

Abstract
The low solubility of many proteins hinders large scale expression and purification as well as biophysical measurements. Here, we devised a general strategy to solubilize a protein by conjugating it at a solvent-exposed position to a 6 kDa protein that was re-engineered to be highly soluble. We applied this method to the CARD domain of Apoptosis-associated speck-like protein containing a CARD (ASC), which represents one member of a class of proteins that are notoriously prone to aggregation. Attachment of the tag to a cysteine residue, introduced by site-directed mutagenesis at its self-association interface, improved the solubility of the ASC CARD over 50-fold under physiological conditions. Although it is not possible to use nuclear magnetic resonance (NMR) to obtain a high quality 2D correlation spectrum of the wild type domain under physiological conditions, we demonstrate that NMR relaxation parameters of the solubilized variant are sufficiently improved to facilitate virtually any demanding measurement. The method shown here represents a straightforward approach for dramatically increasing protein solubility, enabled by ease of labeling as well as flexibility in tag placement with minimal perturbation to the target. © 2013 The Protein Society
Keywords: protein solubility, protein engineering, protein aggregation, nuclear magnetic resonance, protein A, CARD domain
Introduction
Solution nuclear magnetic resonance (NMR) has proven to be a powerful technique for studies of structure, stability, and dynamics of proteins.1 Over the last decade we have observed significant advances in hardware and methodologies that improve the sensitivity of solution NMR.2,3 While nowadays, 1D measurements and 2D 1H-15N and 1H-13C correlation maps can often be achieved at concentrations of tens of micromolar, a number of other experiments, such as backbone and side-chain assignments, studies of dynamics and acquisition of NOESY datasets for structure determination, require protein concentrations in excess of hundreds of micromolar.4,5 While this has not been a problem for a number of model systems, for many proteins achieving solubility in this range has often proved challenging. Even if a protein can be concentrated without visible aggregation, it is rare that these costly samples are stable for the weeks that it would take to enable data collection and analysis for many triple resonance measurements. Moreover, self-association can lead to slower tumbling and shorter relaxation times than would be obtained from a monomeric protein. Sample degradation can change the spectral appearance over time, adding complexity to data analysis. The above factors collectively increase the demand on the time required to conduct NMR measurements.6
Caspase recruitment domains, or Caspase activation and recruitment domains (CARDs), are interaction motifs found in a wide array of proteins, typically those involved in processes relating to inflammation and apoptosis.7,8 These domains mediate the formation of large protein complexes via direct interactions between individual CARDs. Likely because its self-association is inherent to its function to build scaffolds, CARD domains (and death fold domains, in general) are highly insoluble, and only a handful of NMR studies can be performed for a few of the more soluble variants.9–12 Apoptosis-associated speck-like protein containing a CARD (ASC CARD) is a 10 kDa protein with a typical six-helix bundle death domain-fold. A protein construct that consists of the CARD, corresponding to residues 107–195 of ASC, is maximally soluble at 10 µM in a pyroptosis buffer (20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 20 mM KCl pH 7.4) that mimics the environment in which it functions.13
Lack of solubility could be due to: (a) low protein stability, resulting in transient exposure of aggregation-prone regions; (b) presence of an intrinsic dimerization (oligomerization) interface; and (c) performing studies in solutions at a pH that is at or close to the protein's isoelectric point (pI).14,15 Protein stability is usually improved by mutagenesis16 or by optimizing buffer composition using thermal shift assays.17 If identified, the oligomerization interface can be removed using mutagenesis,18 or oligomerization propensity can be reduced by optimizing the ionic strength or pH of the buffer.19,20 Oligomerization properties can also be reduced by changing the pI: by performing the measurements without cleaving the solubility tag used during recombinant protein expression,21 by adding several charged residues at the N- or C- termini,18 by adding short peptides that would bind the solubility-enhancing protein22 or by linking the protein using intein chemistry.23,24
All of the aforementioned methods have had great success in decreasing the oligomerization propensities of proteins, but each of them is specialized and unlikely to provide a general approach. The major limitation of the chimeric fusions that have been proposed is that the solubility tag was attached only at the protein termini,18,21–24 resulting in non-optimal tag placement that is limited to only two positions. Moreover, for some of them, the attachment results in isotopic labeling identical for the tag and the target protein, complicating the NMR spectra with undesired resonances from the tag.21
Here, we introduced mutations to Staphylococcus aureus B domain of protein A (BdpA) to create a highly negatively charged, 6 kDa variant of protein A (PA) that can be attached to, and improve the solubility of, a target partner protein. Specifically, the N-terminus of PA was linked to a cysteine located at a solvent-exposed position in the ASC CARD that was introduced by site-directed mutagenesis using a commercially available heterobifunctional crosslinking molecule, SM-(PEG)2. The ASC CARD was produced with isotopic labeling for NMR, while the PA solubility tag was not, enabling NMR measurements to be conducted selectively for ASC CARD residues without obstruction by the tag. The PA-SM-(PEG)2-ASC CARD fusion which was soluble up to 500 µM, yielded high quality 2D 1H-15N NMR spectra that does not change over time and had relaxation properties consistent with 17 kDa molecular weight species. This method can also be applied to increase the protein solubility of many other targets.
Results
Design of the fusion tag
Our goal was to develop a generalized method for straightforward attachment of a solubility tag that can be made NMR-invisible for protein NMR applications. The design of the tag is based on a 60 residue, three helix bundle fold, exemplified by domains of Staphylococcus aureus PA, which were used as a fusion tag for increasing protein solubility in vivo.25 Protein A has five independent domains, which have over 80% sequence identity and the same structure.26,27 The B domain of PA consists of three helices that span K9-L20 (H1), E25-D38 (H2), Q41-A57 (H3) [Fig. 1(A)]. This domain represents a model system for protein folding, where the thermodynamic stability and kinetics of folding have been characterized for more than 100 mutants.28–31
Figure 1.
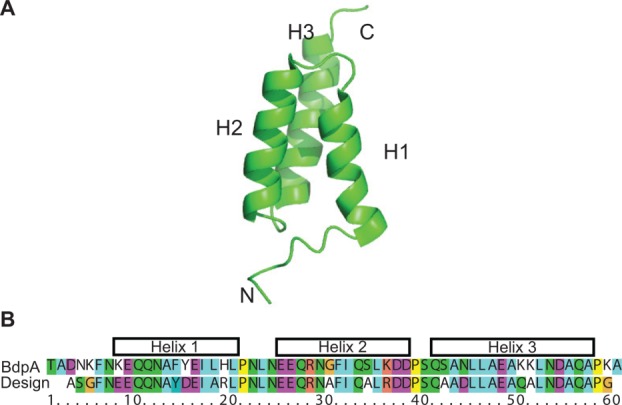
Design of PA solubility tag. A. Structure of BdpA (PDB: 1SS1),28 used as a template for protein re-design. B. Sequence alignment of the designed PA and BdpA. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
We decided to create a highly charged, solubility-enhanced version of the fold that would have greater resistance to aggregation. Charged residues were introduced using available mutagenesis data28,29,31 so that the resulting PA variant: (a) has a low pI; (b) remains folded under solution conditions where most solution NMR studies are performed ((0–50°C), as well as low ionic strength, where cryo-probe benefits are the greatest32); (c) does not contain any lysines (due to the linker chemistry). The details of how the solubility tags increase the soluble protein concentration are not fully understood, but it is well-known that increased negative surface charge correlates with increased solubility.6,14,33 Therefore, many negatively charged residues were introduced into PA to give it a pI that is lower than the vast majority of proteins. The solubility of the protein being targeted is increased with the formation of the PA-target construct because repulsive electrostatic forces between PAs will oppose the driving force for aggregation between targets. Since most of the proteins have pIs in the 5–9 range,34,35 attachment of PA will effectively decrease the pI of the target, which may be beneficial for improving solubility under the solution conditions of interest.
To re-engineer PA, we mutated 12 amino-acids (all at solvent-exposed positions) out of 53 residues available in our construct (23%) using the above principles as a guide [Fig. 1(B)]. Generally, a mutation was made if it: (a) has been found to stabilize the PA fold (G30A, S34A, S42A); (b) likely has negligible effect on stability, but removes a hydrophobic residue from the surface (Y15D, L18A, H19R); (c) likely has negligible effect on stability, but removes a lysine or lowers the pI (K8E, K36R, N44D, K50Q, K51A). In addition, we introduced a F14Y mutation to facilitate concentration determination by ultraviolet absorbance at 280 nm. Since no long-range NOEs have been found between the first five residues of the N-terminus and the rest of the protein as well as the last three on the C-terminus,28 we shortened the construct by three residues on the N-terminus and one residue on the C-terminus. Mutations such as H19R, K36R, and K50Q, which are located at positions within helices, increase helical propensity through which it increases global protein stability. Finally, the H19R and K36R mutations introduce two new types of interactions to stabilize the protein. First, it creates a D-R (i, i + 4) salt bridge; and second, the charged side chain has favorable electrostatic interactions with the helix dipole.36 Similarly, K50Q creates a Q-D (i, i + 4) hydrogen bond.37 The final construct had 57 residues, a predicted pI of 3.9, and an overall net charge of −8 under physiological conditions, in comparison with the WT protein of 60 residues, a pI of 5.2, and a net charge of −2.
A codon-optimized gene corresponding to the above PA construct was synthesized, cloned into the pET29a vector, and expressed in E. coli. The peaks in 1H-15N HSQC spectra of the purified protein had high chemical shift dispersion similar to that of the WT protein,27,28 indicative of structural similarity [Fig. 2(A)]. Circular dichroism (CD) spectrum was typical for a folded, helical protein and similar to that of BdpA [Fig. 2(B)]. To ensure that PA remains folded under the conditions of most NMR experiments, we measured its thermal denaturation mid-point (Tm) using CD spectroscopy [Fig. 2(C)]. Since stability of proteins that have charged residues in close proximity is often affected by ionic strength,38–40 we focused on low and high ionic strength buffers. The Tm of PA in a low ionic strength buffer (10 mM potassium phosphate, pH 7.4) was 70°C and in a high-ionic strength buffer (10 mM potassium phosphate, 500 mM NaCl, pH 7.4) was 78.5°C, therefore the protein will remain >98% folded in the temperature range of most NMR experiments. In comparison, the Tm of BdpA is 77°C.28 The ionic strength dependence of protein stability might result from unfavorable electrostatic interactions between negatively charged residues, which are more effectively screened at the higher ionic strength.39,40
Figure 2.
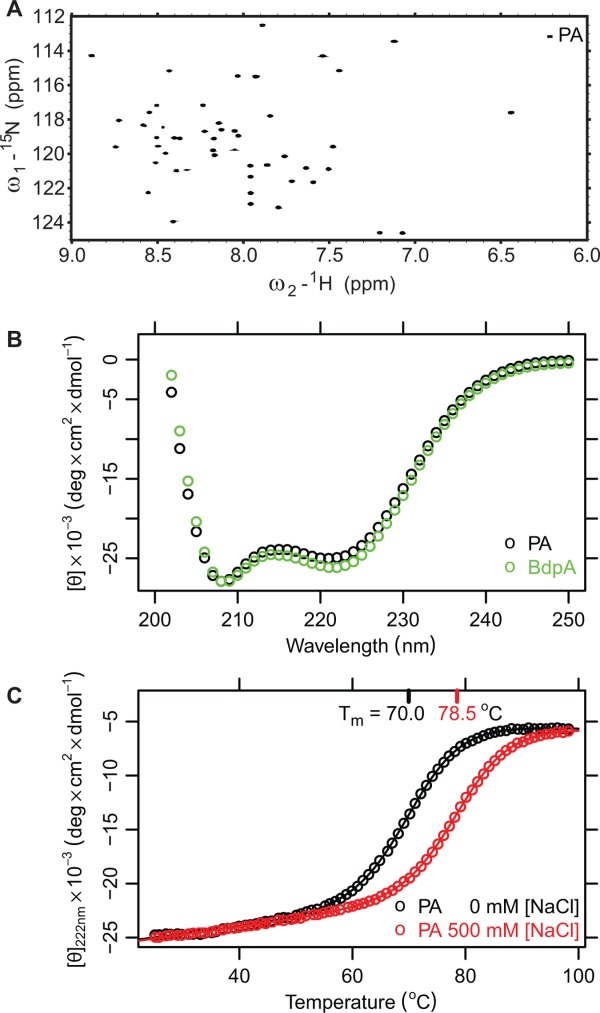
Biophysical properties of the designed PA tag. A. 1H-15N HSQC spectrum of designed PA in pyroptosis buffer. B. Circular dichroism spectrum of the B domain of PA (green) and re-designed PA (black) at low ionic strength. C. Thermal denaturation of designed PA monitored by CD at two ionic strengths.
Applying PA to solubilize an aggregation-prone CARD domain under physiological conditions
Attachment of modified PA to the target protein was accomplished using a heterobifunctional crosslinker, succinimidyl-[(N-maleimidopropionamido)-diethyleneglycol] ester (SM-(PEG)2) (Thermo Scientific) [Fig. 3(A)]. This crosslinker contains N-hydroxysuccinimide (NHS) ester and maleimide groups that allow covalent conjugation of amine- and sulfhydryl-containing molecules, which in this case correspond to PA and the target, respectively. This linker has arm length of 18 Å, equivalent to a N- to C- distance of ∼5–6 disordered amino-acids, which is long enough so that PA does not directly interact with the CARD domain, but short enough, that its negative charge will decrease the aggregation properties of its partner. Numerous other possibilities are available which range in the arm length of 4.4–95 Å if, depending upon the specific need, shorter or longer linkers might be desired.41
Figure 3.
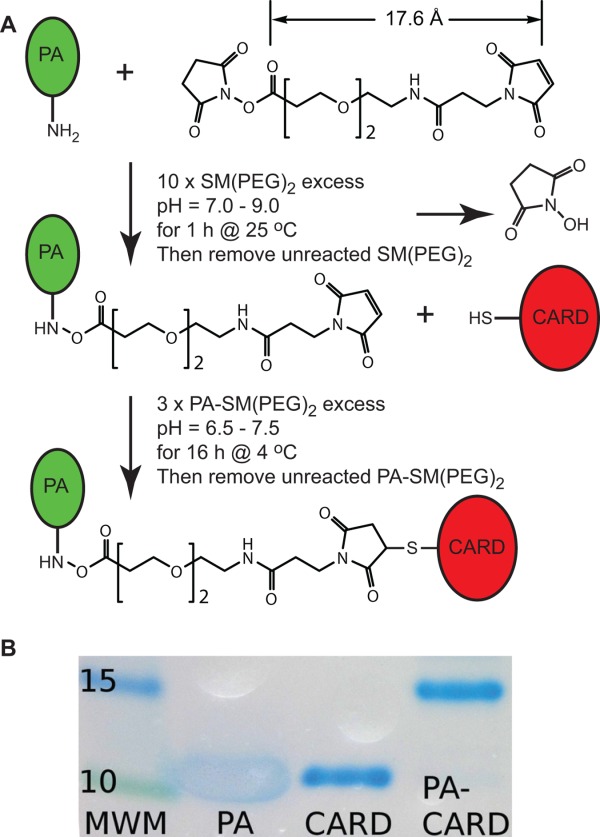
Conjugation of ASC CARD to the PA solubility tag. A. Overview of the labeling reaction. B. 4–20% SDS-PAGE gel showing successful labeling. PA was resistant to staining with Coomassie G-250 stain, and runs at higher apparent molecular weight than expected. Unusual amino acid distribution, with scarcity of positively charged and aromatic residues53 and low pI54 are likely responsible for the observed behavior.
The solubility of the ASC CARD is likely limited by self-association, as it has been observed to assemble into filamentous structures in COS cells.42 The theoretical pI of the domain is 6.9, and therefore the solubility is expected to be low under physiological conditions. Moreover, experiments in living cells have identified residues on the surface on the ASC CARD that are essential for mediating its assembly into signal transduction complexes called inflammasomes. Based on these results, we selected E130, D134 and Y137 as positions for introducing a cysteine,43 where conjugation of PA might sterically block and electrostatically repel self-association as well as lower the pI of the overall complex. Conjugation of PA to all mutants had the desired effect (see below), and ultimately we chose to focus our analysis on the D134C C173S construct.
As mentioned above, PA was linked to ASC CARD D134C C173S using the commercially available crosslinker, SM-(PEG)2. NHS esters react with primary amines at pH 7–9 to form amide bonds, while the maleimides react with sulfhydryl groups at pH 6.5–7.5 to form stable thioether bonds.41 Since the target protein is likely to have a number of primary amine sites, corresponding to lysine side chains and the N-terminus, the SM-(PEG)2 linker is first conjugated to the N-terminus of PA. Specifically, PA was incubated with 10-fold molar excess SM-(PEG)2 at room temperature for 1 h in a buffer at pH 7.3 [Fig. 3(A), details in Materials and Methods]. Unreacted linker was then removed using a centrifugal concentrator (Millipore), as well as exchanged into a buffer conducive to refolding of the ASC CARD domain. Specifically, ASC CARD D134C C173S was denatured in guanidine-HCl and refolded by rapid dilution into a solution containing PA-SM-(PEG)2. The conjugation reaction between the proteins was allowed to proceed overnight at 5°C. The PA-SM-(PEG)2-ASC CARD was purified from unreacted components using size exclusion chromatography (details in Materials and Methods). The labeling was confirmed using SDS-PAGE gel [Fig. 3(B)], where we observed a band corresponding to the combined molecular weights of the ASC CARD and PA (∼17 kDa).
Characterization of the ASC CARD-PA fusion by NMR and fluorescence
The suitability of PA-SM-(PEG)2-ASC CARD was assessed for NMR applications. Enhanced solubility of ASC CARD was immediately obvious for the PA conjugate, since the protein did not aggregate during buffer exchange for NMR applications. A 1H-15N HSQC spectrum acquired at a protein concentration of 500 µM at 25°C in pyroptosis buffer13 has 85 resolved peaks [Fig. 4(A)] of 88 that would be expected on the basis of the primary structure. Moreover, the chemical shifts of the peaks are well dispersed, indicative of a folded protein. Narrow linewidths further suggest that the relaxation properties are satisfactory for most NMR applications. Finally, the sample was stable for weeks at room temperature and the appearance of the 1H-15N HSQC spectrum remained unchanged (data not shown).
Figure 4.
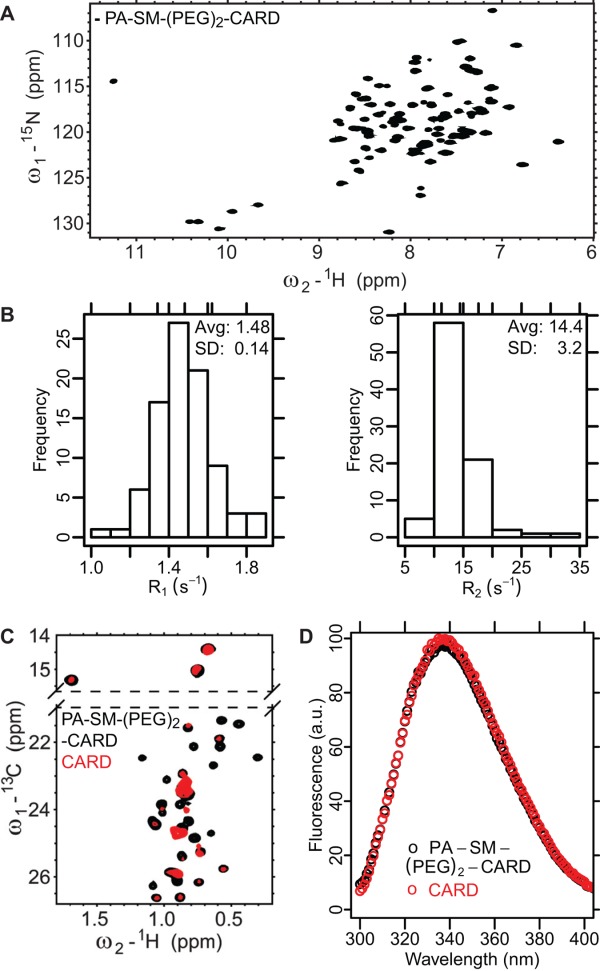
Biophysical analysis of PA-SM-(PEG)2-CARD. A.1H-15N HSQC spectrum of ASC CARD D134C C173S with PA solubility tag. 85 well dispersed resonances are visible in the spectrum, suggesting a well folded protein. B. Distribution of R1 (left) and R2 (right) values for ASC CARD in PA-SM-(PEG)2-CARD. The relaxation properties are consistent with monomeric protein. C. 1H-13C HSQC spectrum of ASC CARD with and without the PA solubility tag. The spectrum of ASC CARD only was acquired for 16 h on a 10 µM sample; the spectrum of CARD with the solubility tag took only 20 min on a 500 µM sample. Signal:noise on the 20 min spectrum is still >10-fold higher. D. Fluorescence emission spectrum of ASC CARD with and without the PA solubility tag. The excitation wavelength was 290 nm to ensure that tyrosines do not contribute significantly to this emission spectrum. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
We measured 15N T1, 15N T2 and {1H}-15N NOE for ASC CARD in PA-SM-(PEG)2-CARD in pyroptosis buffer at 25°C, allowing for protein tumbling time determination.44 The average 15N R2 and R1 values were 14.4 ± 3.2 s−1, and 1.48 ± 0.14 s−1, respectively, on a spectrometer operating at 600 MHz [Fig. 4(B)]. The correlation time estimated from R2/R1 ratios was 9.20 ± 0.05 ns. This is consistent with a monomeric 17 kDa complex. Importantly, it should be possible to acquire all standard triple resonance experiments on this sample.45
In contrast to the PA-SM-(PEG)2-CARD sample, we were unable to concentrate the wild type ASC CARD by itself in pyroptosis buffer above 10 µM, since the protein formed visible aggregates above this concentration. We attempted to obtain 1H-13C HSQC spectrum on a sample where Ile, Leu, Met, and Val methyl groups are 13C, 1H labeled. Methyl groups are some of the most sensitive probes available for NMR analysis of proteins and can be visible in species with unfavorable relaxation properties (such as high molecular weight oligomers) when other types of probes are not.3 While peaks in the spectrum of the free protein are broadened, suggesting self-association even at 10 µM, the chemical shifts of visible resonances for both PA-SM-(PEG)2-CARD and ASC CARD are very similar; thus labeling does not appear to fundamentally change the structure of CARD domain [Fig. 4(C)]. Differences in relative peak intensities between the two samples are most likely due to aggregation of the ASC CARD sample, albeit a dynamic process on a millisecond time scale cannot be excluded. If an NMR spectrum of the native protein cannot be obtained under the same solution conditions, tryptophan fluorescence is an alternative technique for determining the effects of labeling on the target protein structure. PA has been designed to have no tryptophans and a single tyrosine, and therefore tryptophan fluorescence measurements reflect properties of the target protein. Tryptophan fluorescence spectra of the ASC CARD are not affected by conjugation to PA, suggesting that labeling does not alter the conformation of ASC CARD [Fig. 4(D)].
Discussion
The approach presented here for increasing protein solubility has a number of obvious advantages: (1) there are a number of commercially available linkers that can be used that vary in chemical composition and length,41 (2) the solubility tag can be introduced at any position on the target protein, (3) nearly all the target protein was labeled in the conjugation reaction, (4) ability to selectively introduce isotopic labels into the target protein, as well as deuterate the solubility tag, (5) the labeling reactions can be conducted under a wide range of solution conditions that are compatible with downstream NMR applications, and (6) highly sensitive fluorescence-based analysis determining whether or not the native fold is preserved upon labeling. The drawback of the method is the requirement for a solvent-exposed cysteine. If a protein structure is unknown, Ellman's reagent can be used to establish the number of solvent-exposed cysteines.46 PA could potentially be conjugated to any solvent-exposed cysteine that is available. In the case that multiple cysteine side chains are solvent exposed, PA could be conjugated at all of these positions, with a concomitant increase in molecular weight of just over 6 kDa for every cysteine that is labeled. In contrast, if none are naturally available, cysteines could be introduced at positions close to the N- or C-termini immediately adjacent to the protein, where the mutation would not likely affect the protein fold. Positions not conserved in sequence alignments and predicted to be on the surface of the protein would also be excellent choices for cysteine substitution.
Other protein folds aside from the BdpA may provide even better platforms for designing solubility tags that can be attached using heterobifunctional crosslinkers. Small, single domain proteins that have proven to enhance the solubility of its partners in vivo and in vitro are the best targets, such as GB1 domain, SUMO or ubiquitin.25,47 Similar to PA, all of them will require sequence re-design to remove lysines, while lowering or raising the pI to extreme values and maintaining stability of the fold. Extensive mutagenesis data are available for most of them.48,49
We showed that attaching a small, 6 kDa protein to the CARD domain of ASC with a heterobifunctional crosslinker resulted in dramatic improvements in protein solubility, so that effectively any NMR measurement is possible on this protein under physiological conditions. The method used here has the advantage that the solubility tag can be conjugated to the protein at any position, and not just at the N- or C-terminus as a recombinant fusion. The tag would most effectively be conjugated at positions on the target protein that would break an oligomerization interface. Also, the tag effectively lowers the pI of the PA-target protein conjugate, increasing the solubility of proteins that have pIs close to 7 at physiological pH. Finally, selective deuteration of PA in the PA-ASC CARD conjugate will eliminate any background signals arising from the tag that might otherwise complicate interpretation of NMR data. The method is sufficiently simple that it can be applied to any protein solubility problem. It will be of broad and general interest to anybody struggling with proteins that form dimers, oligomers, or aggregates.
Materials and Methods
Protein expression
Re-designed PA was expressed in E. coli Rosetta 2 (DE3) cells (Novagen) from the pET29a vector in either terrific broth (TB) when unlabeled protein was required, or M9 minimal media when 15N labeling was needed. The cells were grown at 37°C in TB or M9 H2O medium supplemented with 100 mg/L of kanamycin until OD600 ∼ 2, when expression was induced by addition of 1 mM IPTG and allowed to proceed overnight at 25°C. The protein was purified using a two-step protocol that included Ni affinity and Q ion exchange columns. Specifically, the bacteria were harvested by centrifugation and resuspended in Ni-A buffer (10 mM imidazole, 50 mM potassium phosphate, 500 mM sodium chloride, pH 8.0) supplemented with PMSF, lysozyme and DNAse I. The cells were lysed by sonication and spun down at 45,000 g for 30 min. The filtered supernatant was applied to a column with Ni Sepharose beads (GE Healthcare), equilibrated and washed with Ni-A buffer. PA was eluted using Ni-B buffer (500 mM imidazole, 50 mM potassium phosphate, 500 mM sodium chloride, pH 8.0). After overnight dialysis into 20 mM Tris pH 8.0 in 3k MWCO dialysis membrane (Spectrum Laboratories) with 1 mg of TEV protease,50 the protein was further purified on the HiTrap Q HP 5 mL column (GE Healthcare) using 1%/mL NaCl gradient from 0 to 1 M in 20 mM Tris, pH 8.0. The purity and identity of the protein was confirmed using SDS-PAGE, NMR and electrospray ionization mass spectrometry.
BdpA was expressed from pRSET A vector, where the domain was fused to lipoyl fusion protein, separated by a thrombin protease cleavage site, and produced as described previously.28
The ASC CARD was expressed in E. coli C43(DE3) cells grown in M9 minimal media containing 15N ammonium chloride to yield protein that is isotopically enriched with 15N. In addition, methyl groups were selectively labeled with 13C at δ1 of isoleucine, δ1 and δ2 of leucine, γ1 and γ2 of valine, and ε1 of methionine by adding 60 mg/L of 2-keto-3,3-D2-4-13C-butyrate (Ile), 80 mg/L 2-keto-3-methyl-D3-3-D1-4-13C-butyrate (Leu,Val), or 80 mg/L 13C-methyl methionine (Met) to the media 1 h before induction, as previously described.18 Protein expression was induced at an OD600 of 0.8 and proceeded at 37°C for 6 h. After centrifugation, cells were resuspended in 7 M guanidine HCl, 50 mM potassium phosphate, 300 mM sodium chloride, 10 mM imidazole, 1 mM tris(2-carboxyethyl)phosphine (TCEP) pH 8.0 (Ni-C) and lysed by freezing overnight at −80°C. Cells were briefly sonicated and spun at 45,000 g for 45 min. The supernatant was applied to a HisTrap FF column (GE Healthcare) equilibrated with Ni-C and washed with ∼200 mL Ni-C. To remove TCEP and shift the pH to 7.0, the column was washed with three column volumes of 7 M guanidine HCl, 50 mM potassium phosphate, 300 mM sodium chloride, 10 mM imidazole, pH 7.0, and then eluted with three column volumes of 7 M guanidine HCl, 50 mM potassium phosphate, 300 mM sodium chloride, 250 mM imidazole, pH 7.0.
Labeling
A stock of SM-(PEG)2 (Thermo Scientific) was prepared in dimethyl sulfoxide (DMSO) as described by the manufacturer. It was diluted into a ∼1 mM solution of PA in 25 mM HEPES pH 7.3, 100 mM NaCl at a 10-fold molar excess of of SM-(PEG)2 to PA and incubated for 1 h at 25°C. Unreacted linker was removed by buffer exchanging into 50 mM potassium phosphate pH 7.0, 300 mM NaCl using a centrifugal concentrator (Millipore, 3 kDa cutoff). The ASC CARD was refolded at 5 μM into a solution containing PA-SM-(PEG)2 at 15 μM, 5°C. The reaction between the linker and ASC CARD was allowed to proceed overnight. The next morning, the reaction was quenched by addition of 1 mM dithiothreitol (DTT), and the N-terminal His tag on the ASC CARD was cleaved by addition of TEV protease for 1 day incubation at 5°C. TEV was removed using a HisTrap FF column equilibrated in the refolding buffer, where the flow through contained the PA-SM-(PEG)2–ASC CARD. The solution was concentrated using a centrifugal concentrator (Millipore, 3 kDa cutoff) and then purified by size exclusion on a HiLoad 16/60 Superdex 200 prep grade gel filtration column (GE Healthcare) equilibrated in 50 mM potassium phosphate pH 7.0, 300 mM NaCl.
This labeling reaction was performed by refolding the protein into the labeling buffer, but the labeling reaction for most of the target proteins should not require refolding. The solubility tags used for protein expression generally lack solvent-exposed cysteines,47 so the reaction could easily be performed when the fusion tag used in protein expression is still attached to the target protein. After successful labeling, the reaction could be quenched with addition of DTT, and the fusion tag removed by adding appropriate protease. The target protein should remain in solution due to the attachment of PA.
SDS-PAGE gel electrophoresis
The labeling was confirmed on Any kD Mini-PROTEAN TGX precast polyacrylamide gel (Bio-rad) run for 90 min at 100 V. The proteins were visualized using Commassie Brilliant Blue G-250 staining protocol.51
NMR
Samples for NMR measurements were prepared in 20 mM HEPES, 20 mM potassium chloride, 0.02% sodium azide, 2% D2O buffer that mimics physiologically-relevant conditions under which ASC self assembles into a regulatory complex called the pyroptosome to initate an inflammatory form of cell death.13 All NMR measurements were performed at 25°C on Bruker Avance 600 MHz spectrometer equipped with 5 mm TXI triple resonance cryo-probe and single-axis gradients. The 2D 15N T1, T2 and {1H}–15N nuclear Overhauser effect (NOE) experiments were acquired using Bruker built-in pulse sequences. The relaxation data was analyzed using Sparky 3.115 and fit using the non-linear least-squares Levenberg-Marquardt algorithm. The correlation time was determined using Tensor 2 software from R2/R1 ratio, considering only residues for which {1H}–15N NOE>0.65 and R1, R2 values are within one standard deviation from the mean.44 This minimized the effect of motions that are faster than the overall rotational diffusion but slower than ∼100 ps (which alter R1 values), and motions occurring on a micro-second to milli-second time scale (which affect R2 values).52
Circular dichroism
Far-ultraviolet CD spectra were acquired on a Aviv Model 215 at 25°C using 50 µM protein in a cuvette with 1-mm path length in two buffers: 10 mM potassium phosphate, pH 7.4 and 10 mM potassium phosphate, 500 mM sodium chloride, pH 7.4. Temperature scans were acquired under above conditions at 222 nm using bandwith of 1.5 nm at 1°C intervals between 25 and 98°C with 30 s equilibration at each temperature. Thermal denaturation curves were fitted to a two state model assuming ΔCp of 650 cal mol−1 K−1 (Refs. 16,28) using R 2.14.1 statistical analysis software.
Fluorescence
Measurements of tryptophan fluorescence were performed on a Tecan Infinite M1000 PRO plate reader in black 96 full-well plates at 25°C. Fluorescence spectra of samples containing 3 µM protein in pyroptosis buffer were recorded with an excitation wavelength of 290 nm and a bandwidth of 2.5 nm.
Glossary
- ASC CARD
Apoptosis-associated speck-like protein containing a CARD
- BdpA
B domain of protein A
- CARD
caspase activation and recruitment domain
- CD
circular dichroism
- DTT
dithiothreitol
- NHS
N-hydroxysuccinimide
- NMR
nuclear magnetic resonance
- PA
protein A
- pI
isoelectric point
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Barrett PJ, Chen J, Cho M-K, Kim J-H, Lu Z, Mathew S, Peng D, Song Y, Van Horn WD, Zhuang T, Sönnichsen FD, Sanders CR. The quiet renaissance of protein nuclear magnetic resonance. Biochemistry. 2013;52:1303–1320. doi: 10.1021/bi4000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kovacs H, Moskau D, Spraul M. Cryogenically cooled probes—a leap in NMR technology. Prog Nucl Mag Res Sp. 2005;46:131–155. [Google Scholar]
- 3.Atreya HS, editor. Isotope labeling in Biomolecular NMR. Adv. Exp. Dordrecht: Springer Netherlands; 2012. [Google Scholar]
- 4.Bieri M, Kwan AH, Mobli M, King GF, Mackay JP, Gooley PR. Macromolecular NMR spectroscopy for the non-spectroscopist: beyond macromolecular solution structure determination. FEBS J. 2011;278:704–715. doi: 10.1111/j.1742-4658.2011.08005.x. [DOI] [PubMed] [Google Scholar]
- 5.Kwan AH, Mobli M, Gooley PR, King GF, Mackay JP. Macromolecular NMR spectroscopy for the non-spectroscopist. FEBS J. 2011;278:687–703. doi: 10.1111/j.1742-4658.2011.08004.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhou P, Wagner G. Overcoming the solubility limit with solubility-enhancement tags: successful applications in biomolecular NMR studies. J Biomol NMR. 2010;46:23–31. doi: 10.1007/s10858-009-9371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kersse K, Verspurten J, Vanden Berghe T, Vandenabeele P. The death-fold superfamily of homotypic interaction motifs. Trends Biochem Sci. 2011;36:541–552. doi: 10.1016/j.tibs.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Park HH, Lo Y-C, Lin S-C, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–586. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou P, Chou J, Olea RS, Yuan J, Wagner G. Solution structure of Apaf-1 CARD and its interaction with caspase-9 CARD: a structural basis for specific adaptor/caspase interaction. Proc Natl Acad Sci USA. 1999;96:11265–11270. doi: 10.1073/pnas.96.20.11265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chou JJ, Matsuo H, Duan H, Wagner G. Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell. 1998;94:171–180. doi: 10.1016/s0092-8674(00)81417-8. [DOI] [PubMed] [Google Scholar]
- 11.Ferrage F, Dutta K, Nistal-Villán E, Patel JR, Sánchez-Aparicio MT, De Ioannes P, Buku A, Aseguinolaza GG, García-Sastre A, Aggarwal AK. Structure and dynamics of the second CARD of human RIG-I provide mechanistic insights into regulation of RIG-I activation. Structure. 2012;20:2048–2061. doi: 10.1016/j.str.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Alba E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC) J Biol Chem. 2009;284:32932–32941. doi: 10.1074/jbc.M109.024273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandes-Alnemri T, Alnemri ES. Assembly, purification, and assay of the activity of the ASC pyroptosome. Methods Enzymol. 2008;442:251–270. doi: 10.1016/S0076-6879(08)01413-4. [DOI] [PubMed] [Google Scholar]
- 14.Kramer RM, Shende VR, Motl N, Pace CN, Scholtz JM. Toward a molecular understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophys J. 2012;102:1907–1915. doi: 10.1016/j.bpj.2012.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schein CH. Solubility as a function of protein structure and solvent components. Biotechnology. 1990;8:308–315. doi: 10.1038/nbt0490-308. [DOI] [PubMed] [Google Scholar]
- 16.Fersht AR. W. H. Freeman and Company. New York, USA: 1999. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. [Google Scholar]
- 17.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 18.Ruschak AM, Kay LE. Proteasome allostery as a population shift between interchanging conformers. Proc Natl Acad Sci USA. 2012;109:E3454–E3462. doi: 10.1073/pnas.1213640109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bagby S, Tong K, Ikura M. Optimization of protein solubility and stability for protein nuclear magnetic resonance. Methods Enzymol. 2001;339:20–41. doi: 10.1016/s0076-6879(01)39307-2. [DOI] [PubMed] [Google Scholar]
- 20.Lepre CA, Moore JM. Microdrop screening: a rapid method to optimize solvent conditions for NMR spectroscopy of proteins. J Biomol NMR. 1998;12:493–499. doi: 10.1023/a:1008353000679. [DOI] [PubMed] [Google Scholar]
- 21.Zhou P, Lugovskoy AA, Wagner G. A solubility-enhancement tag (SET) for NMR studies of poorly behaving proteins. J Biomol NMR. 2001;20:11–14. doi: 10.1023/a:1011258906244. [DOI] [PubMed] [Google Scholar]
- 22.Durst FG, Ou H, Der Löhr F, Dötsch V, Straub WE. The better tag remains unseen. J Am Chem Soc. 2008;130:14932–14933. doi: 10.1021/ja806212j. [DOI] [PubMed] [Google Scholar]
- 23.Kobashigawa Y, Kumeta H, Ogura K, Inagaki F. Attachment of an NMR-invisible solubility enhancement tag using a sortase-mediated protein ligation method. J Biomol NMR. 2009;43:145–150. doi: 10.1007/s10858-008-9296-5. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi H, Swapna GVT, Wu K-P, Afinogenova Y, Conover K, Mao B, Montelione GT, Inouye M. Segmental isotope labeling of proteins for NMR structural study using a protein S tag for higher expression and solubility. J Biomol NMR. 2012;52:303–313. doi: 10.1007/s10858-012-9610-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bogomolovas J, Simon B, Sattler M, Stier G. Screening of fusion partners for high yield expression and purification of bioactive viscotoxins. Protein Exp Purif. 2009;64:16–23. doi: 10.1016/j.pep.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Jansson B, Uhlén M, Nygren P. All individual domains of staphylococcal protein A show Fab binding. FEMS Immunol Med Microbiol. 1998;20:69–78. doi: 10.1111/j.1574-695X.1998.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 27.Zheng D, Aramini J, Montelione G. Validation of helical tilt angles in the solution NMR structure of the Z domain of Staphylococcal protein A by combined analysis of residual dipolar coupling. Prot Sci. 2004;13:549–554. doi: 10.1110/ps.03351704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato S, Religa TL, Daggett V, Fersht AR. Testing protein-folding simulations by experiment: B domain of protein A. Proc Natl Acad Sci USA. 2004;101:6952–6956. doi: 10.1073/pnas.0401396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato S, Religa TL, Fersht AR. Phi-analysis of the folding of the B domain of protein A using multiple optical probes. J Mol Biol. 2006;360:850–864. doi: 10.1016/j.jmb.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 30.Fersht AR, Sato S. Phi-value analysis and the nature of protein-folding transition states. Proc Natl Acad Sci USA. 2004;101:7976–7981. doi: 10.1073/pnas.0402684101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato S, Fersht AR. Searching for multiple folding pathways of a nearly symmetrical protein: temperature dependent phi-value analysis of the B domain of protein A. J Mol Biol. 2007;372:254–267. doi: 10.1016/j.jmb.2007.06.043. [DOI] [PubMed] [Google Scholar]
- 32.Kelly AE, Ou HD, Withers R, Dotsch V. Low-conductivity buffers for high-sensitivity NMR measurements. J Am Chem Soc. 2002;124:12013–12019. doi: 10.1021/ja026121b. [DOI] [PubMed] [Google Scholar]
- 33.Trevino S, Scholtz J, Pace C. Measuring and increasing protein solubility. J Pharm Sci. 2008;97:4155–4166. doi: 10.1002/jps.21327. [DOI] [PubMed] [Google Scholar]
- 34.Kiraga J, Mackiewicz P, Mackiewicz D, Kowalczuk M, Biecek P, Polak N, Smolarczyk K, Dudek MR, Cebrat S. The relationships between the isoelectric point and: length of proteins, taxonomy and ecology of organisms. BMC Genomics. 2007;8:163. doi: 10.1186/1471-2164-8-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwartz R, Ting CS, King J. Whole proteome pI values correlate with subcellular localizations of proteins for organisms within the three domains of life. Genome Res. 2001;11:703–709. doi: 10.1101/gr.gr-1587r. [DOI] [PubMed] [Google Scholar]
- 36.Huyghues-Despointes B, Scholtz JM, Baldwin RL. Helical peptides with three pairs of Asp-Arg and Glu-Arg residues in different orientations and spacings. Protein Sci. 1993;2:80–85. doi: 10.1002/pro.5560020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huyghues-Despointes BMP, Klingler TM, Baldwin RL. Measuring the strength of side-chain hydrogen bonds in peptide helixes: the Gln. cntdot. Asp (i, i+ 4) interaction. Biochemistry. 1995;34:13267–13271. doi: 10.1021/bi00041a001. [DOI] [PubMed] [Google Scholar]
- 38.Horovitz A, Serrano L, Avron B, Bycroft M, Fersht AR. Strength and co-operativity of contributions of surface salt bridges to protein stability. J Mol Biol. 1990;216:1031–1044. doi: 10.1016/S0022-2836(99)80018-7. [DOI] [PubMed] [Google Scholar]
- 39.Religa TL, Markson JS, Mayor U, Freund SMV, Fersht AR. Solution structure of a protein denatured state and folding intermediate. Nature. 2005;437:1053–1056. doi: 10.1038/nature04054. [DOI] [PubMed] [Google Scholar]
- 40.Arbely E, Neuweiler H, Sharpe TD, Johnson CM, Fersht AR. The human peripheral subunit-binding domain folds rapidly while overcoming repulsive Coulomb forces. Prot Sci. 2010;19:1704–1713. doi: 10.1002/pro.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hermanson GT. Bioconjugate techniques. Elsevier London, United Kingdom. 2008 [Google Scholar]
- 42.Masumoto J, Taniguchi S, Sagara J. Pyrin N-terminal homology domain- and caspase recruitment domain-dependent oligomerization of ASC. Biochem Biophys Res Commun. 2001;280:652–655. doi: 10.1006/bbrc.2000.4190. [DOI] [PubMed] [Google Scholar]
- 43.Proell M, Gerlic M, Mace PD, Reed JC, Riedl SJ. The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem J. 2013;449:613–621. doi: 10.1042/BJ20121198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dosset P, Hus JC, Blackledge M, Marion D. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J Biomol NMR. 2000;16:23–28. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- 45.Rossi P, Swapna GVT, Huang YJ, Aramini JM, Anklin C, Conover K, Hamilton K, Xiao R, Acton TB, Ertekin A, Everett JK, Montelione GT. A microscale protein NMR sample screening pipeline. J Biomol NMR. 2010;46:11–22. doi: 10.1007/s10858-009-9386-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riener CK, Kada G, Gruber HJ. Quick measurement of protein sulfhydryls with Ellman's reagent and with 4,4′-dithiodipyridine. Anal Bioanal Chem. 2002;373:266–276. doi: 10.1007/s00216-002-1347-2. [DOI] [PubMed] [Google Scholar]
- 47.Waugh DS. Making the most of affinity tags. Trends Biotechnol. 2005;23:316–320. doi: 10.1016/j.tibtech.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 48.Went HM, Jackson SE. Ubiquitin folds through a highly polarized transition state. Protein Eng Des Sel. 2005;18:229–237. doi: 10.1093/protein/gzi025. [DOI] [PubMed] [Google Scholar]
- 49.Morrone A, Giri R, Toofanny RD, Travaglini-Allocatelli C, Brunori M, Daggett V, Gianni S. GB1 is not a two-state folder: identification and characterization of an on-pathway intermediate. Biophys J. 2011;101:2053–2060. doi: 10.1016/j.bpj.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blommel PG, Fox BG. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Exp Purif. 2007;55:53–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pink M, Verma N, Rettenmeier AW, Schmitz-Spanke S. CBB staining protocol with higher sensitivity and mass spectrometric compatibility. Electrophoresis. 2010;31:593–598. doi: 10.1002/elps.200900481. [DOI] [PubMed] [Google Scholar]
- 52.Tjandra N, Wingfield P, Stahl S, Bax A. Anisotropic rotational diffusion of perdeuterated HIV protease from 15N NMR relaxation measurements at two magnetic fields. J Biomol NMR. 1996;8:273–284. doi: 10.1007/BF00410326. [DOI] [PubMed] [Google Scholar]
- 53.Compton SJ, Jones CG. Mechanism of dye response and interference in the Bradford protein assay. Anal Biochem. 1985;151:369–374. doi: 10.1016/0003-2697(85)90190-3. [DOI] [PubMed] [Google Scholar]
- 54.Shi Y, Mowery Ra, Ashley J, Hentz M, Ramirez AJ, Bilgicer B, Slunt-Brown H, Borchelt DR, Shaw BF. Abnormal SDS-PAGE migration of cytosolic proteins can identify domains and mechanisms that control surfactant binding. Prot Sci. 2012;21:1197–1209. doi: 10.1002/pro.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.