

Abstract
The influenza A M2 protein forms a proton channel for virus infection and mediates virus assembly and budding. While extensive structural information is known about the transmembrane helix and an adjacent amphipathic helix, the conformation of the N-terminal ectodomain and the C-terminal cytoplasmic tail remains largely unknown. Using two-dimensional (2D) magic-angle-spinning solid-state NMR, we have investigated the secondary structure and dynamics of full-length M2 (M2FL) and found them to depend on the membrane composition. In 2D 13C DARR correlation spectra, 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC)-bound M2FL exhibits several peaks at β-sheet chemical shifts, which result from water-exposed extramembrane residues. In contrast, M2FL bound to cholesterol-containing membranes gives predominantly α-helical chemical shifts. Two-dimensional J-INADEQUATE spectra and variable-temperature 13C spectra indicate that DMPC-bound M2FL is highly dynamic while the cholesterol-containing membranes significantly immobilize the protein at physiological temperature. Chemical-shift prediction for various secondary-structure models suggests that the β-strand is located at the N-terminus of the DMPC-bound protein, while the cytoplasmic domain is unstructured. This prediction is confirmed by the 2D DARR spectrum of the ectodomain-truncated M2(21–97), which no longer exhibits β-sheet chemical shifts in the DMPC-bound state. We propose that the M2 conformational change results from the influence of cholesterol, and the increased helicity of M2FL in cholesterol-rich membranes may be relevant for M2 interaction with the matrix protein M1 during virus assembly and budding. The successful determination of the β-strand location suggests that chemical-shift prediction is a promising approach for obtaining structural information of disordered proteins before resonance assignment.
Keywords: membrane protein, influenza M2, chemical shift prediction, conformational change
Introduction
The M2 protein of influenza A viruses is a 97-residue integral membrane protein that both acts as a proton channel for virus entry1–4 and mediates virus assembly and budding.5 The protein can be divided into three domains: an N-terminal ectodomain (residues 1–21), an α-helical central domain encompassing a transmembrane (TM) helix (residues 22–46) connected to an amphipathic helix (AH), and a C-terminal cytoplasmic tail (residues 63–97)6,7 (Supporting Information Fig. S1). M2 oligomerizes into a homotetramer through the TM helix to form the proton channel,8,9 with His37 and Trp41 as the proton-selective residue10 and the channel-gating residue,11 respectively. The protein is targeted by the antiviral drugs amantadine and rimantadine,12,13 which inhibit the proton channel activities.14
Recent high-resolution structural studies using X-ray crystallography,15,16 solution NMR,17 solid-state NMR (SSNMR),18–22 and electron paramagnetic resonance23,24 have revealed the structure of the central TM-AH domain. The TM helix is tilted by 26–35° from the bilayer normal, depending on the membrane thickness and pH.23,25–27 When associated with phospholipid bilayers, the AH lies on the membrane surface, connected by a tight turn from the TM helix.22 Amantadine and rimantadine bind with high affinity to the TM pore near Ser31,16,20,28,29 with the polar amine pointing to the C-terminus.30 A second, low-affinity, binding site unrelated to M2 inhibition31,32 exists on the lipid-facing surface of the protein near Asp44 and Arg45,17,20 and is populated by excess drug.20 Drug binding to the pore, which is the pharmacologically relevant site, is sensitive to the membrane thickness33–35 and composition.36
Analogous to drug binding studies, studies of the proton conduction mechanism of M2 have so far mainly used TM constructs. Two proton conduction mechanisms, a water-wire model37,38 and a His37 shuttle model,39 have been proposed. Direct observation by SSNMR of His37 side chain motion and His37-water proton exchange at low pH21,21,41 show that the shuttle mechanism is operative.
Compared to the TM-AH domain, almost no structural information is available about the N-terminal ectodomain and the cytoplasmic tail. Early oriented-membrane 15N SSNMR data of full-length M2 (M2FL) confirmed the orientations of the TM helix obtained from peptide studies42 but did not determine the orientation of the rest of the protein. Even the secondary structure of the ectodomain and cytoplasmic tail remains unknown. Structural investigation of M2FL is important for understanding the mechanisms of the entire panel of M2 functions, including not only the proton-channel function but also the virus-assembly function. Chimeras of M2 with the Sendai virus F protein indicate that the ectodomain is responsible for incorporating the protein into the virion.43 The ectodomain sequence is highly conserved among influenza A viruses 7,44 and has been the target of anti-flu vaccines.45 Ectodomain-specific antibodies that inhibit virus growth and replication have been reported.7,45,46 Two cysteines in this domain, C17 and C19, form intermolecular disulfide bonds,8 although C17 alone is sufficient to cause tetramer formation in detergent micelles.9,13,47,48 The cytoplasmic tail has been implicated in M2 interactions with the matrix protein M148 and with hemagglutinin and neuraminidase to regulate virus morphology.51 Truncation mutants and alanine mutants of the cytoplasmic tail alter the virus morphology51,52 reduce genome packaging and decrease virus infectivity.53,54
Given the functional relevance of the extramembrane domains, it is important to determine the three-dimensional structure of M2FL. In this work, we report initial characterization of the backbone conformation and mobility of M2FL using 13C magic-angle-spinning (MAS) NMR spectroscopy. Two types of lipid bilayers, without and with cholesterol, were used to mimic the environment of M2 in the host-cell plasma membrane and in the virus envelope, respectively. We show that M2FL contains highly dynamic and unstructured domains in the 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) bilayer at ambient temperature but is significantly immobilized in a cholesterol-containing virus-mimetic (VM+) membrane and in the 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE)/POPG/cholesterol membrane. Moreover, 2D 13C correlation spectra indicate that the extramembrane domains of DMPC-bound M2FL contain β-strand residues, which are absent when the protein is bound to the cholesterol-containing membranes. By fitting the measured 2D 13C correlation spectra with predicted chemical shifts for various secondary-structure models, we show that the β-strand is located at the N-terminus of the protein. This is confirmed using an ectodomain-truncated M2 variant, which does not show any β-strand peaks in the DMPC bilayer. We discuss potential implications of the membrane-induced conformational and dynamical changes of the protein to the influenza virus lifecycle.
Results
M2FL conformation and dynamics in DMPC bilayers
We investigated the conformation and dynamics of uniformly 13C, 15N-labeled M2FL in three phospholipid membranes: the DMPC bilayer, the VM+ membrane, which contains POPC, POPE, cholesterol, and sphingomyelin (SM), and an anionic POPC/POPG/cholesterol membrane. DMPC was chosen because both the TM peptide and a longer TM-AH construct adopt drug- and pH-sensitive conformations in this membrane.36 The VM+ membrane mimics the virus-envelope lipid composition closely,35,36 thus can shed light on the conformation of M2FL in the virus. The POPC/POPG/cholesterol membrane is of interest because it has been used to study the function of M2FL in virus assembly and budding.55
Figure 1 shows the one-dimensional (1D) 13C cross-polarization (CP) MAS spectra of M2FL in the three lipid membranes as a function of temperature. The use of 1H-13C CP to establish the 13C magnetization preferentially enhances the signals of immobile residues. From 243 to 303 K, the Cα intensities of DMPC-bound M2FL decreased fivefold, indicating that a significant fraction of the protein undergoes large-amplitude motion in the liquid-crystalline phase of the membrane. In contrast, in the two cholesterol-containing membranes, the 303-K spectra retain ∼75% of the Cα intensities of the 243 K spectra, indicating that the protein is significantly immobilized when the membrane viscosity is increased by cholesterol.
Figure 1.
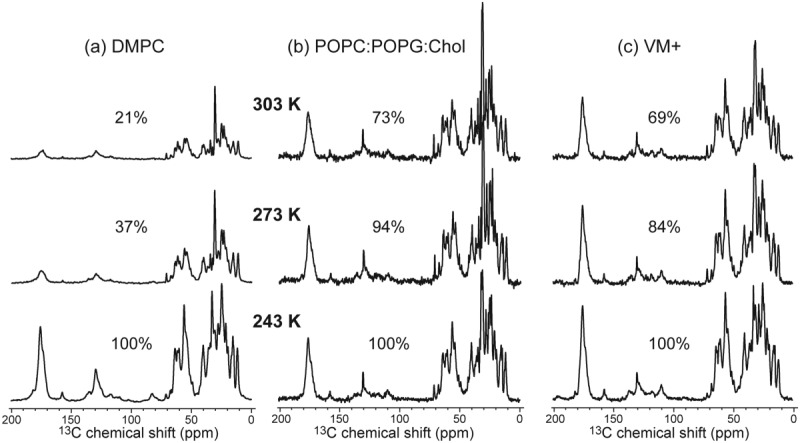
One-dimensional 13C CP-MAS spectra of uniformly 13C, 15N-labeled M2FL in (a) the DMPC membrane, (b) the POPC:POPG:cholesterol membrane, and (c) the VM+ membrane. The spectra were measured at 303, 273, and 243 K from top to bottom. The integrated intensities of the Cα region relative to the 243 K spectra are indicated. DMPC-bound M2FL shows much lower intensities at 303 K than at 243 K, indicating large-amplitude motion of the protein at high temperature when bound to this membrane.
2D 13C-13C correlation experiments were used to better resolve 13C chemical shifts and examine the M2FL conformation. Figure 2(a) shows the 2D DARR spectrum of DMPC-bound M2FL in the gel phase, where the protein motion is suppressed. Spectra measured at 253 K and 273 K showed little difference and thus were co-added. The apparent 13C linewidths are 0.5–1.3 ppm for side chain carbons and ∼1.5 ppm for Cα. These 13C linewidths result from overlapped signals, so single-site linewidths may be smaller. Characteristic chemical shift cross peaks of Leu, Ile, Val, Ala, Ser, Thr, and Pro are observed. Among these residues, Ile, Val, and Leu show prominent signals at α-helical Cα and Cβ chemical shifts, consistent with the high percentage of hydrophobic residues in the helical TM-AH domain of the protein (Supporting Information Fig. S1). Surprisingly, in addition to these α-helical chemical shifts, we also observed cross peaks at strongly β-strand chemical shifts. For example, a β-strand Ser Cβ-Cα peak at (65.2, 53.8) ppm, a β-strand Ala Cα-Cβ peak at (48.8, 21.4) ppm, and a β-strand Leu Cα-Cβ cross peak at (51.6, 43.1) ppm, are detected. To confirm that these Cα-Cβ cross peaks indeed result from Ser, Ala, and Leu, we apply the recently introduced Python program, PLUQ,56 which rapidly predicts the amino-acid type as well as the secondary structure of correlated chemical shifts based on the large protein chemical shift database, PACSY.57 Supporting Information Table S1 shows that indeed, Ser, Ala, and Leu are the top-ranked results for these three cross peaks, and their numbers of occurrences in the database are 4–23-fold higher than those of the second-ranked results. Moreover, the queried torsion angles of the top-ranked residues cluster predominantly in the β-strand region of the Ramachandran diagram (Supporting Information Fig. S2).
Figure 2.

Two-dimensional 13C–13C correlation spectra of DMPC-bound M2FL. (a) Full DARR spectrum with 15-ms 13C spin diffusion. Spectra measured at 253 and 273 K did not show significant differences and were thus co-added to give higher sensitivity. Residue-type assignments for peaks with characteristic chemical shifts are shown in blue for β-sheet, red for α-helix, and green for random coil. Superimposed in orange is the sheared J-INADEQUATE 2D spectrum in Figure 3, indicating the chemical shifts of mobile residues. (b) Water-edited 2D DARR spectrum, measured at 253 K. Shaded bars guide the eye for the preferential retention of the β-sheet signals and suppression of the α-helical peaks. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
To verify if the β-strand chemical shifts of DMPC-bound M2FL result from the two extramembrane domains, we conducted a water-edited 2D 13C correlation experiment using the pulse sequence shown in Supporting Information Figure S3(a).58–60 The experiment preferentially detects water-exposed residues by 1H magnetization transfer from water to the protein. At 253 K, in the gel phase of the DMPC membrane, the lipid 1H signals are broadened and suppressed by the 1H T2 filter, whereas the water 1H magnetization survives due to the residual mobility of interbilayer water. Figure 2(b) shows the water-edited 2D spectrum of DMPC-bound M2FL, measured using a T2 filter of 1.5 ms and a water-protein 1H spin diffusion mixing time of 15 ms. The latter is relatively short in order to distinguish between water-exposed and membrane-embedded residues. The water-edited 2D spectrum shows that the α-helical signals are preferentially suppressed while the β-strand signals with small Cα chemical shifts are preferentially retained. Comparison of the integrated intensities of the full 2D spectrum with the water-edited spectrum indicates that the fraction of remaining intensities of the β-strand peaks is 2.8-fold higher than the fraction of the remaining α-helical intensities. Although the 4-helix bundle formed by the TM domain surrounds a water-filled pore, previous water-protein spin diffusion experiments61 indicated that at short mixing times, water spin diffusion to lipid-facing residues is noticeably slower than water spin diffusion to pore-facing residues. Thus, the lipid-facing residues in the TM and amphipathic helices should be preferentially suppressed by the water-edited experiment at this mixing time. Taken together, the two 2D spectra indicate that the β-strand segments lie in the extramembrane domains.
The DARR spectrum in Figure 2(a) also shows coil chemical shifts (annotated in green), which are consistent with the high mobility of the DMPC-bound protein indicated by the temperature-dependent 1D 13C spectra (Fig. 1). To further investigate the extent of protein motion, we measured a 2D 13C J-INADEQUATE spectrum at 303 K. The experiment combined direct 13C polarization, a short recycle delay, and 13C-13C J-coupling for polarization transfer to selectively detect the signals of highly mobile residues [Supporting Information Fig. S3(b)]. The 2D J-INADEQUATE spectrum of DMPC-bound M2FL is shown in Figure 3. A large number of cross peaks with narrow linewidths of 0.4–0.8 ppm are observed at random-coil chemical shifts and can be readily assigned to residue types such as Val, Asp, Glu, and Thr. Altogether we found 21 spin systems in the spectrum (Supporting Information Table S2), some of which must be assigned to the ectodomain and the cytoplasmic tail based on the protein sequence. For example, the protein contains three Asn residues, two in the ectodomain (N13 and N20) and one in the SNA triplet remaining from His-tag cleavage (Supporting Information Fig. S1). Thus, if the two Asp/Asn spin systems found in the INADEQUATE spectrum contain at least one Asn, then that Asn must be located in the N-terminal portion of the protein. The spectrum shows three Glu/Gln spin systems; 10 out of 11 Glu residues in the protein lie in the N- and C-termini, also supporting the presence of highly mobile segments in these two domains.
Figure 3.

Two-dimensional 13C refocused J-INADEQUATE spectrum of DMPC-bound M2FL, measured at 303 K under 12 kHz MAS. Amino-acid type assignments indicate random coil chemical shifts for most detected residues (Supporting Information Table S2). [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
The 2D INADEQUATE spectrum correlates the double-quantum (DQ) chemical shift with the single-quantum (SQ) 13C chemical shift. To compare this type of spectrum directly with the SQ-SQ DARR correlation spectra shown in Figure 2, one can shear the INADEQUATE spectrum along the ω1 dimension by 45°. Figure 2(a) superimposes the sheared INADEQUATE spectrum (orange) with the low-temperature DARR spectrum of DMPC-bound M2FL. Many peaks with coil-like chemical shifts in the gel-phase membrane have corresponding signals in the high-temperature J-INADEQUATE spectrum. For example, the well resolved Thr spin system at 68.0 ppm in the DARR spectrum is observed in the J-INADEQUATE spectrum. The low-temperature DARR spectrum shows Ser Cα-Cβ cross peaks for all three canonical secondary structures, while the J-INADEQUATE spectrum only displays a Ser peak at the random-coil chemical shift. At the same time, some of the INADEQUATE cross peaks are not detected in the DARR spectrum, suggesting that some of the residues may undergo intermediate-timescale motion at the temperature of the DARR experiments (253–273 K), thus broadening their signals.
M2FL conformation in cholesterol-rich membranes
To investigate whether M2FL conformation depends on the membrane composition, we measured the 2D DARR spectra of the protein bound to the VM+ and anionic membranes. The spectrum of the VM+ bound protein [Fig. 4(a)] shows much higher sensitivity than the DMPC sample due to immobilization of the protein. More importantly, the spectrum indicates a significant change in the secondary structure: the β-strand chemical shifts of Ser, Ala and Leu disappeared, and the intensities of the α-helical signals are higher, indicating that the protein conformation is more helical in the VM+ membrane than in the DMPC bilayer. The anionic POPC/POPG/cholesterol sample gave a similar 2D spectrum as the VM+ membrane (Supporting Information Fig. S4). As these two membranes differ in membrane surface charge and absence or presence of SM, the spectral similarity suggests that cholesterol is most likely responsible for the conformational change of the protein to the more helical state. This membrane-induced conformational change is confirmed by CD spectra (Supporting Information Fig. S5), which show a more pronounced α-helical signature for the VM+ bound protein than for the DMPC-bound protein. Spectral deconvolution gave an estimate of 8% β-strand content for DMPC-bound M2FL but undetectable β-strand content for the VM+ bound protein.
Figure 4.

(a) Two-dimensional 13C DARR correlation spectrum of VM+ bound M2FL, measured at 273K with a 20-ms mixing time. (b) Two-dimensional spectrum of DMPC-bound M2FL reproduced from Figure 2(a) for comparison. Yellow rectangles highlight regions where β-sheet signals are absent in the VM+ membrane but present in the DMPC membrane. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Chemical-shift-guided structure modeling of DMPC-bound M2FL
As the β-strand chemical shifts of DMPC-bound M2FL result from the extramembrane domains, and the broad linewidths make it difficult at present to conduct unambiguous sequential assignment of this protein, we sought to obtain qualitative secondary-structure information for the extramembrane domains by simulating the experimental 2D spectra of DMPC-bound M2FL for various secondary-structure models. The accuracy of current 13C chemical-shift prediction programs has become very high. For example, the SHIFTX2 program predicts 13C chemical shifts with an accuracy of 0.44 ppm for Cα and 0.51 ppm for Cβ.62 This high accuracy is the result of a large high-quality database of training proteins, machine-learning techniques, and knowledge of side chain torsion angles and hydrogen-bond geometry. To test if SHIFTX2 is able to predict “extreme” β-sheet chemical shifts, we calculated the 13C chemical shifts of the 19-stranded β-barrel protein, VDAC-1, based on its high-resolution solution structure (PDB code: 2K4T).63 Supporting Information Figure S6 compares the SHIFTX2-predicted 13C chemical shifts of VDAC-1 with the experimentally measured 13C chemical shifts. Excellent agreement is obtained, with an RMSD of 1.1 ppm for Cα and 1.5 ppm for Cβ between the predicted and measured chemical shifts. Most importantly, strongly β-sheet Cα and Cβ chemical shifts are predicted for Ser, Leu and Ala, as indicated in Supporting Information Figure S6, which are consistent with experimental data. We also tested whether the removal of interstrand hydrogen bonds and other tertiary structure constraints affects the predicted chemical shifts. To do this, we isolated the β-strand fragments spanning residues 25–40, 94–103, and 216–226 in VDAC-1. The predicted chemical shifts for these isolated strands differ from those of the intact protein by an average of 0.72 ppm for Cα and 0.88 ppm for Cβ, indicating that tertiary structure constraints and hydrogen-bonding indeed exert a noticeable effect on the 13C chemical shifts.64
We start by considering limiting-case structural models where the entire protein is assumed to have the same backbone conformation. Although clearly simplistic and unlikely to be correct, these limiting-case simulations give some information on the secondary-structure content of the extramembrane domains. Figure 5(a–c) shows the simulated 2D 13C correlation spectra of all coil, all helix, and all strand models, superimposed with the measured 2D DARR spectrum (gray) as well as the sheared J-INADEQUATE spectrum (orange) of the DMPC-bound protein. The cross peaks from the three domains of the protein are color-coded for clarity. The all-coil model [Fig. 5(a)] completely missed the helical and strand Cα peaks, which lie at the two ends of the Cα band between 65 and 50 ppm, but the model nicely reproduced many J-INADEQUATE peaks such as the Ser and Thr coil signals. Thus, this model indicates the presence of significant unstructured segments in the DMPC-bound protein. The all-helix model [Fig. 5(b)] captured the strong helical cross peaks with large Cα chemical shifts, but completely missed the Cα band between 50 and 55 ppm, the extreme β-sheet Ser, Ala, and Leu peaks, as well as most of the J-INADEQUATE peaks. The all-strand model [Fig. 5(c)] reproduced the small Cα chemical shifts between 55 and 50 ppm well and give cross peaks that approach the extreme β-sheet chemical shifts, but missed all the helix signals as well as most of the J-INADEQUATE peaks. Compared to these three limiting models, a mixed conformational model where the ectodomain and cytoplasmic tail are unstructured while the TM-AH domain is kept helical reproduced most of the helix chemical shifts [Fig. 5(d)] as well as capturing many J-INADEQUATE peaks. However, the 50–55 ppm Cα band and the three extreme β-strand peaks are mostly missed. Comparison of these simulated spectra illustrates two useful points. First, the spectral fit deteriorates when the helical segment is extended from TM-AH to the entire protein. Second, many predicted chemical shifts from the all-strand model partially overlay with the measured chemical shifts, confirming the presence of β-strands in the protein.
Figure 5.

Ideal secondary-structure models and the predicted chemical shifts, superimposed with the measured 2D DARR spectrum (gray) and sheared J-INADEQUATE spectrum (orange) of DMPC-bound M2FL. (a) All-coil model. (b) All-helix model. (c) All-strand model. (d) A coil–helix–coil model. Coil, strand, and helix are denoted by a thin line, a thick line with an arrow, and an oscillation, respectively. The predicted chemical shifts are colored in green for the N-terminal ectodomain, red for the TM-AH domain, and blue for the cytoplasmic tail. Boxes show a few peaks of interest. The simulated spectra plot correlation peaks up to three bonds.
To determine the location of the β-strand segments, we next considered more complex secondary-structure models. Figure 6(a,b) shows the simulated spectra with either the cytoplasmic tail or the ectodomain completely β-strand while keeping the rest of the protein helical. The two simulated spectral patterns are similar, but the cytoplasmic-strand model agrees better with the measured spectral intensities at (ω1 = 52 ppm, ω2 = 29–34 ppm). On the other hand, both models miss the J-INADEQUATE peaks completely. In addition, these two models disagree with the CD result (Supporting Information Fig. S5) that the β-strand content is less than ∼10% for the DMPC-bound protein. To create a more realistic model that includes all three secondary structures, we used the PSIPRED software, which predicted a short β-strand at the C-terminus, a helical segment in the middle of the cytoplasmic tail, and coil for the rest of the extramembrane domains [Fig. 6(c)]. The resulting secondary-structure content agrees well with the CD result. The corresponding simulated spectrum reproduces both the J-INADEQUATE peaks and the helical chemical shifts in the DARR spectrum well. However, the short C-terminal β-strand includes an Ile, which is clearly absent in the DARR spectrum (indicated by blue arrows), and excludes a β-strand Ala, which is clearly present in the DARR spectrum. Thus, this model still does not agree well with the measured β-strand chemical shifts.
Figure 6.

Predicted 2D spectra of mixed secondary-structure models, superimposed with the experimental spectra of DMPC-bound M2FL. (a) The cytoplasmic tail as strand while the rest of the protein as helix. (b) The ectodomain as strand while the rest of the protein as helix. (c) PSIPRED-predicted secondary structure. Blue arrows indicate the positions of β-strand Ile peaks. (d) A model with a β-strand at the N-terminus, coil for the rest of the ectodomain, helix for TM-AH, and coil for the cytoplasmic tail.
The final structural model is shown in Figure 6(d). We placed the β-strand at the first eight N-terminal residues (MSLLTEVE), together with the SNA triplet remaining from the His-tag cleavage. This is the only short segment in the protein that contains all three β-strand residue types, Ser, Ala, and Leu. As can be seen, the model partially reproduced the β-strand Ala and Leu chemical shifts, although the β-strand Ser peak is still not well fit. The two Glu residues in this segment contributed some of the β-strand intensities in the (ω1 = 52 ppm, ω2 = 29–34 ppm) region. The presence of Thr5 in this segment is also consistent with the measured Thr peaks in the DARR spectrum. The length of this proposed strand segment (11 residues) also agrees well with the β-strand content estimated from the CD spectrum (Supporting Information Fig. S5), suggesting that there are unlikely additional β-strand segments in the protein. Furthermore, no residues in this N-terminal segment give peaks that violate the measured chemical shifts. We modeled the rest of the extramembrane domains as unstructured. The resulting coil chemical shifts show excellent agreement with the J-INADEQUATE peaks [Fig. 7(d)]. The unstructured nature of the cytoplasmic tail is also suggested by three protein-disorder prediction programs, FoldIndex©,65 PONDR®,66,67 and DISOPRED,68 all of which predicted residues 62–86 to be disordered. To further verify the fraction and location of the unstructured segments, we simulated the J-INADEQUATE spectra of three other coil models. Figure 7(a,b) shows that models with only the ectodomain or only the cytoplasmic tail being unstructured do not have enough coil peaks to match the experimental spectrum. A model where coil residues are placed in both termini but several sections of the cytoplasmic tail are kept structured [Fig. 7(c)] also gave somewhat inferior fit than the final model. For example, the peaks near (56, 37) ppm are not reproduced. Taken together, the simulations in Figures 6 and 7 indicate that the best secondary-structure model of DMPC-bound M2FL contains an N-terminal β-strand, a central α-helical TM-AH domain, and is unstructured for the rest of the extramembrane domains.
Figure 7.

Predicted coil chemical shifts and one-bond cross peaks for several structural models, superimposed with the sheared 2D J-INADEQUATE spectrum (dark orange) of DMPC-bound M2FL. A solid orange line represents the coil segment, while dotted black lines indicate residues that are not simulated. (a) Coil for the full ectodomain. (b) Coil for the cytoplasmic tail. (c) Coil for the ectodomain and two segments in the cytoplasmic tail. (d) Coil for a part of the ectodomain and the entire cytoplasmic tail. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Verification of the β-strand location in the protein using M2(21–97)
To verify the above secondary-structure model of M2FL, we expressed and purified an ectodomain-truncated M2 variant spanning residues 21–97. The recombinant protein contained the same SNA triplet before the first native protein's residue, D21. Figure 8 shows the 2D DARR spectrum of DMPC-bound M2(21–97). Indeed, the three “extreme” β-strand peaks of Ser, Ala and Leu clearly disappeared from the spectrum, and the side chain signals of the β-strand Leu are also absent, confirming that the β-strand signals in the DMPC-M2FL result from residues in the ectodomain.
Figure 8.

Experimental 2D 13C DARR spectrum (gray contours) of DMPC-bound M2(21–97), superimposed with the predicted chemical shifts using the secondary-structure model of Figure 6(d) except that the ectodomain is removed. The SNA triplet is assumed to be random coil, the TM-AH domain is kept α-helical, and the cytoplasmic tail is kept as random coil. Yellow rectangles highlight the frequency positions of β-strand peaks that are present in the DMPC-M2FL spectrum but absent in the M2(21–97) spectrum. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Discussion
The above chemical shift analysis indicate that the conformation and dynamics of the extramembrane domains of M2FL depend on the membrane composition. The DMPC bilayer promoted partial β-strand conformation and significant mobility, while the two cholesterol-containing membranes shifted the protein conformational equilibrium to predominantly α-helical. The chemical-shift-constrained secondary structure in Figure 6(d) is in good agreement with the CD data (Supporting Information Fig. S5): the former predicted a helix:coil:strand ratio of 42%:49%:9%, while the latter estimated a helix:coil:strand ratio of 48%:44%:8%. Chemical-shift prediction and spectral simulation suggest that the β-strand is located at the N-terminus of the protein, which is borne out by the spectrum of the ectodomain-truncated protein, which no longer exhibits β-strand peaks in the DMPC-bound state.
The presence of a β-strand segment in DMPC-bound M2FL does not appear to be a specific phenomenon of the DMPC membrane, because the same “extreme” β-strand cross peaks were also reported in the spectra of M2FL bound to E. coli membranes and 1,2-dioleoyl-sn-glycero-3-phosphocholine/1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPC/DOPE) membranes,69 but they were not interpreted. The full-length protein used in this previous study retained the 24-residue His-tag that ended with the SNA triplet before the N-terminus of the native M2 sequence. Thus, the segment predicted and confirmed to be β-strand here, SNA-MSLLTEVE, was fully present in the previous study. In other words, all three noncholesterol membranes promote a β-strand segment in M2FL.
In principle, factors other than cholesterol can contribute to the conformational changes of M2. For example, the VM+ and POPC/POPG/cholesterol membranes both contain unsaturated phospholipids whereas DMPC is a saturated lipid. The two cholesterol-containing membranes are also slightly thicker than the DMPC bilayer. However, in the context of the influenza virus lifecycle, the absence or presence of cholesterol is likely the most important factor regulating the M2 conformation. The lipid envelope of flu viruses is preferentially enriched in cholesterol compared to the host-cell plasma membrane from which the viruses bud.70 The high order of the virus lipid envelope has been implicated as the reason for the small number (5–15) of M2 tetramers in the virion,49 even though M2 is expressed at much higher levels in the host-cell membrane.6 M2 is also known to interact with the influenza protein M1 in a temporally and spatially controlled fashion during virus assembly and budding,51,71 and the site of M1-M2 interaction has been attributed to the M2 cytoplasmic domain based on mutagenesis data.53,54 Thus, it is conceivable that the unstructured nature of the cytoplasmic domain when M2 is bound to the fluid and low-cholesterol regions of the host plasma membranes serves the purpose of preventing premature interactions between M2 and M1. But after M2 is recruited to the cholesterol-rich region of the membrane during virus budding, conformational changes to a more α-helical form may be important for facilitating M2 interaction with the predominantly helical M1.72 The functional relevance of the short β-strand in the ectodomain is less obvious. We speculate that it may serve a structural role by promoting intermolecular association of the four protomers of the tetrameric channel. Exactly how the lipid composition changes the conformation of the extramembrane segments is not yet understood and will be of interest to study in the future.
The verification of the chemical-shift-predicted β-strand location by the truncated M2(21–97) construct suggests that chemical-shift prediction and back-calculation of experimental spectra represent a viable approach for obtaining global conformational constraints of disordered proteins prior to full resonance assignment. This chemical-shift-prediction approach has recently seen increasing use in SSNMR.73,74 Our simulations are not able to fully reproduce the extreme β-sheet Cα and Cβ chemical shifts of Ser, Ala and Leu. We attribute this to the difficulty of creating a true β-sheet with the relevant hydrogen bonds and other tertiary structure constraints,64 since the limited chemical shifts and the lack of intermolecular distances make it too speculative to model a multistrand β-sheet. At the same time, the fact that each M2 protomer does not have enough β-strand content to create interstrand hydrogen bonds suggests that the β-sheet chemical shifts can only originate from intermolecular association of the N-terminus of the four protomers of the tetrameric channel. Future experiments using more selective isotopic labeling and distance measurements will be necessary to elucidate the structures and conformational states of this protein in lipid membranes.
Materials and Methods
Expression and isotopic labeling of full-length M2
E. Coli BL21 (DE3) cells containing the plasmid with the M2FL gene were provided by Professor Tim Cross. The cDNA of the M2 protein from influenza A H3N2 virus, modified by having a N-terminal His-tag and a TEV protease cleavage site (with an amino acid sequence of MHHHHHHSSGVDLGTENLYFQSNA), was inserted into plasmid pET37.75 C19 and C50 were mutated to Ser while residue 17 was kept as Cys [Supporting Information Fig. S1(a)]. The latter stabilizes tetramer formation in detergents,9,47,48 which are used during membrane reconstitution of the protein.
E. coli cells were cultured on ampicilin-containing LB agar. A fresh colony was used to inoculate 100 mL of LB media. The cells were grown in a shaker overnight at 37°C. A 10-mL aliquot was used to inoculate 1 L of LB media until OD600 reached 0.7–0.8. The cells were spun down (20°C, 7000 rpm, 10 min) and resuspended in 500 mL M9 medium (48 mM Na2HPO4, 22 mM KH2PO4, 8.55 mM NaCl, 1 g/L 15NH4Cl, 4 g/L U-13C glucose, 2 mL/L of 1M MgSO4, 0.1 mL/L of 1M CaCl2, 100 mg/mL ampicilin). After half an hour, isopropyl β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 0.4 mM to induce protein expression. The cells grew for 3–4 h until the OD600 reached 1.7–1.8. The cells were then stored in lysis buffer (50 mM Tris-HCl, 50 mM NaCl) at −20°C overnight.
Purification of full-length M2
M2FL is purified using Ni affinity column. Cells were thawed and lysed on ice using a probe sonicator with 5 s on and 5 s off for 90 s. Benzonase nuclease and phenylmethylsulfonyl fluoride were added to prevent DNA binding and protein degradation. After incubation for 30 min at 4°C, cells were sonicated at amplitude 4 with 30 s on and 5 min off for 16 cycles. The soluble fraction was removed by centrifugation (10,000g, 8°C, 30 min). The M2-containing inclusion bodies were dissolved in lysis buffer with 6M urea and 2 wt% octyl-β-d-glucopyranoside (OG). After the suspension was shaken for 2 hours, it was centrifuged (10,000g, 8°C, 30 min) to remove the cell debris. The 12-mL supernatant containing the protein was added to a 3-mL Ni-NTA affinity column, shaken overnight at 4°C to allow protein binding to the column. About 100 mL of wash buffer I (50 mM Tris-HCl, 100 mM NaCl, 20 mM imidazole, 4M urea, 1% OG) and 50 mL of wash buffer II (50 mM Tris-HCl, 100 mM NaCl, 20 mM imidazole, 2M urea, 1% OG) were then used to wash the column. About 50 mL of refold buffer I (50 mM Tris-HCl, 100 mM NaCl, 20 mM imidazole, 0.5% OG, 20% glycerol) and 100 mL of refold buffer II (50 mM Tris-HCl, 50 mM NaCl, 0.5% OG, 20% glycerol) were then applied to the column to refold the protein. Subsequently, 50 mL of elution buffer (50 mM Tris-HCl, 50 mM NaCl, 300 mM imidazole, 0.5% OG, 20% glycerol) was used to elute M2FL from the Ni-NTA column. The amount of the protein was quantified from OD280 using an extinction coefficient of 15,220/M/cm. SDS-PAGE gel (Supporting Information Fig. S7) showed that most of the His-tagged protein exists as a dimer of 28 kDa after purification.
The His-tag was cleaved using TEV protease. About 30 mg purified His-tagged M2 was mixed with 2 mg of TEV protease and incubated at 4°C for 20 h on a Thermo Scientific platform rocker. The completeness of cleavage was verified by SDS-PAGE (Supporting Information Fig. S7), HPLC and electrospray ionization (ESI) mass spectrometry (data not shown). The cleaved protein was separated from TEV protease using the Ni-NTA column. The eluted fraction containing M2FL was concentrated to 2 mL using an Amicon Ultra centrifugal filter (Millipore) with a molecular weight cut off of 3 kDa.
Membrane reconstitution of M2FL by detergent dialysis
OG was added to purified M2FL to a final concentration of 36 mM. Lipid vesicles were prepared by eight freeze-thaw cycles and added to the protein/detergent solution to reach a protein:lipid mass ratio of ∼1:1.5, which corresponds to a protein:lipid molar ratio of about 1:27. The protein–lipid-detergent mixture was dialyzed against Tris buffer (10 mM Tris-Base, 1 mM EDTA, 0.1 mM NaN3, pH 7.5) at 4°C for 3 days with six buffer changes. HPLC-evaporative light scattering detection (ELSD) and ESI-MS confirmed that the amount of OG in the proteoliposome after dialysis was miniscule, <0.01% that of the protein (Supporting Information Figs. S8 and S9). Proteoliposomes were spun at 55,000 rpm for 4 h using a SW60 Ti rotor (Beckman Instruments) to yield a membrane pellet, which was then transferred to MAS rotors through a pipette tip. Typical 4-mm MAS samples contained ∼50-mg hydrated membrane, while 2.5-mm MAS samples contained ∼10 mg sample. It has been shown that lipid-bilayer-reconstituted M2FL predominantly forms tetramers.75
Three lipid membranes were used to reconstitute M2FL and test the conformational dependence of the protein on the membrane composition. All lipids were purchased from Avanti Polar Lipids (Alabaster, Alabama). DMPC was used because it retains the drug binding function of both M2(22–46) and M2(21–61) constructs.20,28,36 A chain-unsaturated VM+ membrane containing POPC:POPE: egg SM:cholesterol at a molar ratio of 25.6%:25.6%:25.6%:23%35,36,76 was used to mimic the virus lipid envelope. Finally, a POPC:POPG:cholesterol (62%:15%:23%) membrane was used because this mixture has been used to study the virus-budding function of M2.55
Expression, purification, and membrane reconstitution of M2(21–97)
To verify the chemical-shift based secondary-structure determination of M2FL, we also investigated an ectodomain-truncated M2 construct that spans residues 21–97. The cDNA of M2(21–97) with the N-terminal His-tag was inserted into a pET21 plasmid. After expression and purification, the His-tag was cleaved by TEV protease, leaving the SNA triplet before the native sequence starts at residue D21. M2(21–97) was purified by reversed-phase HPLC, and its mass was checked using ESI-MS. M2(21–97) was reconstituted into DMPC bilayers at a protein:lipid molar ratio of 1:22.5. The proteoliposomes in pH 7.5 Tris buffer were spun down to yield a membrane pellet with a hydration level of ∼40%, which was transferred to a 4-mm MAS rotor.
Solid-state NMR spectroscopy
All SSNMR experiments were carried out on a Bruker AVANCE 600 MHz (14.1 T) wide-bore NMR spectrometer using a 4-mm and a 2.5-mm MAS probe. All 13C chemical shifts are reported on the TMS scale, and are externally referenced through the α-Gly 13C′ signal at 176.49 ppm. This calibration is equivalent to putting the adamantane CH2 chemical shift at 38.48 ppm.77
One-dimensional 13C CP-MAS spectra were measured as a function of temperature for membrane-bound M2FL to examine the mobility of the protein. Two-dimensional 13C–13C DARR correlation spectra78 were measured using spin diffusion mixing times of 15–40 ms under 8 kHz MAS for the 4 mm samples and 10 kHz for the 2.5 mm samples. Maximum t1 evolution times were 5.6–6.6 ms. The number of scans per t1 slice were 128 and 256 for the 4 mm samples and 384 and 496 for the 2.5-mm MAS samples. The 2D spectra were measured between 243 and 273 K.
To selectively detect the signals of extramembrane residues, we carried out a water-edited 2D experiment58,59 on DMPC-bound M2FL. The experiment was conducted at 253 K under 8-kHz MAS. A 1H T2 filter of 1.5 ms was used to suppress the rigid protein 1H magnetization and most of the lipid 1H magnetization [Supporting Information Fig. S3(a)]. A 1H spin diffusion period of 15 ms transferred the water 1H magnetization to the protein and detected as 13C signals after CP.
To identify mobile residues, we conducted a 2D refocused J-INADEQUATE experiment that correlates DQ and SQ chemical shifts [Supporting Information Fig. S3(b)].77 The experiment was carried out on DMPC-bound M2FL at 303 K. The DQ excitation and reconversion period τCC was 2 ms, corresponding to 24 rotor periods under 12 kHz MAS.
Circular dichroism experiments
A small amount of proteoliposomes were removed from the MAS rotors for circular dichroism (CD) measurements. The membranes were dissolved in trifluoroethanol, dried under nitrogen gas, then resuspended in pH 7.5 Tris-buffer and sonicated. The solution was diluted to a final protein concentration of 0.08–0.09 mg/mL. The solution was transferred to a quartz cuvette of 0.1 cm path length and measured on a Jasco J-715 CD spectropolarimeter at room temperature. Three scans were collected from 260 to 200 nm. Protein-free DMPC and VM+ membranes were also measured to serve as controls, and their signals were subtracted from the protein-containing CD spectra. The secondary-structure content was calculated using the CDPro package.
Secondary structure modeling by chemical shift prediction
To obtain secondary-structure constraints for DMPC-bound M2FL, we generated a number of structural models and predicted their 13C chemical shifts using SHIFTX2 and CamCoil to compare with the experimental spectra. These models were designed to find secondary-structure elements that agreed with the data, but do not exclude other possible models to fit the 2D spectra, nor does the chemical-shift prediction provide tertiary structure information.
A Python script using a YASARA79 module was used to build models with ideal secondary structures (Helix: ϕ = −57°, ψ = −47°; Strand: ϕ = −139°, ψ = +135°). 13C chemical shifts were calculated from these structures using SHIFTX2.62 For random coil segments, CamCoil was used to calculate the backbone chemical shifts.80 Since CamCoil does not predict side chain chemical shifts, we calculated the side chain chemical shifts of a completely extended backbone using SHIFTX2, then combined these with the backbone CamCoil shifts. For all ideal models, the helix, coil and strand chemical shifts were combined to give the 2D spectral patterns of the proposed secondary-structure models.
More complex structural models contained the existing M2(22–62) structure (PDB: 2L0J) for the central TM-AH domain. PSIPRED,81 a secondary-structure prediction program, was used for one model. The placement of the β-strand at the N-terminus in the final model was governed by the need to have at least one Ala, Leu, and Ser in the strand. The 2D 13C J-INADEQUATE spectrum also required a coil Ala, which could result from the cytoplasmic domain or from the SNA triplet remaining from the His-tag cleavage. Further details about the various structural models are described in the Results section and the figure captions.
The predicted chemical shifts were plotted in a 2D correlation fashion using MATLAB. A 2D matrix with 0.1-ppm bins was populated with the calculated chemical shifts by creating correlations for all atoms within three bonds. To find correlations within a specific number of bonds regardless of side chain branching, each amino acid was represented as a bond distance matrix. The peaks were Gaussian-broadened to a full-width at half maximum of 0.71 ppm, then plotted and superimposed over the experimental 2D spectra.
Acknowledgments
The authors are grateful to Tim Cross and Huajun Qin for providing the M2 plasmid and expression protocol. They also thank Sarah Cady for help with the initial protein expression.
Glossary
- AH
amphipathic helix
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- M2FL
full-length M2
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- TM
transmembrane.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Cady SD, Luo WB, Hu FH, Hong M. Structure and function of the influenza A M2 proton channel. Biochemistry. 2009;48:7356–7364. doi: 10.1021/bi9008837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pinto LH, Holsinger LJ, Lamb RA. Influenza virus M2 protein has ion channel activity. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- 3.Pinto LH, Lamb RA. The M2 proton channels of infhluenza A and B viruses. J Biol Chem. 2006;281:8997–9000. doi: 10.1074/jbc.R500020200. [DOI] [PubMed] [Google Scholar]
- 4.Hong M, Degrado WF. Structural basis for proton conduction and inhibition by the influenza M2 protein. Protein Sci. 2012;21:1620–1633. doi: 10.1002/pro.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rossman JS, Lamb RA. Influenza virus assembly and budding. Virology. 2011;411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamb RA, Zebedee SL, Richardson CD. Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell. 1985;40:627–633. doi: 10.1016/0092-8674(85)90211-9. [DOI] [PubMed] [Google Scholar]
- 7.Zebedee SL, Richardson CD, Lamb RA. Characterization of the influenza virus M2 integral membrane protein and expression at the infected-cell surface from cloned cDNA. J Virol. 1985;56:502–511. doi: 10.1128/jvi.56.2.502-511.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holsinger LJ, Lamb RA. Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology. 1991;183:32–43. doi: 10.1016/0042-6822(91)90115-r. [DOI] [PubMed] [Google Scholar]
- 9.Sugrue RJ, Hay AJ. Structural characteristics of the M2 protein of influenza A viruses: evidence that it forms a tetrameric channel. Virology. 1991;180:617–624. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C, Lamb RA, Pinto LH. Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue. Biophys J. 1995;69:1363–1371. doi: 10.1016/S0006-3495(95)80003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang Y, Zaitseva F, Lamb RA, Pinto LH. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J Biol Chem. 2002;277:39880–39886. doi: 10.1074/jbc.M206582200. [DOI] [PubMed] [Google Scholar]
- 12.Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J Virol. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4:3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bright RA, Medina MJ, Xu X, Perez-Oronoz G, Wallis TR, Davis XM, Povinelli L, Cox NJ, Klimov AI. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366:1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- 15.Acharya A, Carnevale V, Fiorin G, Levine BG, Polishchuk A, Balannick V, Samish I, Lamb RA, Pinto LH, DeGrado WF, Klein ML. Structural mechanism of proton transport through the influenza A M2 protein. Proc Natl Acad Sci USA. 2010;107:15075–15080. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andreas LB, Eddy MT, Pielak RM, Chou JJ, Griffin RG. Magic angle spinning NMR investigation of influenza A M218−60: support for an allosteric mechanism of inhibition. J Am Chem Soc. 2010;132:10958–10960. doi: 10.1021/ja101537p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cady SD, Hong M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proc Natl Acad Sci USA. 2008;105:1483–1488. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu F, Luo W, Hong M. Mechanisms of proton conduction and gating by influenza M2 proton channels from solid-state NMR. Science. 2010;330:505–509. doi: 10.1126/science.1191714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath D, Zhou HX, Cross TA. Atomistic mechanism of the influenza A proton channel from a structure solved in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duong-Ly KC, Nanda V, DeGrado WF, Howard KP. The conformation of the pore region of the M2 proton channel depends on lipid bilayer environment. Protein Sci. 2005;14:856–861. doi: 10.1110/ps.041185805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen PA, Soto CS, Polishchuk A, Caputo GA, Tatko CD, Ma C, Ohigashi Y, Pinto LH, DeGrado WF, Howard KP. pH-induced conformational change of the influenza M2 protein C-terminal domain. Biochemistry. 2008;47:9934–9936. doi: 10.1021/bi801315m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cady SD, Goodman C, Tatko C, DeGrado WF, Hong M. Determining the orientation of uniaxially rotating membrane proteins using unoriented samples: a 2H, 13C, and 15N solid-state NMR investigation of the dynamics and orientation of a transmembrane helical bundle. J Am Chem Soc. 2007;129:5719–5729. doi: 10.1021/ja070305e. [DOI] [PubMed] [Google Scholar]
- 26.Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Kim S, Kovacs F, Cross TA. Structure of the the transmembrane region of the M2 protein H+ channel. Prot Sci. 2001;10:2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cady SD, Mishanina TV, Hong M. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J Mol Biol. 2009;385:1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pielak RM, Oxenoid K, Chou JJ. Structural investigation of rimantadine inhibition of the AM2-BM2 chimera channel of influenza viruses. Structure. 2011;19:1655–1663. doi: 10.1016/j.str.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cady SD, Wang J, Wu Y, DeGrado WF, Hong M. Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy. J Am Chem Soc. 2011;133:4274–4284. doi: 10.1021/ja102581n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jing X, Ma C, Ohigashi Y, Oliveira FA, Jardetzky TS, Pinto LH, Lamb RA. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc Natl Acad Sci USA. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenberg MR, Casarotto MG. Coexistence of two adamantane binding sites in the influenza A M2 ion channel. Proc Natl Acad Sci USA. 2010;107:13866–13871. doi: 10.1073/pnas.1002051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu F, Luo W, Cady SD, Hong M. Conformational plasticity of the influenza A M2 transmembrane peptide in lipid bilayers under varying pH, drug binding and membrane thickness. Biochim Biophys Acta. 2011;1808:415–423. doi: 10.1016/j.bbamem.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li C, Qin H, Gao FP, Cross TA. Solid-state NMR characterization of conformational plasticity within the transmembrane domain of the influenza A M2 proton channel. Biochim Biophys Acta. 2007;1768:3162–3170. doi: 10.1016/j.bbamem.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo W, Cady SD, Hong M. Immobilization of the influenza A M2 transmembrane peptide in virus-envelope mimetic lipid membranes: a solid-state NMR investigation. Biochemistry. 2009;48:6361–6368. doi: 10.1021/bi900716s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cady SD, Wang T, Hong M. Membrane-dependent effects of a cytoplasmic helix on the structure and drug binding of the influenza virus M2 protein. J Am Chem Soc. 2011;133:11572–11579. doi: 10.1021/ja202051n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sansom MSP, Kerr ID. Influenza virus M2 protein: A molecular modeling study of the ion channel. Protein Eng. 1993;6:65–74. doi: 10.1093/protein/6.1.65. [DOI] [PubMed] [Google Scholar]
- 38.Chen H, Wu Y, Voth GA. Proton transport behavior through the influenza A M2 channel: insights from molecular simulation. Biophys J. 2007;93:3470–3479. doi: 10.1529/biophysj.107.105742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc Natl Acad Sci USA. 1997;94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu F, Schmidt-Rohr K, Hong M. NMR detection of pH-dependent histidine-water proton exchange reveals the conduction mechanism of a transmembrane proton channel. J Am Chem Soc. 2012;134:3703–3713. doi: 10.1021/ja2081185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong M, Fritzsching KJ, Williams JK. Hydrogen-bonding partner of the proton-conducting histidine in the influenza M2 proton channel revealed from 1H chemical shifts. J Am Chem Soc. 2012;134:14753–14755. doi: 10.1021/ja307453v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian C, Gao PF, Pinto LH, Lamb RA, Cross TA. Initial structural and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Protein Sci. 2003;12:2597–2605. doi: 10.1110/ps.03168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park EK, Castrucci MR, Portner A, Kawaoka Y. The M2 ectodomain is important for its incorporation into influenza A virions. J Virol. 1998;72:2449–2455. doi: 10.1128/jvi.72.3.2449-2455.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ito T, Gorman OT, Kawaoka Y, Bean WJ, Webster RG. Evolutionary analysis of the influenza A virus M gene with comparison of the M1 and M2 proteins. J Virol. 1991;65:5491–5498. doi: 10.1128/jvi.65.10.5491-5498.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neirynck S, Deroo T, Saelens X, Vanlandschoot P, Jou WM, Fiers W. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat Med. 1999;5:1157–1163. doi: 10.1038/13484. [DOI] [PubMed] [Google Scholar]
- 46.Liu W, Li H, Chen YH. N-terminus of M2 protein could induce antibodies with inhibitory activity against influenza virus replication. FEMS Immunol Med Microbiol. 2003;35:141–146. doi: 10.1016/S0928-8244(03)00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holsinger LJ, Shaughnessy MA, Micko A, Pinto LH, Lamb RA. Analysis of the posttranslational modifications of the influenza virus M2 protein. J Virol. 1995;69:1219–1225. doi: 10.1128/jvi.69.2.1219-1225.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakaguchi T, Tu Q, Pinto LH, Lamb RA. The active oligomeric state of the minimalistic influenza virus M2 ion channel is a tetramer. Proc Natl Acad Sci USA. 1997;94:5000–5005. doi: 10.1073/pnas.94.10.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zebedee SL, Lamb RA. Influenza A virus M2 protein: monoclonal antibody restriction of virus growth and detection of M2 in virions. J Virol. 1988;62:2762–2772. doi: 10.1128/jvi.62.8.2762-2772.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zebedee SL, Lamb RA. Growth restriction of influenza A virus by M2 protein antibody is genetically linked to the M1 protein. Proc Natl Acad Sci USA. 1989;86:1061–1065. doi: 10.1073/pnas.86.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossman JS, Jing X, Leser GP, Balannik V, Pinto LH, Lamb RA. Influenza virus M2 ion channel protein is necessary for filamentous virion formation. J Virol. 2010;84:5078–5088. doi: 10.1128/JVI.00119-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iwatsuki-Horimoto K, Horimoto T, Noda T, Kiso M, Maeda J, Watanabe S, Muramoto Y, Fujii K, Kawaoka Y. The cytoplasmic tail of the influenza A virus M2 protein plays a role in viral assembly. J Virol. 2006;80:5233–5240. doi: 10.1128/JVI.00049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen BJ, Leser GP, Jackson D, Lamb RA. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J Virol. 2008;82:10059–10070. doi: 10.1128/JVI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCown MF, Pekosz A. Distinct domains of the influenza a virus M2 protein cytoplasmic tail mediate binding to the M1 protein and facilitate infectious virus production. J Virol. 2006;80:8178–8189. doi: 10.1128/JVI.00627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rossman JS, Jing X, Leser GP, Lamb RA. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell. 2010;142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fritzsching KJ, Yang Y, Schmidt-Rohr K, Hong M. Practical use of chemical shift databases for protein solid-state NMR: 2D chemical shift maps and amino-acid assignment with secondary-structure information. J Biomol NMR. 2013;56:155–167. doi: 10.1007/s10858-013-9732-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee W, Yu W, Kim S, Chang I, Lee W, Markley JL. PACSY, a relational database management system for protein structure and chemical shift analysis. J Biomol NMR. 2012;54:169–179. doi: 10.1007/s10858-012-9660-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ader C, Schneider R, Seidel K, Etzkorn M, Becker S, Baldus M. Structural rearrangements of membrane proteins probed by water-edited solid-state NMR spectroscopy. J Am Chem Soc. 2009;131:170–176. doi: 10.1021/ja806306e. [DOI] [PubMed] [Google Scholar]
- 59.Huster D, Yao XL, Hong M. Membrane protein topology probed by 1H spin diffusion from lipids using solid-state NMR spectroscopy. J Am Chem Soc. 2002;124:874–883. doi: 10.1021/ja017001r. [DOI] [PubMed] [Google Scholar]
- 60.Kumashiro KK, Schmidt-Rohr K, Murphy OJ, Ouellette KL, Cramer WA, Thompson LK. A novel tool for probing membrane protein structure: solid-state NMR with proton spin diffusion and X-nucleus detection. J Am Chem Soc. 1998;120:5043–5051. [Google Scholar]
- 61.Luo W, Hong M. Conformational changes of an ion channel detected through water-protein interactions using solid-state NMR spectroscopy. J Am Chem Soc. 2010;132:2378–2384. doi: 10.1021/ja9096219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han B, Liu Y, Ginzinger SW, Wishart D. SHIFTX2: significantly improved protein chemical shift prediction. J Biomol NMR. 2011;50:43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321:1206–1210. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shu I, Scian M, Stewart JM, Kier BL, Andersen NH. 13C structuring shifts for the analysis of model beta-hairpins and beta-sheets in proteins: diagnostic shifts appear only at the cross-strand H-bonded residues. J Biomol NMR. 2013:1–17. doi: 10.1007/s10858-013-9749-3. [DOI] [PubMed] [Google Scholar]
- 65.Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 66.Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform Ser Workshop Genome Inform. 1999;10:30–40. [PubMed] [Google Scholar]
- 67.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 68.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 69.Miao Y, Qin H, Fu R, Sharma M, Can TV, Hung I, Luca S, Gor'kov PL, Brey WW, Cross TA. M2 proton channel structural validation from full-length protein samples in synthetic bilayers and E. coli membranes. Angew Chem Int Ed Engl. 2012;51:8383–8386. doi: 10.1002/anie.201204666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scheiffele P, Rietveld A, Wilk T, Simons K. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J Biol Chem. 1999;274:2038–2044. doi: 10.1074/jbc.274.4.2038. [DOI] [PubMed] [Google Scholar]
- 71.Leser GP, Lamb RA. Influenza virus assembly and budding in raft-derived microdomains: a quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology. 2005;342:215–227. doi: 10.1016/j.virol.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 72.Harris A, Forouhar F, Qiu S, Sha B, Luo M. The crystal structure of the influenza matrix protein M1 at neutral pH: M1-M1 protein interfaces can rotate in the oligomeric structures of M1. Virology. 2001;289:34–44. doi: 10.1006/viro.2001.1119. [DOI] [PubMed] [Google Scholar]
- 73.Gradmann S, Ader C, Heinrich I, Nand D, Dittmann M, Cukkemane A, van Dijk M, Bonvin AMJJ, Engelhard M, Baldus M. Rapid prediction of multi-dimensional NMR data sets. J Biomol NMR. 2012;54:377–387. doi: 10.1007/s10858-012-9681-y. [DOI] [PubMed] [Google Scholar]
- 74.Renault M, Pawsey S, Bos MP, Koers EJ, Nand D, Tommassen-van Boxtel R, Rosay M, Tommassen J, Maas WE, Baldus M. Solid-state NMR spectroscopy on cellular preparations enhanced by dynamic nuclear polarization. Angew Chem Int Ed Engl. 2012;51:2998–3001. doi: 10.1002/anie.201105984. [DOI] [PubMed] [Google Scholar]
- 75.Tian C, Tobler K, Lamb RA, Pinto LH, Cross TA. Expression and initial structural insights from solid-state NMR of the M2 proton channel from influenza A virus. Biochemistry. 2002;41:11294–11300. doi: 10.1021/bi025695q. [DOI] [PubMed] [Google Scholar]
- 76.Wang T, Cady SD, Hong M. NMR determination of protein partitioning into membrane domains with different curvatures and application to the influenza M2 peptide. Biophys J. 2012;102:787–794. doi: 10.1016/j.bpj.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morcombe CR, Zilm KW. Chemical shift referencing in MAS solid state NMR. J Magn Reson. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 78.Takegoshi K, Nakamura S, Terao T. C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 79.Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G. Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins. 2004;57:678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- 80.Simone AD, Cavalli A, Hsu SD, Vranken W, Vendruscolo M. Accurate random coil chemical shifts from an analysis of loop regions in native states of proteins. J Am Chem Soc. 2009;131:16332–16333. doi: 10.1021/ja904937a. [DOI] [PubMed] [Google Scholar]
- 81.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.