

Abstract
The accumulation of cross-β-sheet amyloid fibrils is the hallmark of amyloid diseases. Recently, we reported the discovery of amyloid disaggregase activities in extracts from mammalian cells and Caenorhabditis elegans. However, we have discovered a problem with the interpretation of our previous results as Aβ disaggregation in vitro. Here, we show that Aβ fibrils adsorb to the plastic surface of multiwell plates and Eppendorf tubes. This adsorption is markedly increased in the presence of complex biological mixtures subjected to a denaturing air-water interface. The time-dependent loss of thioflavin T fluorescence that we interpreted previously as disaggregation is due to increased adsorption of Aβ amyloid to the surfaces of multiwell plates and Eppendorf tubes in the presence of biological extracts. As the proteins in biological extracts denature over time at the air-water interface due to agitation/shaking, their adsorption increases, in turn promoting adsorption of amyloid fibrils. We delineate important control experiments that quantify the extent of amyloid adsorption to the surface of plastic and quartz containers. Based on the results described in this article, we conclude that our interpretation of the kinetic fibril disaggregation assay data previously reported in Bieschke et al., Protein Sci 2009;18:2231–2241 and Murray et al., Protein Sci 2010;19:836–846 is invalid when used as evidence for a disaggregase activity. Thus, we correct the two prior publications reporting that worm or mammalian cell extracts disaggregate Aβ amyloid fibrils in vitro at 37°C (see Corrigenda in this issue of Protein Science). We apologize for misinterpreting our previous data and for any confounding experimental efforts this may have caused.
Keywords: amyloid, fibril, amyloid disaggregation, Abeta, Alzheimer's disease, amyloid adsorption, thioflavin T
Broader Impact
In previous work, we provided evidence that Caenorhabditis elegans and mammalian extracts disaggregate and degrade amyloid fibrils in vitro at 37°C. Here, we show that amyloid fibril adsorption to plastic surfaces is enhanced in the presence of biological extracts undergoing air-water interface denaturation, a phenomenon that led us to misinterpret adsorption of amyloid as disaggregation. We also describe important quantitative control experiments for handling amyloid fibrils in multiwell plates and Eppendorf tubes.
Introduction
Many degenerative disorders, such as Alzheimer's disease and the transthyretin amyloidoses, appear to be caused by the misassembly of protein(s) into aggregates, including amyloid fibrils.1–5 Amyloid diseases are named after the amyloid fibril deposits exhibiting a cross-β-sheet structure that are found in patients afflicted with these maladies.6–8 The high stability of the amyloid quaternary structure renders amyloid difficult to degrade.9,10 It is likely that macrophages and similar cells can take up amyloid fibrils into their lysosomes for degradation by a combination of acid denaturation and lysosomal protease cleavage.11,12 Yeast are able to disaggregate amyloid fibrils through the actions of the ATP-dependent chaperone Hsp104, a protein that is not found in metazoans.13,14 We and others are actively searching for analogous mammalian disaggregase machinery, because a disaggregase activity, if it exists, should be useful for clearing intracellular and possibly extracellular protein aggregates, including amyloid fibrils.14–20
Recently, we published that mammalian cell post-nuclear supernatant (PNS) and Caenorhabditis elegans post-debris supernatant (PDS) possess machinery capable of amyloid disaggregation at 37°C, independent of proteolysis activities.15,21,22 We characterized the disaggregase activity described in these articles in 96-well plates using an Aβ amyloid fibril kinetic disaggregation assay. In this assay, thioflavin T (ThT), a dye that exhibits an increase in fluorescence quantum yield and red-shifted emission maximum when bound to amyloid fibrils,23 is used to monitor the amount of amyloid present; a decrease in ThT fluorescence was interpreted as a decrease in amyloid.
We now report that the disaggregase activity detected in PDS of nematodes and PNS of human cells and mouse cells/tissues at 37°C was a misinterpretation of our data. The decrease in ThT fluorescence is caused by time-dependent adsorption of Aβ fibrils to the walls of 96-well plates, a process that is exacerbated by time-dependent denaturation and adsorption of proteins in C. elegans or mouse extracts to the plate walls. The ThT fluorescence signal decreases with time because the fluorescent ThT-Aβ amyloid fibril complex is removed from the light path of the plate reader excitation beam by co-adsorption with denatured murine or C. elegans proteins to the plate walls. This denaturation likely occurs at the air-water interface as a function of time and as a consequence of plate shaking before each fluorescence reading.
Herein, we systematically explore the tendency of amyloid fibrils to adsorb to the plastic surfaces of standard 96-well plates, Eppendorf tubes, and even quartz spectroscopy cells. We quantify the extent to which Aβ fibril adsorption is exacerbated by the presence of complex protein/lipid/carbohydrate/nucleic acid mixtures that are subjected to denaturation by an air-water interface as a consequence of agitation/shaking. We present important control experiments to quantify the extent of this fibril adsorption phenomenon that should become part of each aggregation or disaggregation experiment performed. It is our strong recommendation that disaggregation and aggregation assays performed in the presence of biological extracts be carried out in containers with verified low-binding surfaces. Verification requires quantification of amyloid adsorption to the surfaces of containers used in these types of experiments, both in the presence and absence of biological extracts.
Results and Discussion
We studied the apparent disaggregation of Aβ1–40 amyloid fibrils by cellular and tissue extracts in our previous papers by monitoring the decrease in ThT fluorescence over time at 37°C.15,21,22 In those studies, and in the current study, chemically synthesized Aβ1–40 was aggregated into amyloid fibrils in vitro as the starting point for this assay. Aβ amyloidogenesis was accomplished by incubating Aβ1–40 monomers (173 µg/mL; 40 µM) in aggregation buffer (50 mM sodium phosphate, pH 7.4, 150 mM NaCl, 0.02% NaN3) at 37°C for 4 days with rotary agitation (24 rpm). The aggregates were classified as amyloid fibrils by their ThT binding-associated fluorescence, their Congo red (CR) binding and associated absorbance changes [Fig. 1(A)], and their structure by electron microscopy [Fig. 1(B)]. Preformed Aβ1–40 amyloid fibrils were sonicated for 30 min to afford a uniform fibril length distribution of 50 to 100 nm just before they were added to each well of a 96-well clear-bottom plate (Costar #3631, referred to as standard plates) in aggregation buffer. Amyloid fibrils in standard plates were incubated in the absence or presence of worm PDS or mouse brain PNS in aggregation buffer + 20 µM ThT at a final volume of 100 µL/well. Plates were incubated at 37°C for 2 days, and ThT fluorescence (excitation at 440 nm, emission at 485 nm) was monitored in a Spectramax Gemini EM plate reader every 10 min following 5 s of shaking. The ThT fluorescence of the Aβ fibrils decreased over time in this apparent kinetic disaggregation assay in the presence of C. elegans PDS [Fig. 1(C)] or mouse brain PNS at 37°C [Supporting Information Fig. S1(A)].15,21,22 After 2 days, the amount of amyloid fibrils in solution for samples containing PDS or PNS was less than samples with buffer alone, as measured by atomic force microscopy, Western blot analysis, reversed phase high performance liquid chromatography (RP-HPLC), and filter retention assays.15,21,22
Figure 1.
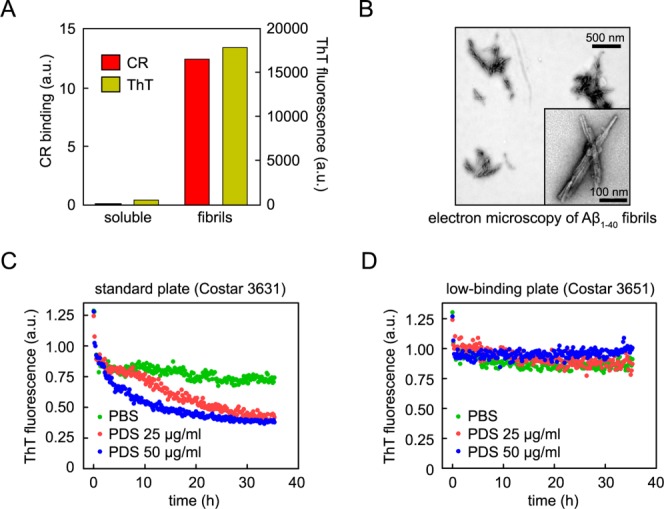
Apparent disaggregase activity of PDS depends on 96-well plate surface properties. (A and B) Characterization of the amyloid properties of the Aβ1–40 fibrils used in the disaggregation kinetic assay, as analyzed by ThT and CR binding (A) and electron microscopy (B). (C and D) Preaggregated, ThT-labeled Aβ1–40 fibrils were incubated with PBS buffer (green) or worm PDS at 25 µg/mL (red) or 50 µg/mL (blue), and the apparent disaggregase activity was monitored as a function of the fluorescence of ThT using standard (C) or low-binding (D) plates. The apparent disaggregase activity was present in standard plates and absent in low-binding plates.
Chromatographic fractionation of PNS or PDS, coupled with the ThT-based Aβ1–40 amyloid fibril kinetic disaggregation assay analysis, allowed us to identify fractions with apparent disaggregase activity. The protein(s) responsible for the apparent disaggregation activity in each fraction were then identified by Multidimensional Protein Identification Technology24,25 (Ya-Juan Wang, John Yates unpublished results). Purified recombinant versions of these candidate disaggregase proteins were subsequently studied using our ThT-based Aβ1–40 amyloid fibril kinetic disaggregation assay. We started to become suspicious when most of these candidates exhibited apparent disaggregase activity. In the process of these experiments, an ordering error occurred in which we purchased low-binding 96-well plates (Costar #3651) instead of the standard plates (Costar #3631). Using these low-binding 96-well plates, we discovered that the reduction in ThT fluorescence that had been attributed to disaggregase activity of PDS or PNS was no longer observed [Fig. 1(D), Supporting Information S1(B)]. This led us to suspect that the reduction in ThT fluorescence observed in standard 96-well plates is due to adsorption of the Aβ1–40 fibril-ThT complex to the well surfaces of the standard plates. With a 100 µL sample, the surface area of the sides of the well exposed to the Aβ1–40 amyloid fibrils is two times that of the bottom of the well. An even greater surface area is exposed upon shaking for 5 s every 10 min in the plate reader. This would allow at least twice as much Aβ1–40 to be adsorbed to the sides as to the bottom of the well, leading to the observed decrease in ThT fluorescence from the volume excited by the plate reader laser through the bottom of the well.
To address whether the reduction in ThT fluorescence observed herein [Fig. 1(C)] and in previous Aβ1–40 amyloid fibril kinetic disaggregation assays15,21,22 is due to disaggregation followed by proteolysis or due to adsorption of Aβ1–40 to the walls of the plate at 37°C, we incubated Aβ1–40 amyloid fibrils in the presence of PDS at different concentrations (25 and 50 µg/mL) for 35 h with 5 s of plate shaking every 10 min before each fluorescence reading. As a control, we used fibrils incubated in PBS buffer for 35 h. We processed the samples at the end of the 35 h time course as described in the scheme at the top of Figure 2. For the solution samples (S), the solution was removed from each well and placed in Eppendorf tubes to which guanidine HCl (GndCl) powder was added to a final concentration of 8M. The wall wash samples (W) were prepared by adding a solution of 8M GndCl to the empty wells to dissolve adsorbed Aβ1–40 fibrils. Both the S-sample tubes and the W-sample plates were sonicated in a Fisher Scientific FS60 Sonic Cleaner for 1 h to monomerize any Aβ1–40 fibrils or oligomers present. Samples were then analyzed by RP-HPLC [Fig. 2(A)] and by Western blotting [Fig. 2(B)] to quantify Aβ1–40. For the Aβ fibrils incubated with PBS, the majority of the Aβ1–40 was in solution [Fig. 2(A,B) Solution (S)]. Conversely, for the Aβ fibrils incubated with PDS, the majority of the Aβ1–40 was adsorbed to the walls of the plate wells [Fig. 2(A), B Wash (W)]. Experiments performed with mouse brain PNS afforded analogous results [Supporting Information Fig. S1(A), right panel]. Notably, much less Aβ1–40 from fibrils incubated with PNS was adsorbed to the walls of low-binding plates [Supporting Information Fig. S1(B), right panel]. This is in agreement with the finding that ThT fluorescence in the Aβ1–40 disaggregation assay did not decrease in the presence of PDS [Fig. 1(D)] or PNS [Supporting Information Fig. S1(B), left panel] in the low-binding plates. Interestingly, as can be seen in Supporting Information Figure S1(B), HPLC quantification shows a slight loss in total Aβ1–40 (S + W) incubated with PNS without a loss in ThT-bound Aβ1–40, indicating that nonfibrillar Aβ1–40 is proteolyzed whereas fibrillar Aβ1–40 is resistant to PNS-mediated proteolysis. Aβ1–40 adsorption in standard plates increases as a function of time over a 48 h time course [Supporting Information Fig. S1(C)], which parallels the loss in ThT fluorescence over time.
Figure 2.
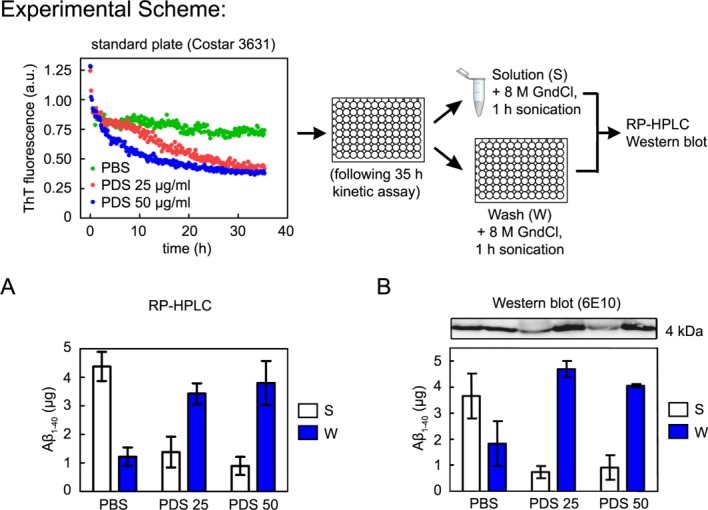
Loss of ThT fluorescence corresponds to Aβ1–40 adsorption to the walls of plate wells. Aβ1–40 fibrils were incubated in the presence of 25 or 50 µg/mL worm PDS (PDS 25 and PDS 50, respectively), or PBS alone, as described in Figure 1(C). The Solution was removed from each well, and guanidine HCl powder was added to Solution samples to a final concentration of 8M; 8M guanidine HCl was then added to each well. Both sets of samples, Solution and Wash, were sonicated for 1 h before being analyzed by RP-HPLC or Western blot. For Western blot the samples were diluted ×100 in 2% SDS and boiled for 10 min. (A) RP-HPLC quantification of Aβ1–40 in Solution (S) and in Wash (W). A significant amount of Aβ1–40 adsorbs to the well walls in the absence of PDS. Adsorption is enhanced in the presence of PDS. (B) The same profile is observed when the quantification of Aβ1–40 is performed by Western blot using a specific Aβ antibody (6E10). Error bars indicate standard deviations of three independent experiments using different Aβ1–40 fibril preparations.
Collectively, these data suggest that the reduction in ThT fluorescence observed in the Aβ1–40 amyloid fibril kinetic assay conducted in standard plates with PNS or PDS (complex biological samples) was misinterpreted as a disaggregase activity. In reality, the time-dependent decrease in the ThT signal attributed to disaggregation is due to adsorption of Aβ1–40 fibrils to the hydrophobic walls of standard plate wells, which increases in the presence of complex biological extracts as a function of time. This is most likely a consequence of denaturation of proteins present in biological extracts occurring at the air-water interface resulting from the 5 s of shaking that occurs every 10 min. While we were previously aware that a minor amount of Aβ1–40 fibrils adsorbs to the walls, what we had not considered was that this would increase as a function of incubation time in the presence of complex biological samples. We hypothesize that time-dependent protein denaturation facilitates increased Aβ1–40 fibril adsorption, explaining the incorrectly interpreted disaggregation time courses. (Lipids and poly-anions could also play a role.)
We previously quantified the amount Aβ1–40 present in solution at the end of the kinetic disaggregation assay in the presence versus the absence of Roche Protease Inhibitor Cocktail (PIC) and found more Aβ1–40 to be present with protease inhibition.15,21 We attributed a decrease of Aβ1–40 in the absence of PIC to proteolysis by proteases in biological extracts. For samples incubated with biological extract and PIC, we saw a decrease in ThT but little loss of Aβ1–40 in solution, as measured by RP-HPLC. We interpreted this as a decoupling of disaggregation from proteolysis activities. However, we now realize that much of the decrease in Aβ1–40 present in solution is a result of adsorption rather than proteolysis. We believe that our HPLC quantification of total Aβ1–40 in previous studies was confounded by components of the Roche PIC, which have similar HPLC elution profiles to that of the Aβ1–40 peptide. In our original studies, we subtracted the HPLC absorbance curve of Roche PIC alone from those of fibrils + Roche PIC + PDS/PNS to take into account any peak area contribution from the Roche PIC. Despite this precaution, it is likely that something present in the Roche PIC gave us a false indication that we had recovered more Aβ1–40 than was actually retrieved, causing us to incorrectly interpret that we were decoupling perceived disaggregation from perceived proteolysis.
To further scrutinize the biological extract induced Aβ1–40 fibril adsorption hypothesis, we glutaraldehyde cross-linked the proteins present in PDS, which would disable protein-mediated activities found within the PDS. We observed a similar loss of ThT fluorescence [Fig. 3(A)] and a substantial amount of Aβ1–40 adsorbed to the walls of the wells [Fig. 3(B)], irrespective of PDS protein cross-linking. These results corroborate the artifactual nature of the apparent ThT-based Aβ1–40 amyloid fibril kinetic disaggregase activity.
Figure 3.

The apparent disaggregase activity remains after cross-linking of PDS proteins. (A) Aβ1–40 fibrils were incubated in the absence (green) or presence of non-cross-linked (blue) or cross-linked (orange) PDS. After chemical cross-linking, the PDS retains apparent disaggregase activity. Inset: SDS-PAGE and silver stain of worm PDS before (−) and after (+) cross-linking. The strong high molecular weight band indicates cross-linking of PDS proteins by glutaraldehyde. (B) Western blot quantification of Aβ1–40 adsorption for samples from (A) shows that adsorption is unaffected by PDS cross-linking. Error bars indicate standard deviations of three independent experiments using different Aβ1–40 fibril preparations.
To address whether the Aβ1–40 adsorbed to the walls of standard 96-well plates retains its amyloid fibril architecture, we carried out a ThT-based Aβ1–40 amyloid fibril kinetic disaggregation assay for 35 h and then sonicated the plates for 1 h. This was sufficient to recover the majority of the starting ThT fluorescence from PDS-treated fibrils, suggesting that intact amyloid fibrils bound to ThT were adsorbed to the walls of the plate wells and that these were resolubilized by sonication (Fig. 4). Analogous resolubilization of Aβ1–40 fibrils by sonication was observed after the ThT-based Aβ1–40 amyloid fibril kinetic disaggregation assay was performed in the presence of mouse brain PNS (Supporting Information Fig. S2). Collectively, these results suggest that intact amyloid fibrils bound to ThT adsorb to the walls of the plate wells in the presence of C. elegans PDS or mouse brain PNS, providing strong evidence that significant disaggregation is not occurring.
Figure 4.
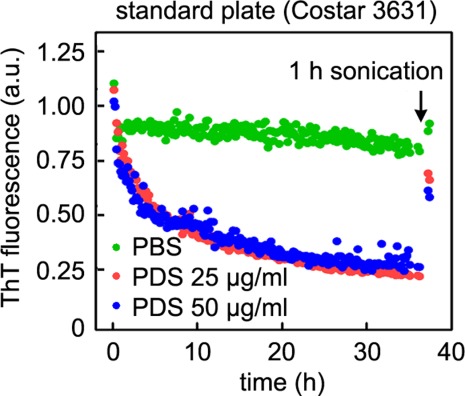
PDS facilitates the adsorption of intact amyloid fibrils to plate well walls. Aβ1–40 fibrils were incubated in the absence (green) or in the presence of 25 or 50 µg/mL PDS (red and blue, respectively), as described in Figure 1(C). After 35 h of incubation, the plate was sonicated for 1 h, and the ThT fluorescence measured again. Most of the ThT fluorescence of PDS samples is recovered, indicating that Aβ fibrils are adsorbed to the well walls.
In order to evaluate whether the adsorption of Aβ1–40 amyloid fibrils to plastic surfaces in the presence of complex biological extracts is restricted to 96-well plates, we performed experiments using Eppendorf tubes (Axygen # MCT-175-C) (Fig. 5). Aβ1–40 amyloid fibrils were incubated in the absence or presence of PDS at 37°C for 35 h in aggregation buffer with rotary agitation at 24 rpm. Congo red (CR) was then added to a final concentration of 50 µM, and the samples were incubated for 30 min with rotary agitation at 24 rpm. The samples were spun at 16,000g for 30 min, and the supernatant was discarded [Fig. 5(A)]. A red pellet (Aβ amyloid fibrils) was observed in samples with no PDS added, whereas no red pellet was observed in the samples incubated with 25 µg/mL PDS, even in low-binding Eppendorf tubes (Costar #3207). Instead, we observed extensive CR staining of the walls of the Eppendorf tubes when C. elegans PDS was present. The CR pellet, along with any CR adsorbed to the tube walls, was dissolved in DMSO [Fig. 5(B)], and CR absorbance was measured at 530 nm [Fig. 5(C)]. (Background CR absorbance for buffer alone and for buffer + PDS was subtracted from signals reported for fibrils and fibrils + PDS, respectively). The same CR signal was present for Aβ1–40 fibrils incubated in the absence or presence of PDS; the roughly equal amounts of CR recovered came mostly from the pellet for samples without PDS and mostly from the walls for samples with PDS. The low-binding tubes did not prevent adsorption of amyloid fibrils. These results demonstrate that, as in the case of the standard 96-well plates, the presence of PDS facilitates the adsorption of CR-binding Aβ1–40 amyloid fibrils onto the walls of the plastic surface of Eppendorf tubes.
Figure 5.
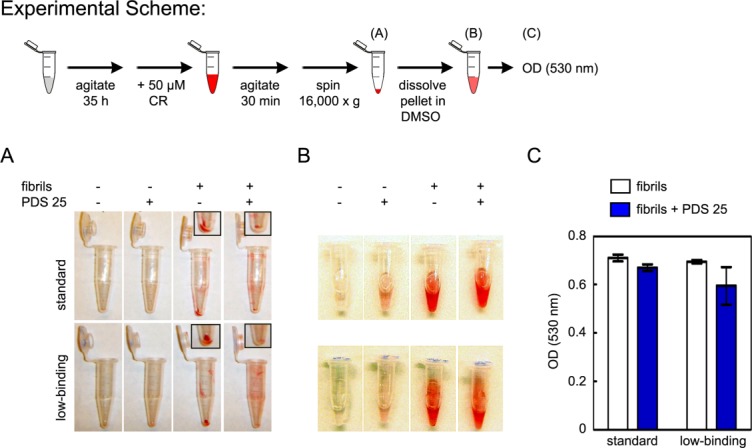
PDS facilitates Aβ1–40 fibril adsorption in Eppendorf tubes. Aβ1–40 amyloid fibrils were rotary agitated (24 rpm) at 37°C in the absence or presence of 25 µg/mL PDS. Control tubes contained buffer alone or 25 µg/mL PDS alone, without Aβ1–40 fibrils. After 35 h incubation, CR was added to a final concentration of 50 µM, and the samples were rotary agitated for 30 min at 37°C to allow CR binding to the amyloid fibrils. The samples were spun at 16,000g for 30 min in order to obtain a pellet composed of amyloid fibrils bound to CR (A). The Eppendorf tubes were washed with DMSO in order to dissolve the CR (B) and the absorbance was measured at 530 nm (C). (A) After centrifugation, Aβ1–40 fibrils + CR form a pellet in both standard and low-binding tubes (Column 3). In the presence of PDS, Aβ1–40 fibrils + CR do not form a pellet in either standard or low-binding tubes (Column 4). The absence of pellet in these samples can be explained by Aβ1–40 adsorption to the walls of the tubes. Inset: magnification of pellets. (B) Tubes containing adsorbed/pelleted Aβ1–40 + CR dissolved in DMSO. (C) Spectroscopic quantification of CR from (B) shows equal amounts of CR in samples incubated in the absence or presence of PDS; thus, adsorbed Aβ species are CR-competent fibrils. Error bars indicate standard deviations of three independent experiments using different Aβ1–40 fibril preparations.
ThT fluorescence of Aβ1–40 fibrils in the kinetic disaggregation assay in standard plates does not significantly decrease in the presence of a highly stable protein like RNase A (Sigma-Aldrich #R6513) (Fig. 6). This suggests that, as is the case with PNS and PDS, loss of ThT fluorescence correlates with Aβ1–40 adsorption to a denatured protein and not with a disaggregation activity. To explore this further, we incubated Aβ1–40 fibrils with RNAse A ± β-mercaptoethanol (BME), which destabilizes RNase A by reducing its disulfide bonds.26 Incubation with destabilized RNase A, unlike incubation with properly-folded RNase A, leads to a decrease in ThT fluorescence (Fig. 6). Thus, we hypothesize that denatured or partially denatured proteins with exposed hydrophobic residues adsorb to hydrophobic surfaces, increasing hydrophobic surface area and facilitating Aβ1–40 adsorption.
Figure 6.
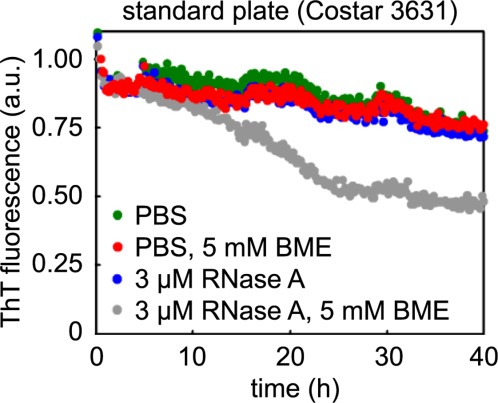
Incubation of Aβ1–40 fibrils with RNase A destabilized by reducing agent leads to loss of ThT fluorescence. Aβ1–40 fibrils (preaggregated from 20 µM monomerized Aβ1–40) were incubated with PBS or 3 µM RNase A ± 5 mM β-mercaptoethanol (BME). Only RNase A + BME (grey), which reduces its disulfide bonds to destabilize the protein, leads to loss of fibril ThT fluorescence.
An air-water interface is present in all our experiments, and it is well established that protein denaturation occurs at amphiphilic air-water interfaces, especially upon agitation.27–32 We propose that the proteins present in the C. elegans PDS or mammalian PNS are slowly denatured at the air-water interface and then adsorb to the surface of Eppendorf tubes or standard 96-well plates, leading to adsorption of amyloid fibrils and an apparent decrease in ThT fluorescence as a function of time. To monitor protein denaturation in the kinetic assay, we used Sypro Orange, which fluoresces strongly when bound to hydrophobic regions of denatured or partially denatured proteins.33 Incubation of C. elegans PDS alone in low-binding plates for 20 h with agitation for 5 s every 10 min resulted in an increase in Sypro Orange fluorescence over time [Supporting Information Fig. S3(A)], indicating protein denaturation. To test whether the denatured proteins in PDS adsorb to the walls of Eppendorf tubes, we incubated buffer or PDS for 35 h with rotary agitation (24 rpm) at 37°C. Coomassie blue was then added to the samples, which were agitated for an additional 30 min. Samples were centrifuged at 16,000g for 30 min, and supernatants were decanted. The walls of the tubes incubated with PDS were more strongly stained with Coomassie blue than tubes incubated with buffer, indicating PDS protein adsorption [Supporting Information Fig. S3(B)]. To measure the amount of wall-adsorbed PDS in 96-well plates, we incubated PDS (50 µg/mL) in both standard and low-binding plates for 35 h at 37°C, with shaking every 10 min for 5 s. We then followed the same experimental scheme outlined in Figure 2, using 8M urea to solubilize the adsorbed protein instead of 8M GndCl, as GndCl interferes with the bicinchonic acid assay (BCA) for protein quantification. We observed that roughly 25% of the PDS proteins adsorb to the walls of standard plates, whereas less than 5% absorb to low binding plates [Supporting Information Fig. S3(C)] after 35 h.
To assess whether air-water interface protein denaturation, which increases upon agitation, is required for Aβ1–40 fibril adsorption, we performed the Aβ1–40 kinetic disaggregation assay in standard 96-well plates in the absence of agitation. There is no decrease in ThT fluorescence in the absence of agitation, indicating no Aβ1–40 wall adsorption [Fig. 7(A)]. The increase in ThT fluorescence is likely due to sedimentation of larger aggregates in the bottom of wells in the fluorescence read volume. To explore the requirement for agitation further, we incubated Aβ1–40 fibrils with PDS (25 µg/mL) or with disaggregation assay buffer in standard Eppendorf tubes without or with rotary agitation. We then decanted the solution to a quartz cuvette, added ThT (25 µM), and measured ThT fluorescence as an indication of the amount of soluble amyloid in the supernatant. We added fresh PBS to the empty Eppendorf tubes, sonicated them for 1 h to facilitate Aβ1–40 fibril desorption, added ThT to these adsorbed samples, and measured ThT fluorescence in quartz cuvettes. Only those samples subjected to agitation exhibited a decrease in ThT fluorescence in solution and an increase in ThT fluorescence of the Aβ1–40 adsorbed samples [Fig. 7(B)]. Moreover, adsorption of Aβ1–40 fibrils was increased by the presence of PDS only upon rotary agitation [Fig. 7(B)], supporting our hypothesis that a denaturing air-water interface facilitates adsorption of proteins present in biological extracts and Aβ1–40 amyloid fibrils. We subjected the Aβ1–40 adsorbed to the walls of Eppendorf tubes during rotary agitation in the presence of PDS to sonication in PBS to dislodge the fibrils, which were then analyzed by electron microscopy to identify the type of Aβ1–40 species adsorbed. We found that the adsorbed Aβ1–40 was indeed fibrillar and not disaggregated [Fig. 7(B), inset]. These results support our model that denatured proteins present in complicated organismal extracts like PDS and PNS adsorb to hydrophobic surfaces. This in turn allows more Aβ1–40 fibril adsorption to the well walls or tube surfaces (Fig. 8), which we incorrectly interpreted as disaggregation and proteolysis.15,21,22
Figure 7.
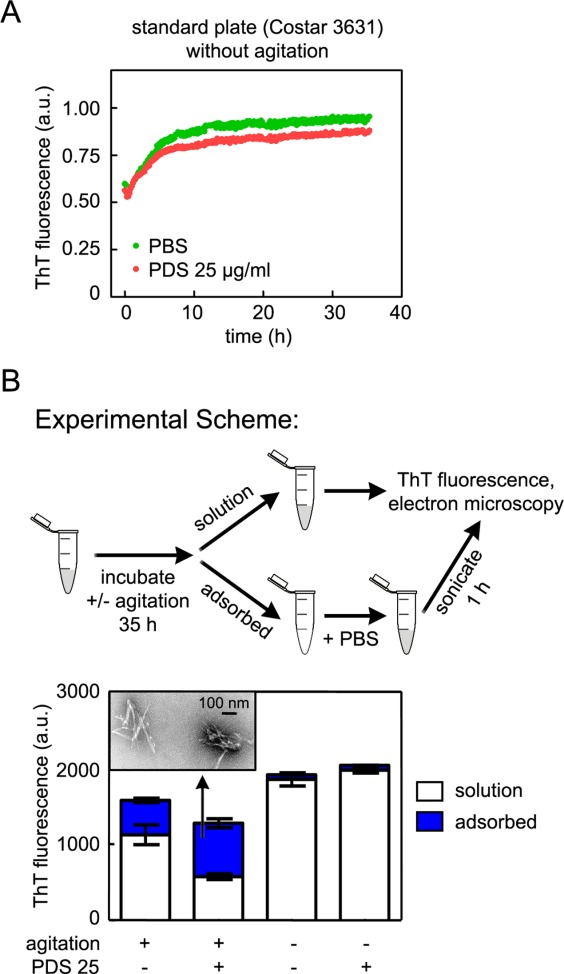
A denaturing air-water interface, created by rotary agitation, facilitates the adsorption of fibrillar, ThT-reactive Aβ1–40. (A) Aβ1–40 fibrils were incubated in the absence (green) or in the presence of 25 µg/mL PDS (red) in standard 96-well plates without shaking. There is no discernible Aβ1–40 wall adsorption in standard plates in the absence of agitation. (B) Aβ1–40 amyloid fibrils were incubated quiescently or with rotary agitation (24 rpm) at 37°C in the absence or presence of 25 µg/mL PDS. After 35 h, the solution was removed from each sample, and fresh buffer was added to each tube. Tubes were then sonicated for 1 h to dislodge adsorbed Aβ1–40. ThT was added to Solution and Adsorbed samples, and fluorescence was measured. ThT fluorescence was observed in Adsorbed agitated samples but not Adsorbed quiescent samples, indicating that adsorption of Aβ fibrils only occurred in the presence of agitation. PDS increased the amount of Adsorbed fibrils in agitated samples. The adsorbed Aβ1–40 in the presence of agitation and PDS is fibrillar, by electron microscopy (inset).
Figure 8.

PDS and PNS facilitate the adsorption of Aβ1–40 amyloid fibrils to plastic surfaces. (A) In standard 96-well plates or Eppendorf tubes, a small fraction of Aβ1–40 fibrils adsorb to the plastic surface. (B) This adsorption is enhanced in the presence of certain proteins. The agitation and constant contact with the air/water interface induces protein denaturation, enhancing binding of the denatured proteins to the plastic surface. These proteins then drive the adsorption of the Aβ1–40 fibrils to the plastic surface, probably via hydrophobic interactions.
To date, the only eukaryotic protein known to disaggregate amorphous aggregates and amyloid fibrils in vitro and in vivo is the yeast chaperone Hsp104.34,35 Hsp104 appears to fragment Sup35 amyloid and Ure2 yeast prion fibrils within 30 min,36 a time frame drastically shorter than the 35 h employed in our search for an amyloid disaggregase activity15,21,22 [Fig. 1(C)]. No metazoan homolog or analog of Hsp104 has been identified thus far.13 No activity capable of refolding aggregated luciferase has been detected in fractionated extracts of human cells; however, human cells transfected with yeast Hsp104 are able to refold luciferase.37 Recently, Shorter showed that mammalian Hsp110 synergizes with Hsp70 and Hsp40 to promote disaggregation of amorphous protein but not alpha-synuclein amyloid or the yeast Sup35 prion.14 The search for functional equivalents of Hsp104 continues, but to date no amyloid disaggregase has been identified in metazoa.
Conclusions
We apologize to the scientific community for our serious mistake of misinterpreting a decrease in ThT fluorescence in the presence of biological extracts as Aβ1–40 fibril disaggregation. We did not realize that the C. elegans and mammalian proteins denatured at the air-water interface over the course of our assay mediate an increase in adsorption of Aβ1–40 fibrils in standard multiwell plates (Fig. 8). It is important to note that other Aβ1–40 species, including oligomers and monomers, may also be adsorbed under these conditions. We recommend that this type of Aβ1–40 amyloid fibril kinetic disaggregation assay monitored by ThT fluorescence be conducted in low-binding 96-well plates, which minimize adsorption. Agitation is not recommended for these experiments, as this will very likely denature proteins that could carry out such activity. Quantification of the amount of adsorbed amyloid fibrils should be conducted with all aggregation and disaggregation assays, especially when additional proteins are present, and even when low-binding plates are used. Furthermore, amyloid adsorption control experiments should be conducted with every new batch of multiwell plates or Eppendorf tubes purchased, as manufacturing differences in low-binding plates or tubes could also lead to protein-mediated absorption of Aβ1–40 fibrils. We even have observed such an adsorption phenomenon in quartz cuvettes. Eppendorf tubes used commonly in aggregation and disaggregation experiments are problematic from the perspective of amyloid fibril adsorption, and it is clear from our experiments that adsorption of amyloid fibrils should be suspected and controlled for. Any introduction of potential hydrophobic surfaces in these experiments, such as polymers, lipids, amphipathic proteins, or denatured proteins, should be carefully controlled for, and all Aβ1–40 should be accounted for.
It is important to stress that our results do not call into question the existence of a disaggregase activity in multicellular organisms. However, the ThT-based Aβ1–40 amyloid fibril kinetic disaggregation assay carried out in standard plates at 37°C is invalid when used as evidence for a disaggregation activity. Based on these results, we correct two Protein Science manuscripts15,21 reporting C. elegans PDS and mammalian cell PNS disaggregase activity (Corrigenda in this issue of Protein Science).
Materials and Methods
Preparation of Aβ1–40 fibrils
Aβ1–40 fibrils were prepared as previously described.15 The Aβ1–40 fibrils were sonicated for 30 min in a Fisher Scientific FS60 Sonic Cleaner. All experiments were performed with at least three different Aβ1–40 amyloid fibrils preparations.
Preparation of C. elegans and mammalian extracts
Wild type worms (N2) were grown at 20°C, and crude extracts were spun at 3000 rpm to obtain postdebris supernatant (PDS), as previously described.21 The brain postnuclear supernatant (PNS) was also prepared as previously described.15 Total protein concentrations were determined using the Pierce BCA assay. In glutaraldehyde cross-linking experiments, PDS was incubated with 0.1% glutaraldehyde for 5 min at 25°C. The reaction was stopped by addition of an excess of Tris-HCl, and cross-linking was verified by SDS-PAGE using pre-cast 4 to 12% gradient gels (Invitrogen #NP0343BOX) and Bio-Rad Silver Stain Plus kit (Bio-Rad #161-0449).
In vitro Aβ1–40 kinetic disaggregation assay
The disaggregation assay was performed as previously described.15,21,22 Briefly, 15 µM sonicated Aβ1–40 fibrils were incubated in the absence or presence of PDS or PNS in aggregation buffer (50 mM sodium phosphate, pH 7.4, 150 mM NaCl, 0.02% NaN3) containing ThT (20 µM) at 37°C. Every 10 min, the plates were shaken for 5 s, and fluorescence (excitation at 440 nm, emission at 485 nm) was monitored using a Spectra Gemini EM fluorescence plate reader.
Western blotting
SDS-PAGE was performed under reducing conditions using 16% tris-tricine gels. The 8M GndCl samples were diluted 100× in PBS with Laemmli buffer and boiled for 10 min. Samples were transferred to nitrocellulose membranes and probed with 6E10 antibody (1:10,000). Blots were then probed with goat anti-mouse secondary antibody conjugated to IRDye 800CW (1:10,000) and developed/quantified using an Odyssey Infrared Imaging System.
Electron microscopy
Copper grids (carbon- and formvar-coated 400 mesh) (Electron Microscopy Sciences, Hatfield PA) were glow discharged and inverted on a 5 µL aliquot of sample for 3 min. Excess sample was removed and the grids immediately placed briefly on a droplet of double distilled water, followed by 2% uranyl acetate solution for 2 min. Excess stain was removed and the grid allowed to dry thoroughly. Grids were then examined on a Philips CM100 electron microscope (FEI, Hillsbrough OR) at 80 kV and images collected using a Megaview III CCD camera (Olympus Soft Imaging Solutions, Lakewood CO).
HPLC quantification
The samples were analyzed by RP-HPLC using a Gemini-NX 5µ C18 column in a water (0.1% NH4OH)/acetonitrile (0.1% NH4OH) gradient and quantified by UV absorption at 220 nm, as previously described.15
Congo red assay
After incubation on a rotary shaker at 37°C for 35 h, a CR solution was added to the samples to give a final concentration of 50 µM. Then, the samples were rotated for 30 min at 37°C and spun at 16,000g for 30 min. The supernatant was removed, and 400 µL DMSO was added to the pellet. Samples were rotated in DMSO for an additional 30 min at 37°C, after which the absorbance of samples was measured spectroscopically at 530 nm. Nonspecific CR binding to the tubes with PBS or PDS without amyloid fibrils was subtracted from the optical density plotted in the graphic presented in Figure 5(C).
Sypro Orange Assay
Sypro Orange (Molecular Probes # S-6650) was diluted to 1×, according to the manufacturers' instructions, in aggregation buffer containing 50 µg/mL of PDS in low-binding plates. As a control, we incubated 1× Sypro Orange in aggregation buffer in the absence of PDS (PBS). Plates were incubated at 37°C with 5 s agitation every 10 min, and fluorescence (excitation at 490 nm, emission at 600 nm) was monitored using a Spectra Gemini EM fluorescence plate reader.
Acknowledgments
The authors thank Dr. Colleen Fearns for critical reading of and assistance with the preparation of this publication.
Glossary
- BME
β-mercaptoethanol
- CR
Congo red
- DMSO
dimethyl sulfoxide
- GndCl
guanidine HCl
- PBS
phosphate buffered saline
- PDS
post-debris supernatant
- PIC
protease inhibitor cocktail
- PNS
post-nuclear supernatant, RP-HPLC, reversed-phase high performance liquid chromatography
- ThT
thioflavin T.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
Figure S1. Apparent disaggregase activity of PNS depends on the type of 96-well plate used for the assay and corresponds to the amount of Aβ1-40 adsorbed to the wells. (A) Incubation with 60 µg/ml mouse brain PNS leads to loss of ThT fluorescence of Aβ1-40 fibrils (15 µM) in standard plates. More Aβ1-40 is lost from Solution and recovered by 8 M GndCl Wash for samples incubated with PNS than for samples containing fibrils and PBS alone, as quantified by RP-HPLC. (B) No loss of ThT fluorescence is observed in low-binding plates, and little Aβ1-40 is lost from Solution or recovered in the 8 M GndHCl Wash for either PBS- or PNS-treated samples. The slight loss in overall amount of Aβ1-40 incubated with PNS is attributed to proteolysis. (C) Time course of adsorption of Aβ1-40 fibrils. Aβ1-40 fibrils were incubated in the absence or in the presence of 50 µg/ml PDS (upper left) or 60 µg/ml mouse brain PNS (lower left). At four time points throughout the incubation (indicated by vertical arrows), samples were processed as described in the experimental scheme of Figure 2. The amount of Aβ1-40 in Solution and Wash was determined by RP-HPLC for PDS (upper right) and PNS (lower right) experiments. Note that even after only 4-5 h incubation in the presence of PDS or PNS, we observed an increase in adsorption of Aβ1-40 to plate wells compared to PBS controls. (Error bars indicate standard deviations of three independent experiments.)
Figure S2. Mouse brain PNS facilitates the adsorption of intact amyloid fibrils to plate wells. Aβ1-40 fibrils (pre-aggregated from 15 µM monomerized Aβ1-40) were incubated in the absence (PBS) or in the presence of 15 or 45 µg/ml mouse brain PNS. After 40 h of incubation, the plate was sonicated for 1 h, and the ThT fluorescence measured again. ThT fluorescence of PNS samples is recovered, indicating that Aβ fibrils are adsorbed to the wells.
Figure S3. Denaturation and adsorption of the C. elegans PDS proteins. (A) Incubation of 50 µg/mL PDS alone, without Aβ1-40 fibrils, with Sypro Orange in standard plates for 22 h with agitation for 5 s every 10 min resulted in increased fluorescence over time, indicating denaturation of PDS proteins. (B) PDS alone, without Aβ1-40 fibrils, was incubated with rotary shaking (24 rpm) in Eppendorf tubes for 35 h at 37°C. The supernatants in the Eppendorf tubes were then discarded and the protein adsorbing to the walls visualized with Coomassie blue. (C) PDS alone, without Aβ1-40 fibrils, was incubated with agitation for 5 s every 10 min in 96-well plates for 35 h. The amount of protein in the Solution (S) and in Wash (W) was quantified by BCA. A significant amount of the PDS adsorbs to the well walls in the standard plates as compared to the low-binding plates.
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Levine H., III . Imaging of misfolded proteins. In: Ramirez-Alvarado M, Kelly JW, Dobson CM, editors. Protein misfolding diseases. Hoboken, NJ: John Wiley & Sons, Inc; 2010. pp. 647–671. [Google Scholar]
- 3.Abedini A, Raleigh DP. Islet amyloid polypeptide. In: Ramirez-Alvarado M, Kelly JW, Dobson CM, editors. Protein misfolding diseases. Hoboken, NJ: John Wiley & Sons, Inc; 2010. pp. 517–541. [Google Scholar]
- 4.Fandrich M, Schmidt M, Grigorieff N. Recent progress in understanding Alzheimer's beta-amyloid structures. Trends Biochem Sci. 2011;36:338–345. doi: 10.1016/j.tibs.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith DP, Ashcroft AE, Radford SE. Hemodialysis-related amyloidosis. In: Ramirez-Alvarado M, Kelly JW, Dobson CM, editors. Protein misfolding diseases. Hoboken, NJ: John Wiley & Sons, Inc; 2010. pp. 347–379. [Google Scholar]
- 6.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer's amyloid-beta(1-42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer's beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenwald J, Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18:1244–1260. doi: 10.1016/j.str.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 10.Smith JF, Knowles TPJ, Dobson CM, MacPhee CE, Welland ME. Characterization of the nanoscale properties of individual amyloid fibrils. Proc Natl Acad Sci USA. 2006;103:15806–15811. doi: 10.1073/pnas.0604035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majumdar A, Capetillo-Zarate E, Cruz D, Gouras GK, Maxfield FR. Degradation of Alzheimer's amyloid fibrils by microglia requires delivery of ClC-7 to lysosomes. Mol Biol Cell. 2011;22:1664–1676. doi: 10.1091/mbc.E10-09-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majumdar A, Chung H, Dolios G, Wang R, Asamoah N, Lobel P, Maxfield FR. Degradation of fibrillar forms of Alzheimer's amyloid beta-peptide by macrophages. Neurobiol Aging. 2008;29:707–715. doi: 10.1016/j.neurobiolaging.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vashist S, Cushman M, Shorter J. Applying Hsp104 to protein-misfolding disorders. Biochem Cell Biol. 2010;88:1–13. doi: 10.1139/o09-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One. 2011;6:e26319. doi: 10.1371/journal.pone.0026319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray AN, Solomon JP, Wang YJ, Balch WE, Kelly JW. Discovery and characterization of a mammalian amyloid disaggregation activity. Protein Sci. 2010;19:836–846. doi: 10.1002/pro.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arimon M, Grimminger V, Sanz F, Lashuel HA. Hsp104 targets multiple intermediates on the amyloid pathway and suppresses the seeding capacity of Abeta fibrils and protofibrils. J Mol Biol. 2008;384:1157–1173. doi: 10.1016/j.jmb.2008.09.063. [DOI] [PubMed] [Google Scholar]
- 17.Ben-Zvi AP, Goloubinoff P. Review: mechanisms of disaggregation and refolding of stable protein aggregates by molecular chaperones. J Struct Biol. 2001;135:84–93. doi: 10.1006/jsbi.2001.4352. [DOI] [PubMed] [Google Scholar]
- 18.Calamai M, Canale C, Relini A, Stefani M, Chiti F, Dobson CM. Reversal of protein aggregation provides evidence for multiple aggregated states. J Mol Biol. 2005;346:603–616. doi: 10.1016/j.jmb.2004.11.067. [DOI] [PubMed] [Google Scholar]
- 19.Helsen CW, Glover JR. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein (Hsp104) J Biol Chem. 2012;287:542–556. doi: 10.1074/jbc.M111.302869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mamikonyan G, Necula M, Mkrtichyan M, Ghochikyan A, Petrushina I, Movsesyan N, Mina E, Kiyatkin A, Glabe CG, Cribbs DH, Agadjanyan MG. Anti-Abeta 1–11 antibody binds to different beta-amyloid species, inhibits fibril formation, and disaggregates preformed fibrils but not the most toxic oligomers. J Biol Chem. 2007;282:22376–22386. doi: 10.1074/jbc.M700088200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bieschke J, Cohen E, Murray A, Dillin A, Kelly JW. A kinetic assessment of the C. elegans amyloid disaggregation activity enables uncoupling of disassembly and proteolysis. Protein Sci. 2009;18:2231–2241. doi: 10.1002/pro.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 23.Levine H. Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., III Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 25.Washburn MP, Wolters D, Yates JR., III Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 26.Klink TA, Woycechowsky KJ, Taylor KM, Raines RT. Contribution of disulfide bonds to the conformational stability and catalytic activity of ribonuclease A. Eur J Biochem. 2000;267:566–572. doi: 10.1046/j.1432-1327.2000.01037.x. [DOI] [PubMed] [Google Scholar]
- 27.Noskov BA, Mikhailovskaya AA, Lin SY, Loglio G, Miller R. Bovine serum albumin unfolding at the air/water interface as studied by dilational surface rheology. Langmuir. 2010;26:17225–17231. doi: 10.1021/la103360h. [DOI] [PubMed] [Google Scholar]
- 28.Andrews AT. Partial denaturation and renaturation of beta-lactoglobulin at air-water interfaces. Biochem Soc Trans. 1991;19:272S. doi: 10.1042/bst019272s. [DOI] [PubMed] [Google Scholar]
- 29.Charman SA, Mason KL, Charman WN. Techniques for assessing the effects of pharmaceutical excipients on the aggregation of porcine growth hormone. Pharm Res. 1993;10:954–962. doi: 10.1023/a:1018994102218. [DOI] [PubMed] [Google Scholar]
- 30.Cohen H, Shen BW, Snyder WR, Law JH, Kezdy FJ. The surface pressure dependency of the enzymic hydrolysis of lipid monolayers. Enzyme denaturation at the air-water interface. J Colloid Interface Sci. 1976;56:240–250. [Google Scholar]
- 31.Lechevalier V, Croguennec T, Pezennec S, Guerin-Dubiard C, Pasco M, Nau F. Evidence for synergy in the denaturation at the air-water interface of ovalbumin, ovotransferrin and lysozyme in ternary mixture. Food Chem. 2005;92:79–87. [Google Scholar]
- 32.Postel C, Abillon O, Desbat B. Structure and denaturation of adsorbed lysozyme at the air-water interface. J Colloid Interface Sci. 2003;266:74–81. doi: 10.1016/s0021-9797(03)00571-x. [DOI] [PubMed] [Google Scholar]
- 33.Layton CJ, Hellinga HW. Thermodynamic analysis of ligand-induced changes in protein thermal unfolding applied to high-throughput determination of ligand affinities with extrinsic fluorescent dyes. Biochemistry. 2010;49:10831–10841. doi: 10.1021/bi101414z. [DOI] [PubMed] [Google Scholar]
- 34.Lee S, Sielaff B, Lee J, Tsai FT. CryoEM structure of Hsp104 and its mechanistic implication for protein disaggregation. Proc Natl Acad Sci USA. 2010;107:8135–8140. doi: 10.1073/pnas.1003572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desantis ME, Leung EH, Sweeny EA, Jackrel ME, Cushman-Nick M, Neuhaus-Follini A, Vashist S, Sochor MA, Knight MN, Shorter J. Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell. 2012;151:778–793. doi: 10.1016/j.cell.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shorter J, Lindquist S. Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol Cell. 2006;23:425–438. doi: 10.1016/j.molcel.2006.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mosser DD, Ho S, Glover JR. Saccharomyces cerevisiae Hsp104 enhances the chaperone capacity of human cells and inhibits heat stress-induced proapoptotic signaling. Biochemistry. 2004;43:8107–8115. doi: 10.1021/bi0493766. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.