

Abstract
Previously, we reported that hBD-3 can both antagonize CXCR4 function on T cells, and promote receptor internalization in the absence of activation. In the present study, we explored the important structural elements of hBD-3 that are involved in blocking CXCR4 activation by its natural ligand, stromal derived factor 1α (SDF-1α; CXCL12). Results from site-directed mutagenesis studies suggest that the ability of hBD-3 to inhibit SDF-1α/CXCR4 interaction, as assayed either by blocking SDF-1 binding to CXCR4 or antagonizing SDF-1 induced Ca2+ mobilization, is correlated with the presence of hBD-3 cysteine residues, specific surface-distributed cationic residues, and the electrostatic properties and availability of both hBD-3 termini. Specifically, hBD-3 activity against CXCR4 is reduced by: 1) substituting all six cysteine residues; 2) substituting the cationic residues with acidic ones in the N- and C- termini; 3) removal of the first 10 N-terminal residues; and 4) substituting surface-exposed basic residues K8, K32 and R36 with neutral ones. The hBD-3/CXCR4 interaction has potentially wide ranging implications for HIV-related biology as well as for a host of CXCR4-dependent activities including hematopoiesis, neurogenesis, angiogenesis, carcinogenesis, and immune cell trafficking. CXCR4 is highly expressed on T cells, monocytes, and epithelial cells. Therefore, understanding the structure-function relationship between hBD-3 and CXCR4 that accounts for the antagonistic interaction between the two molecules may provide new insights into HIV/HAART-related pathology as well as novel insights into the interaction between innate and adaptive immunity at mucosal sites.
Keywords: Defensin, HBD-3, CXCR4, structure, antagonism
Introduction
Human β defensins (hBDs) are epithelial cell-derived, cationic peptides, primarily recognized for their antimicrobial properties (2). Recent findings indicate that human β-defensin-3 (hBD-3), the β defensin exclusively expressed in the highly-proliferating, non-differentiated stratum basale of the oral mucosa (3), mediates important immunoregulatory functions, in addition to its antimicrobial properties. These include, but are not limited to, chemotaxis of myeloid cells via CCR2 (3–5), interaction with TLR-1 and -2 that induces maturation of myeloid dendritic cells and monocytes (6), and non-activating internalization of CXCR4 (the co-receptor of HIV-1), which leads to protection from HIV-1 X4 tropic viral infections (1,7).
Previously we reported that hBD-3 can: 1) rapidly antagonize activation of CXCR4 by its natural ligand, stromal derived factor 1 (SDF-1α); and 2) promoteCXCR4 internalization without activating the receptor (1). This suggests that the hBD-3/CXCR4 interaction has potentially wide ranging implications for HIV-related biology as well as for a host of CXCR4-dependent activities including hematopoiesis, neurogenesis, angiogenesis, carcinogenesis, and immune cell trafficking (8–11). CXCR4 is highly expressed on T cells, monocytes, and epithelial cells. Therefore, understanding the structure-function relationship between hBD-3 and CXCR4 that accounts for the dynamic interaction between the two molecules may provide new insights into HIV-related pathology, its modification by HAART, and the interplay between innate and adaptive immunity at mucosal sites.
In the present study, we explored the structural elements of hBD-3 that are involved in blocking the productive interaction between CXCR4 and SDF-1α. Our findings suggest that the presence of cysteine and surface-exposed cationic residues are important for this antagonistic activity. In addition, the cationic/uncharged C-and N-termini also contribute to the CXCR4-blocking activity of hBD-3. Finally, and probably most importantly, truncating the N-terminus by removing the first 10 residues substantially reduces hBD-3 activity.
Results
Generation of hBD-3 mutant proteins for analysis of antagonism of SDF-1α/CXCR4 signaling
Standard methods were used to generate recombinant hBD3 (rhBD-3) and a series of twenty-six hBD-3 mutants with substitutions, deletions, or insertions of specific residues or domains. The sequence of wildtype hBD-3 is shown in Figure 1 as a template for the comparison with the mutant proteins which included: 1) six constructs with single or multiple cysteine substitutions (C11S, C18S, C23G, C33I, C40G/C41Y, C11S/C18S/C23G/C31I/C40G/C41Y); 2) five constructs with single or multiple substitutions in surface-exposed cationic residues (K8A, K32S, R36I, K8A/K32S, K8A/K32S/R36I); 3) three constructs with C-terminal insertions of α-helical domains from the C-terminus of SDF-1α (EYLEKALN insert, EYLE insert, EKGAYNEL insert); 4) six constructs with C-terminal substitutions or insertions of cationic residues (R42S/R43S, K44G/K45G, R42A/R43A/K44G/K45G, RRRK insert, R42E/R43E/K44S/K45S, N4R/T5R/R42E/R43E/K44S/K45S; 5) three constructs with N-terminal substitutions or insertions (GIGDPVT substitution for wildtype residues 1–10, N4E/T5E/K8A, N4R/T5R); 6) three constructs with N-terminal and/or C-terminal deletions (Δ1–5, Δ1–10, Δ1–5/Δ42–45). These hBD-3 mutants were compared for their relative abilities to target SDF-1α/CXCR4 signaling at the level of: 1) inhibition of direct SDF-1α binding to CXCR4-expressing CEM T cells; and 2) attenuation of SDF-1α-induced Ca2+ mobilization in CEM cells.
Figure 1. Sequence of wild-type (W/T) hBD-3.
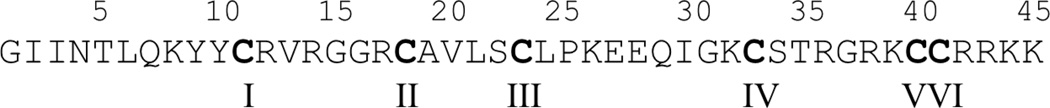
Residues are numbered 1 through 45 from the N-terminus and respective cysteine residues are identified with roman numbers (I through VI).
Inhibition of SDF-1 binding by hBD-3
We previously reported that hBD-3 is an non-classical antagonist of CXCR4, i.e., hBD-3 can inhibit SDF-1 binding to, and activation of, CXCR4 but can also cause CXCR4 internalization without activating G protein signaling responses. These results suggest that the binding of hBD-3 to CXCR4 induces conformational changes in that receptor which: 1) occlude the binding sites for SDF-1; 2) suppress productive interaction with downstream G proteins; and 3) facilitate productive interaction with downstream proteins (e.g., arrestins) involved in receptor internalization. We have now further studied the mechanism of hBD-3/ CXCR4 interaction by characterizing the saturatable binding of SDF-1 to CXCR4-expressing CEM cells in the absence or presence of hBD-3. The SDF-1 binding assay was performed with 0.1 nM 125I-SDF-1α plus increasing concentrations of unlabeled SDF-1α in the absence or presence of 5µg/ml hBD-3. The amount of bound SDF-1α was calculated from the different specific activities (based on the ratios of 125I-SDF-1α to unlabeled [SDF-1α]) The amount of SDF-1α specifically bound to CEM cells progressively increased as extracellular [SDF-1α] was raised from 0.1 to 5 nM and reached a plateau value at [SDF-1α]> 10 nM (Figure 2). In the presence of 5 µg/ml hBD-3, SDF-1α binding was decreased by ~2-fold (or more) over the entire range of tested [SDF-1α], indicating that hBD-3 acts to decrease the maximum number of SDF-1 binding sites. The SDF-1 binding assay illustrated in Figure 2 was performed at 37°C but similar SDF-1α binding and suppressive effects of hBD-3 values were observed at 4°C (data not shown).
Figure 2. Effects of hBD-3 on saturatable SDF-1α binding to CEM cells.

The SDF-1α binding assay was performed as described in the Materials and Methods. 125I labeled SDF-1α (10nM) was added to each tube, together with 0 to 30 nM unlabeled SDF-1α in the absence or presence of 5µg/ml hBD-3. Cells were incubated at 37°C for 20min prior to termination of binding by rapid sedimentation and washing.
Role of disulfide bonds
We started to identify which regions of hBD-3 contribute to the molecule’s ability to inhibit CXCR4 function by systematically replacing each cysteine with uncharged residues. The rationale was based on the fact that hBD-3 has six conserved cysteines (Figure 1) that promote three intramolecular disulfide bonds that stabilize the molecule’s distinctive β-sheeted conformation (12). Figure 3 shows that wild type hBD-3 (20µg/ml) completely blocks calcium mobilization triggered by 10nM SDF-1α. Substituting one or two cysteines in hBD-3 reduced, but did not totally abrogate this activity (Figure 3B–3F). However, substituting all 6 cysteines with uncharged residues was able to completely reverse the blocking effect of hBD-3 on SDF-1α triggered Ca2+ mobilization (Figure 3 G). Similarly, the ability of this linearized cysteine-free mutant of hBD-3 to block high-affinity binding of SDF-1α to CXCR4 was markedly reduced when compared to the wild type molecule or a saturating concentration (10nM) of unlabeled SDF-1α (Figure 3 H).
Figure 3. Effect of Cys mutations on the activity of hBD-3.

Panels A through H show the ability of hBD-3 and its mutants to inhibit SDF-1α-dependent Ca2+ mobilization. A: wild type (wt); B: mutant CM1 (C11S-hBD3); C: mutant CM2 (C18S-hBD3); D: mutant CM3 (C23G-hBD3); E: mutant CM4 (C33I-hBD3); F: mutant CM56 (C40G/C41Y-hBD3); G: CM1–6 (C11S/C18S/C23G/C31I/C40G/C41Y-hBD3); H: inhibition of SDF-1α binding by CM1–6. For Ca2+ mobilization assays (panels A through G), 1 (black) or 2 (colored, blue here) indicate control or experimental (pretreated with wt hBD-3 or mutants) fura2 fluorescence traces respectively; arrows beneath the fluorescence traces indicate the time when respective reagents were added with, black for control, colored for experimental groups. Similar labeling was used for the fura2 fluorescence traces in all subsequent figures.
Effects of surface cationic residue mutations
Previous studies have indicated that electrostatic interactions between SDF-1α and CXCR4 play an important role in CXCR4 activation by SDF-1α (13,14). Based on the coordinates of SDF-1α (ID: 1SDF) and hBD-3 (ID: 1KJ6), available from the Protein Data Bank (PDB; http://www.rcsb.org/pdb/home/home.do), we modeled the structures of both proteins using the program DS ViewerPro 6.0 (Accelrys, http://accelrys.com/). Figure 4 A shows the SDF-1α (left) and the hBD-3 (right) structures, respectively. Three N-terminal surface cationic residues (K1, R8 and R12) are believed to be important for SDF-1α binding to, and activation of, CXCR4 (13). Three similar cationic residues are present on the surface of hBD-3: K8, K32, and R36. We generated several mutants targeting these three residues and tested their function. As shown in Figures 4B–4E, single or double substitution (s) did not diminish the mutants’ ability to block SDF-1α triggered Ca2+ mobilization. However, substitution of all three cationic residues with neutral ones resulted in substantial reversal of this inhibitory hBD-3 function; i.e., approximately 50% (Fig 4 F). In contrast, this mutant effectively reduced high-affinity SDF-1α binding (Fig 4 G).
Figure 4. Effect of mutation of surface-exposed cationic residues.

A: The comparison of surface-exposed cationic residues of SDF-1 (left) and hBD-3 (right). Panels B through F show the inhibition of SDF-1α-triggered Ca2+ mobilization by hBD-3 mutants. B: mutant K8A; C: mutant K32S; D: mutant R36I; E: mutant 8–32 (K8A/K32S); F: mutant 8-32-36 (K8A/K32S/R36I); G: inhibition of high-affinity SDF-1α binding by 8-32-36.
Effects of inserting SDF-1α C-terminal α-helix into the C terminus of hBD-3
Further comparison of the structures of hBD-3 and SDF-1α indicates that hBD-3 lacks the α-helix of SDF-1α (Figure 5 A). Cai et al. found that the C-terminal α-helix is important for SDF-1α triggered CXCR4 signaling and chemotaxis, but is less critical for CXCR4 internalization (15). Therefore, we inserted the C-terminal α-helix-forming residues of SDF-1α (EYLEKALN) into the C-terminus of hBD-3 and found that this mutant, not only did not activate CXCR4 (Ca2+ mobilization) independently, but was unable to block SDF-1α-triggered Ca2+ mobilization (Fig 5 B). This suggests that the α-helix of SDF-1α, positioned in the C-terminus of hBD-3, abrogated the ability of hBD3 to antagonize CXCR4. Binding assay experiments indicated that inserting the helical structure only slightly diminished the molecule’s ability to inhibit SDF-1α binding (Figure 5 E). To further investigate the role of the C-terminal α-helix insertion in reducing hBD-3 activity, we generated two additional mutants. In one, the C-terminus of hBD-3 was extended by four residues, EYLE (Figure 5C), and in the second, by eight residues, EKGAYNEL (Figure 5 D). Neither of these latter C-terminal extensions was predicted to have a helical structure, as determined by “PORTER” (http://distill.ucd.ie/porter/). Both mutants retained the ability to block both SDF-1α-triggered Ca2+ mobilization and high-affinity SDF-1α binding (Figure 5C–5E). These data indicate that the insertion of an α-helix onto, and not mere extension of, the C-terminus of hBD3 reduces its ability to block CXCR4 functions, suggesting that the C-terminus may be important for optimal hBD-3 interaction with CXCR4. Therefore, we generated more variants of hBD-3 with mutated C-termini.
Figure 5. Activity changes resulting from the attachment of α-helix from SDF-1α to the C-terminus of hBD-3.

A: Structural models of SDF-1α (left) and hBD-3 (right). B, C and D: Inhibition of the SDF-1α-triggered Ca2+-mobilization by respective mutants. B: mutant Helix (insert EYLEKALN at C-terminus of hBD-3); C: mutant 4AAs (insert EYLE at C-terminus of hBD3); D: mutant 8AAs (insert EKGAYNEL at C-terminus of hBD3); E: inhibition of high-affinity SDF-1α binding by mutants Helix and 8AAs.
Effects of C-terminal mutations in hBD-3
HBD-3 is the most cationic of the four most widely expressed human β-defensins, bearing a net charge +11 (12). At the C-terminus, there are four consecutive cationic residues (R42RKK45) (Figure 1). To test their involvement in hBD-3-induced CXCR4 antagonism, we generated several mutants by substituting uncharged or anionic residues for the positively-charged ones. Substituting RR with SS (Figure 6 A), KK with GG (Figure 6 B), or all four with AAGG (Figure 6 C) did not reduce the ability of hBD-3 to inhibit the SDF-1α-induced Ca2+ mobilization. Insertion of four additional cationic residues (RRRK) into the wild type C terminus did not abrogate hBD-3 activity (Figure 6 D). However, a modest loss of hBD-3 functional antagonism of CXCR4 was observed when the native tetra-peptide (RRKK) was replaced with EESS (Figure 6 E) and this was correlated with the reduced ability of this mutant to suppress high-affinity SDF-1α binding (Figure 6 G). We speculated that an overall cationic nature of the hBD-3 molecular surface is important for antagonism of CXCR4 and thus, designed a construct to compensate the negative charge of the anionic EESS C-terminus by introducing two additional cationic residues at the N-terminus of hBD-3 (mutations N4R and T5R). Notably, this compensated mutant regained the ability to completely block both CXCR4 functional activation of CXCR4by SDF-1α (Figure 6 F) and high-affinity SDF-1α binding (Figure 6 G). Taken together, these observations confirmed that the introduction of a negative charge at the C-terminus of hBD-3 significantly reduced the ability of this mutant to inhibit the SDF-1α binding, suggesting an important role for the positively-charged C-terminus in hBD-3 antagonism of CXCR4.
Figure 6. Effect of the C-terminal mutations on activity of hBD3.

Panels A through F illustrate inhibition of the SDF-1α-induced Ca2+ mobilization by several C-terminal hBD-3 mutants. A: mutant SSKK (R42S/R43S-hBD3); B: mutant RRGG (K44G/K45G-hBD3); C: mutant AAGG (R42A/R43A/K44G/K45G-hBD3); D: mutant RRKKRRRK (insert RRRK at C-terminus of hBD3); E: mutant CA (R42E/R43E/K44S/K45S-hBD3); F: mutant CA-NC (N4R/T5R/R42E/R43E/K44S/K45S-hBD3); G: inhibition of high-affinity SDF-1α binding by several hBD-3 mutants.
Effects of N-terminal mutations in hBD-3
To examine the involvement of the N-terminus of hBD-3 in blocking SDF-1α-dependent Ca2+ mobilization and inhibiting SDF-1α binding, we took advantage of the fact that hBD-2 does not antagonize CXCR4 as does hBD-3 (1) and that the N-termini of hBD-2 and -3 are highly divergent, with that of hBD-3 being cationic, while that of hBD-2 being anionic (12,16). We generated a chimera by replacing the first 10 residues of hBD-3 preceding the first cysteine (GIINTLQKYY) with the equivalent N-terminal sequence of hBD-2 (GIGDPVT). Figure 7 A shows that this chimera substantially lost the ability to block SDF-1α-dependent Ca2+ mobilization. To further characterize the importance of N-terminal positive charges in hBD-3 interaction with CXCR4, we introduced three positive-charge neutralizing mutations (K8A, N4E, and T5E) into the hBD-3 N-terminus. This mutant (termed NA2) demonstrated reduced activity in blocking Ca2+ mobilization (Figure 7 B). Furthermore, a mutant (NC) with additional cationic residues (N4R and T5R, Figure 7 C) and a mutant (K8A) with a neutral N-terminus (Figure 7 D) retained CXCR4 antagonistic activity. Binding assays also showed that both chimeras H2N and NA2 had reduced ability to inhibit high-affinity SDF-1α binding (Figure 7 E). In contrast, NC and K8A inhibited SDF-1α binding similarly to wildtype hBD-3 (data not shown).
Figure 7. Effect of the N-terminal mutations on activity of hBD3.

Panels A through D describe inhibition of the SDF-1α-dependent Ca2+-mobilization by several N-terminal hBD-3 mutants. A: mutant H2N (replace G1 through Y10 of hBD3 with GIGDPVT)); B: mutant NA2 (N4E/T5E/K8A-hBD3); C: mutant NC (N4R/T5R-hBD3); D: mutant K8A; E: inhibition of high-affinity SDF-1α binding by mutants H2N and NA2.
Determining the minimal hBD-3 motif retaining CXCR4 antagonistic activity
We sought to identify the minimal hBD-3 structure that retains CXCR4 antagonistic activity. An N-terminal truncation mutant (6–45) was generated by deleting residues 1 through 5. This mutant was active in inhibiting both SDF-1α-dependent Ca2+ mobilization (Figure 8 B) and high-affinity SDF-1α binding (Figure 8 E). When this N-terminal truncation was combined with the truncation of the C terminal RRKK cationic residues, the resulting mutant protein (termed 6–41) was less efficacious than the 6–45 construct in suppressing SDF-1α-induced Ca2+ mobilization (Figure 8 D) but retained the ability to inhibit high-affinity SDF-1α binding (Figure 8 E). Notably, further truncation of the N-terminus by deletion of the first 10 residues generated a mutant protein (termed 11–45) with a markedly reduced ability to antagonize both SDF-1α-induced Ca2+ mobilization (Figure 8 C) and high-affinity SDF-1α binding (Figure 8 E). We also generated four mutants with even more extensive truncations (6–39, 6–37, 6–38, 6–36), and found that none were active in blocking CXCR4 (data not shown).
Figure 8. Determining minimal hBD-3 structure for blocking CXCR4.

Inhibition of the SDF-1α-induced Ca2+-mobilization by three hBD-3 mutants, 6–45 (delete G1 through T5 of hBD3), 11–45 (delete G1 through Y10 of hBD3), and 6–41 (delete G1 through T5 and R42 through K45 of hBD3), with various truncations of the N-terminal region. Bar graph shows inhibition of high-affinity SDF-1α-binding by these mutants.
The 6–45 mutant is as potent as wild type hBD3 in blocking CXCR4
The truncation studies revealed that, at 20µg/ml, the efficacy of the 6–45 construct was equivalent to wildtype hBD-3 as an antagonist of CXCR4 function. (Figures 8B and 8E). We further compared the potencies of the 6–45 mutant (Figures 9D, 9E, 9F, 9G) versus wildtype hBD-3 (Figures 9A, 9B, 9C, 9G) as inhibitor of both SDF-1α-triggered Ca2+ mobilization and high-affinity SDF-1-binding. Inhibition of the Ca2+ mobilization response by hBD-3 begins to diminish at 10 µg/ml (1.94 µM), whereas 6–45 retains maximal antagonistic action at 7.5 µg/ml (1.61 µM), but exhibits reduced activity at 3.75 µg/ml (0.80 µM). The binding assay confirmed that 6–45 and hBD-3(wt) are equally potent in suppressing high-affinity SDF-1α binding to CXCR4 (Figure 9G).
Figure 9. N-terminal deletion mutant (6–45) is more active than hBD-3(wt) in blocking CXCR4.

Dose dependent inhibition of the SDF-1α-induced Ca2+-mobilization by hBD-3(wt) and mutant 6–45. Bar graph shows dose-dependent inhibition of high-affinity SDF-1α-binding by mutant 6–45.
Discussion
Three disulfide bonds are central structural features of β-defensins. Through cysteine(s) substitution (Figure 3), we found that a ‘no-Cys’ hBD-3 lost all activity as a CXCR4 antagonist, suggesting that retention of molecular conformation is needed for hBD-3 interaction with CXCR4. We also observed that substituting one or two cysteine residues did not change the activity of hBD-3, suggesting that hBD-3 maintains the required conformation needed to block CXCR4, even with fewer cysteine residues. Since all defensins described in this study were generated using recombinant methods in a bacterial system (E. coli), it is possible that they represent mixtures of variants with different topologies of disulfides. As previously reported by Wu and coworkers, such a mixture results from the in vitro refolding of hBD-3 (17). Further studies are necessary to assign specific CXCR4-blocking activity to particular topological variants of hBD-3. We are in the process of chemically synthesizing hBD3 and its mutants using orthogonal protection of cysteine residues [see ref. (17)] in order to provide more detailed insight into the structure-function analysis of hBD-3/CXCR4 antagonism.
Because previous reports have indicated that three surface-exposed cationic residues of SDF-1 play a critical role in the molecule’s interaction with its cognate receptor CXCR4 (13), we tested whether three similar surface-exposed basic residues, K8, K32, and R36 in hBD3 were critical for its interaction with the same receptor (Figure 4A). Interestingly, only mutagenesis of all three positive residues, but not one or two of them, altered the activity of hBD-3 against CXCR4 (Figure 4), suggesting that the net surface charge of the molecule, rather any individual cationic residue, is important for this activity. In contrast, the observation that replacement of four basic residues at the C terminus did not change the antagonistic efficacy (Figure 6C) suggests that the C-terminal basic residues are not critical for hBD-3 interaction with CXCR4.
Certain mutants, such as the 8-32-36 construct with reduced surface cationic charge (Figures 4F and 4G), the SDF-1α helix insert construct (Figures 5B and 5E), and the 6–41 truncation mutant (Figures 8D and 8E), retained the ability to suppress high-affinity SDF-1α binding (assayed with 0.1 nM SDF-1α) but were unable to markedly attenuate the Ca2+ mobilization response triggered by a saturating (10 nM) concentration of SDF-1α. The differential efficacies of these mutants in the two assays suggest that, at the 20µg/ml test concentration, these hBD-3 variants effectively compete with 0.1 nM SDF-1α for presumably overlapping binding sites on CXCR4 but are unable to compete for these CXCR4 sites in the presence of the 10 nM chemokine. Consistent with this scenario, the 8-32-36 construct was able to completely suppress the Ca2+ mobilization response triggered by 1 nM SDF-1α (data not shown).
Mutations targeting both termini of hBD-3 revealed that anionic residues in these segments substantially reduce the ability of hBD3 to antagonize CXCR4 (Figures 6 and 7). Data for the mutants truncated at the N-terminus indicate that residues 1 through 5 are not necessary for interactions between hBD3 and CXCR4 (Figures 8 and 9). However, removal of 10 N-terminal residues eliminated activity of hBD-3, indicating a central role of this domain in hBD-3 antagonism (Figure 8). Moreover, observations that either the insertion of an α-helix into the hBD-3 C terminus (Figure 5) or a truncation of the last four residues of the C terminus (Figure 8) results in a loss of activity, suggests that an accessible and unmodified C-terminus is important for interaction of hBD-3 with CXCR4.
Kofuka and coworkers suggested that SDF-1α binds to CXCR4 according to a two-step model (14). In the first step, the β-sheet, 50-s loop, and N-loop of SDF-1α bind rapidly to exposed extracellular loop regions of CXCR4, and the second, slower step leads to an interaction between the N terminus of SDF-1α and the transmembrane segments of this heptihelical receptor. A similar two-step model was proposed by Wu et al., who suggested that electrostatic interactions between SDF-1α and the extracellular loop domains of CXCR4 are important for the initial binding (18). Structural analysis indicates a number of similarities between the molecules of SDF-1 and hBD-3, which include cationic properties, a presence of the β-sheet head and a flexible N terminus. These similarities hint at possible factors that direct both hBD-3 and SDF-1α molecules to overlapping recognition epitopes on CXCR4 and are consistent with more specific findings reported in this study. Further study is needed to elucidate whether hBD-3 disrupts the two-step SDF-1α/CXCR4 interaction.
CXCR4 is involved in many physiological processes including hematopoiesis, neurogenesis, angiogenesis, cardiogenesis, immune cell trafficking, and cancer metastasis (8–11,19). It is also one of the co-receptors HIV uses for cell entry (20). Previously, we have shown that hBD-3 can effectively block the CXCR4 function (i.e. its interaction with SDF-1α) by the internalization of this receptor without its activation (1). The structure-function analysis, described in this study, initiates description of determinants of the interaction between hBD3 and CXCR4. Use of this information may aid in development of more potent antagonists of CXCR4, originating from an hBD3 scaffold, with a future goal of utilizing them for anti-HIV treatment or prevention, or in cancer therapy to control processes of angiogenesis and/or metastasis.
Materials and Methods
Cells
CEM X4/R5 cells (expressing both CXCR4 and CCR5) were obtained from the NIH AIDS Research and Reference Reagent Program, maintained in RPMI 1640, supplemented with 5% FBS and 400 µg/ml Geneticin (Invitrogen, Carlsbad, CA).
Cloning hBD-3 mutant plasmids
Site-directed mutagenesis was carried out to generate hBD-3 mutants. Primers were designed according to the Stratagene (La Jolla, CA) QuickChange Site-Directed Mutagenesis Instruction Manual. The Site Find v4.0 program (http://www.utmb.edu/scccb/software/sitefind.html) was used to design primers to generate mutants. Unless specified otherwise, expression plasmids encoding mutated variants of hBD-3 were created by PCR using predesigned primers and the Expand High Fidelity PCR System (Roche, IN) with pET-30c-hBD3 (from J. Harder and J. Schröder, Kiel University, Germany) (12) as the template. The PCR program was as follows: 95°C 3min, followed by 20 cycles of 94°C 1min, 52°C 1 min, and 68°C 10 min, followed by 1 cycle of 94°C 1min, 52°C 1min, and 68°C 1hr. PCR products were kept at 4°C until further use. A small aliquot of the PCR products was subjected to gel electrophoresis to examine the PCR efficiency. Purified PCR products (QIAquick PCR purification kit, QIAGEN, MD) were used for transformation of competent BL21 (DE3) cells (Novagen, WI) and kanamycin-resistant clones were selected and sequenced to verify the mutations. Table 1 lists the mutations and associated primers used to generate plasmids encoding the mutants used in this study (supplemental Table 1).
Generation of recombinant hBD3 (rhBD-3) and its mutants
The wild-type recombinant hBD3 (rhBD-3) and its mutants were expressed and purified as described previously (1,12). Briefly, His6-tagged fusion protein was expressed in BL21 (DE3) cells by 1 mM IPTG induction. Ni2+-affinity columns (Ni-NTA resin from QIAGEN) and RP-HPLC (C-18 column;Phenomenex, Torrance, CA) were used to isolate hBD-3/mutants fusion proteins, followed by digestion with enterokinase (Novagen), to release mature peptides. The mature forms of rhBD-3 and/or mutated variants were purified with RP-HPLC. The molecular weights of recombinant proteins were verified by mass spectrometry.
Ca2+ mobilization assay
SDF-1α-induced increases in cytosolic free [Ca2+] were determined using the fura2 fluorescent Ca2+ indicator dye. Briefly, CEM X4/R5 cells were suspended at 5 × 106 cells/ml in calcium assay buffer (CAB: 130 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 20 mM HEPES, 5 mM glucose and 0.1% BSA). After addition of 1 µM Fura-2 AM (Molecular Probes, Eugene, OR), cell suspensions were incubated at 37°C for 30 min, pelleted by brief centrifugation, washed twice with CAB, and resuspended in CAB at 1 × 106 cells /ml. Aliquots (1.5 ml) of fura2-loaded cell suspension were assayed in UV-transparent plastic cuvets with continuous magnetic stirring and thermostatting at 37°C. Excitation illumination was from a 75 watt xenon arc lamp filtered through a 340 nm narrow bandpass optical filter. Fura2 fluorescence emission through a 500 nm narrow bandpass optical filter was detected quantified with an EMI photomultiplier tube. Unless noted otherwise, CEM cells were treated with 20 µg/ml hBD-3 or hBD-3 mutant (or vehicle control) for 2–3 min prior stimulation with 10 nM SDF-1α. The SDF-1α induced transient changes in fura2-fluorescence were recorded for another 2–3 min prior to plasma membrane permeabilization by addition of 33 µg/ml digitonin and release of the cytosolic dye into the extracellular CAB. The 1 mM [Ca2+] of the CAB saturates the high-affinity Ca2+-binding sites of the released fura2 to provide a maximum Ca2+-saturated fura2 fluorescence signal (Fmax). The permeabilized cell suspension was then supplemented with 16.6 mM EGTA (to chelate Ca2+ bound to the fura2) and 66.6 mM Tris base (to alkalinize the medium to ~pH8.5 for increased affinity of the Ca-EGTA complex); this results in a minimum Ca2+-free fura2 fluorescence signal (Fmin). The resulting Fmax and Fmin signals were used to calibrate in the intracellular fura2 fluorescence signals (F) recorded prior to permeabilization according the equation: [Ca2+] = 224 nM X (F-Fmin)/(Fmax – F), where 224 nM is the Kd for the Ca2+-fura2 complex in the [Mg2+], pH and ionic strength that defines the cytosolic milieu. The calculated [Ca2+] values corresponding to the intracellular fura2 fluorescence were used to define the y-axes on the representative SDF-1α induced fura2 fluorescence transients illustrated in the figures.
125I labelled SDF-1α binding assays
As described previously (1), CEM X4/R5 cells were washed and resuspended in CAB. One million cells in a final volume of 300 µl were aliquoted into microfuge tubes and supplemented with 0.1 nM 125I-SDF-1α (PerkinElmer NEN). For analysis of saturation binding (Figure 2), unlabeled SDF-1α (0–30 nM) was also included plus or minus 5 µg/ml hBD-3. Cell suspensions were incubated at 37°C for 20 min and rapidly centrifuged to pellet the cells. Following aspiration of the supernatant, the cell pellets were washed by resuspension in PBS and recentrifugation. 125I-labeled SDF-1α in the final washed cell pellet was quantified by the γ-counting. For analysis of high-affinity SDF-1 binding in the presence of hBD-3 or hBD-3 mutants (Figures 3H, 4G, 5E, 6G, 7E, 8E, 9G), CEM cells were incubated for 20 min with 0.1 nM 125I-SDF-1α in the absence or presence of 20µg/ml wildtype hBD-3, mutant hBD-3, or 10 nM unlabeled SDF-1α prior to sedimentation, washing, and quantification by γ-counting. For each experiment, the amount of SDF-1α bound to the cells in the presence of hBD-3 mutants was normalized to the amount bound in the absence of any competing ligands (as 100%) and compared to the blocking efficacies of wildtype hBD-3 or saturating unlabeled SDF-1α. All experiments were performed in triplicate.
Supplementary Material
Acknowledgements
This study is supported by NIH/NIDCR PO1DE019759 (AW) and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (JL).
Abbreviation list
- HBD-3
human β-defensin-3
- SDF-1α
stromal derived factor 1α. For the whole list of hBD-3 mutants, please see Table S1
References
- 1.Feng Z, Dubyak GR, Lederman MM, Weinberg A. J Immunol. 2006;177:782–786. doi: 10.4049/jimmunol.177.2.782. [DOI] [PubMed] [Google Scholar]
- 2.McCormick TS, Weinberg A. Periodontol. 2010;54:195–206. doi: 10.1111/j.1600-0757.2010.00373.x. 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawsar HI, Weinberg A, Hirsch SA, Venizelos A, Howell S, Jiang B, Jin G. Oral Oncology. 2009;45:696–702. doi: 10.1016/j.oraloncology.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Jin G, Kawsar HI, Hirsch SA, Zeng C, Jia X, Feng Z, Ghosh SK, Zheng QY, Zhou A, McIntyre TM, Weinberg A. PLoS One. 2010;5:e10993. doi: 10.1371/journal.pone.0010993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rohrl J, Yang D, Oppenheim JJ, Hehlgans T. J Immunol. 2010;184:6688–6694. doi: 10.4049/jimmunol.0903984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Funderburg N, Lederman MM, Feng Z, Drage MG, Jadlowsky J, Harding CV, Weinberg A, Sieg SF. Proc Natl Acad Sci U S A. 2007;104:18631–18635. doi: 10.1073/PNAS.0702130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinones-Mateu ME, Lederman MM, Feng Z, Chakraborty B, Weber J, Rangel HR, Marotta ML, Mirza M, Jiang B, Kiser P, Medvik K, Sieg SF, Weinberg A. Aids. 2003;17:F39–F48. doi: 10.1097/00002030-200311070-00001. [DOI] [PubMed] [Google Scholar]
- 8.Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ, Zhang J, Ratajczak J, Ratajczak MZ. J Mol Histol. 2004;35:233–245. doi: 10.1023/b:hijo.0000032355.66152.b8. [DOI] [PubMed] [Google Scholar]
- 9.Horuk R. Nature. 1998;393:524–525. doi: 10.1038/31116. [DOI] [PubMed] [Google Scholar]
- 10.Lazarini F, Tham TN, Casanova P, Arenzana-Seisdedos F, Dubois-Dalcq M. Glia. 2003;42:139–148. doi: 10.1002/glia.10139. [DOI] [PubMed] [Google Scholar]
- 11.Lataillade JJ, Domenech J, Le Bousse-Kerdiles MC. Eur Cytokine Netw. 2004;15:177–188. [PubMed] [Google Scholar]
- 12.Harder J, Bartels J, Christophers E, Schroder JM. J Biol Chem. 2001;276:5707–5713. doi: 10.1074/jbc.M008557200. [DOI] [PubMed] [Google Scholar]
- 13.Huang X, Shen J, Cui M, Shen L, Luo X, Ling K, Pei G, Jiang H, Chen K. Biophys J. 2003;84:171–184. doi: 10.1016/S0006-3495(03)74840-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kofuku Y, Yoshiura C, Ueda T, Terasawa H, Hirai T, Tominaga S, Hirose M, Maeda Y, Takahashi H, Terashima Y, Matsushima K, Shimada I. J Biol Chem. 2009;284:35240–35250. doi: 10.1074/jbc.M109.024851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai SH, Tan Y, Ren XD, Li XH, Cai SX, Du J. Acta Pharmacol Sin. 2004;25:152–160. [PubMed] [Google Scholar]
- 16.Harder J, Bartels J, Christophers E, Schroder JM. Nature. 1997;387:861. doi: 10.1038/43088. [DOI] [PubMed] [Google Scholar]
- 17.Wu Z, Hoover DM, Yang D, Boulegue C, Santamaria F, Oppenheim JJ, Lubkowski J, Lu W. Proc Natl Acad Sci U S A. 2003;100:8880–8885. doi: 10.1073/pnas.1533186100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zlotnik A. J Pathol. 2008;215:211–213. doi: 10.1002/path.2350. [DOI] [PubMed] [Google Scholar]
- 20.Fenyo EM, Schuitemaker B, Asjo B, McKeating J. The History of HIV-1 Biological Phenotypes Past, Present and Future. Los Alamos: Theoretical Biology and Biophysics Group; 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.