3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER. Very Slow Retrograde and Wallerian Degeneration in the CNS of C57BL/Ola Mice. Eur J Neurosci. 1991;3:102–5. doi: 10.1111/j.1460-9568.1991.tb00815.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Lunn ER, Brown MC, Cahusac S, Gordon S. Evidence that the Rate of Wallerian Degeneration is Controlled by a Single Autosomal Dominant Gene. Eur J Neurosci. 1990;2:408–13. doi: 10.1111/j.1460-9568.1990.tb00433.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- Prasanna G, Hulet C, Desai D, Krishnamoorthy RR, Narayan S, et al. Effect of elevated intraocular pressure on endothelin-1 in a rat model of glaucoma. Pharmacol Res. 2005;51:41–50. doi: 10.1016/j.phrs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Quigley H, Anderson DR. The dynamics and location of axonal transport blockade by acute intraocular pressure elevation in primate optic nerve. Invest Ophthalmol. 1976;15:606–16. [PubMed] [Google Scholar]
- Quigley HA. The number of persons with glaucoma worldwide. Br J Ophthalmol. 1996;80:389–93. doi: 10.1136/bjo.80.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Addicks EM. Chronic experimental glaucoma in primates I. Production of elevated intraocular pressure by anterior injection of autologous ghost red blood cells. Invest Ophthalmol Vis Sci. 1980a;19:126–36. [PubMed] [Google Scholar]
- Quigley HA, Addicks EM. Chronic experimental glaucoma in primates II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest Ophthalmol Vis Sci. 1980b;19:137–52. [PubMed] [Google Scholar]
- Quigley HA, Addicks EM. Chronic experimental glaucoma in primates. II Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest Ophthalmol Vis Sci. 1980c;19:137–52. [PubMed] [Google Scholar]
- Quigley HA, Addicks EM. Regional differences in the structure of the lamina cribrosa and their relation to glaucomatous optic nerve damage. Arch Ophthalmol. 1981;99:137–43. doi: 10.1001/archopht.1981.03930010139020. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Addicks EM, Green WR, Maumenee AE. Optic nerve damage in human glaucoma. II The site of injury and susceptibility to damage. Arch Ophthalmol. 1981;99:635–49. doi: 10.1001/archopht.1981.03930010635009. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Anderson DR. Distribution of axonal transport blockade by acute intraocular pressure elevation in the primate optic nerve head. Invest Ophthalmol Vis Sci. 1977;16:640–4. [PubMed] [Google Scholar]
- Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90:262–7. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Cone FE, Gelman SE, Yang Z, Son JL, et al. Lack of neuroprotection against experimental glaucoma in c-Jun N-terminal kinase 3 knockout mice. Exp Eye Res. 2011;92:299–305. doi: 10.1016/j.exer.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Guy J, Anderson DR. Blockade of rapid axonal transport. Effect of intraocular pressure elevation in primate optic nerve. Arch Ophthalmol. 1979;97:525–31. doi: 10.1001/archopht.1979.01020010269018. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Hohman RM, Addicks EM, Massof RW, Green WR. Morphologic changes in the lamina cribrosa correlated with neural loss in open-angle glaucoma. Am J Ophthalmol. 1983;95:673–91. doi: 10.1016/0002-9394(83)90389-6. [DOI] [PubMed] [Google Scholar]
- Quigley HA, McKinnon SJ, Zack DJ, Pease ME, Kerrigan-Baumrind LA, et al. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol vis Sci. 2000;41:3460–6. [PubMed] [Google Scholar]
- Radius RL, Anderson DR. Rapid axonal transport in primate optic nerve. Arch Ophthalmol. 1981;99:650–4. doi: 10.1001/archopht.1981.03930010650010. [DOI] [PubMed] [Google Scholar]
- Ribchester RR, Tsao JW, Barry JA, Asgari-Jirhandeh N, Perry VH, Brown MC. Persistence of neuromuscular junctions after axotomy in mice with slow Wallerian degeneration (C57BL/WldS) Eur J Neurosci. 1995;7:1641–50. doi: 10.1111/j.1460-9568.1995.tb01159.x. [DOI] [PubMed] [Google Scholar]
- Ritch R, Shields MB, Krupin T. The Glaucomas. St. Louis: Mosby; 1996. [Google Scholar]
- Roberts MD, Grau V, Grimm J, Reynaud J, Bellezza AJ, et al. Remodeling of the connective tissue microarchitecture of the lamina cribrosa in early experimental glaucoma. Invest Ophthalmol Vis Sci. 2009;50:681–90. doi: 10.1167/iovs.08-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen AM, Stevens B. The role of the classical complement cascade in synapse loss during development and glaucoma. Adv Exp Med Biol. 2010;703:75–93. doi: 10.1007/978-1-4419-5635-4_6. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ederra J, Garcia M, Hernandez M, Urcola H, Hernandez BE, et al. The pig eye as a novel model of glaucoma. Exp Eye Res. 2005;81:561–9. doi: 10.1016/j.exer.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Salinas-Navarro M, Alarcon-Martinez L, Valiente-Soriano FJ, Jimenez-Lopez M, Mayor-Torroglosa S, et al. Ocular hypertension impairs optic nerve axonal transport leading to progressive retinal ganglion cell degeneration. Exp Eye Res. 2010;90:168–83. doi: 10.1016/j.exer.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Saragovi HU, Hamel E, Di Polo A. A neurotrophic rationale for the therapy of neurodegenerative disorders. Curr Alzheimer Res. 2009;6:419–23. doi: 10.2174/156720509789207912. [DOI] [PubMed] [Google Scholar]
- Sasaoka M, Taniguchi T, Shimazawa M, Ishida N, Shimazaki A, Hara H. Intravitreal injection of endothelin-1 caused optic nerve damage following to ocular hypoperfusion in rabbits. Exp Eye Res. 2006;83:629–37. doi: 10.1016/j.exer.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006;7:66. doi: 10.1186/1471-2202-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta Ghosh A, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol. 2011;194:751–64. doi: 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields MB. Textbook of Glaucoma. Baltimore: Williams & Wilkins; 1992. [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009 doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele MR, Inman DM, Calkins DJ, Horner PJ, Vetter ML. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Invest Ophthalmol Vis Sci. 2006;47:977–85. doi: 10.1167/iovs.05-0865. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Shimazawa M, Sasaoka M, Shimazaki A, Hara H. Endothelin-1 impairs retrograde axonal transport and leads to axonal injury in rat optic nerve. Curr Neurovasc Res. 2006;3:81–8. doi: 10.2174/156720206776875867. [DOI] [PubMed] [Google Scholar]
- Tezel G, Yang X, Yang J, Wax MB. Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res. 2004;996:202–12. doi: 10.1016/j.brainres.2003.10.029. [DOI] [PubMed] [Google Scholar]
- Whitmore AV, Libby RT, John SWM. Glaucoma: Thinking in new ways - a role for autonomous axonal self-destruction and compartmentalised processes? Prog Retin Eye Res. 2005;24:639–62. doi: 10.1016/j.preteyeres.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Yang X, Luo C, Cai J, Pierce WM, Tezel G. Phosphorylation-dependent interaction with 14-3-3 in the regulation of bad trafficking in retinal ganglion cells. Invest Ophthalmol Vis Sci. 2008;49:2483–94. doi: 10.1167/iovs.07-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Quigley HA, Pease ME, Yang Y, Qian J, et al. Changes in gene expression in experimental glaucoma and optic nerve transection: the equilibrium between protective and detrimental mechanisms. Invest Ophthalmol Vis Sci. 2007;48:5539–48. doi: 10.1167/iovs.07-0542. [DOI] [PubMed] [Google Scholar]
- Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30:81–114. doi: 10.1016/j.preteyeres.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]