

Abstract
Understanding the neurochemical basis for cognitive function is one of the major goals of neuroscience, with a potential impact on the diagnosis, prevention and treatment of a range of psychiatric and neurological disorders. In this review, the focus will be on a biochemical pathway that remains under-recognized in its implications for brain function, even though it can be responsible for moderating the activity of two neurotransmitters fundamentally involved in cognition – glutamate and acetylcholine. Since this pathway – the kynurenine pathway of tryptophan metabolism – is induced by immunological activation and stress, it also stands in a unique position to mediate the effects of environmental factors on cognition and behaviour. Targeting the pathway for new drug development could, therefore, be of value not only for the treatment of existing psychiatric conditions, but also for preventing the development of cognitive disorders in response to environmental pressures.
Keywords: kynurenine, kynurenic acid, tryptophan, quinolinic acid, schizophrenia, Huntington's disease, cognition
The neurochemistry of cognition
The neural complexity of cognitive function makes it exceptionally difficult to identify specific neurochemical substrates, especially those that could provide pharmacologically relevant targets for the prevention or treatment of cognitive dysfunction (Millan et al., 2012). Most of the recognized neurotransmitters in the CNS have been linked with some aspect of cognition either as primary factors or as systems whose activity is modified as a secondary consequence of a neurochemically distinct previous event. Among the major candidates with a putatively primary role in cognition are the monoamines norepinephrine, dopamine and 5-hydroxytryptamine (5HT) (Dalley et al., 2004; Sarter et al., 2007; Robert and Benoit, 2008; Tadaiesky et al., 2008), although dopamine has received most attention because of its potential links to the cognitive dysfunction seen in schizophrenia and Parkinson's disease (Tadaiesky et al., 2008; Xu et al., 2012), with roles in executive functioning and perceptual response speed that are affected in both disorders (Backman et al., 2010). Several other neurotransmitter systems, such as those releasing histamine and neuropeptides have been implicated in cognitive performance or its modification by drugs (Minzenberg and Carter, 2008).
The two systems which have received by far the greatest attention to date are those releasing glutamate or acetylcholine as their transmitters. Both these systems have well-established roles in various aspects of cognitive function, with the focus of interest being on glutamate receptors sensitive to the synthetic ligand NMDA (Castellano et al., 2001; Newcomer and Krystal, 2001; Neill et al., 2010). These systems are of particular relevance to this review since the primary compounds of interest – quinolinic acid and kynurenic acid – both have prominent actions on them. Conversely, little is known of the possible interactions between the kynurenine pathway and other systems contributing to cognitive function, so the focus of this review will be limited to glutamatergic and cholinergic neurons.
The biomedical relevance of these two systems is borne out by the ability of agonists and antagonists to affect cognitive behaviour and for some of these compounds to appear as potential therapeutic agents. Compounds that promote activation of NMDA receptors, especially those such as d-cycloserine which act via the co-agonist strychnine-insensitive Gly-B-site for glycine on the NMDA receptor, facilitate learning in humans (Kalisch et al., 2009; Onur et al., 2010) whereas antagonists such as ketamine, dizocilpine [MK-801] and phencyclidine are detrimental to learning and cognitive function (Jentsch and Roth, 1999; Newcomer et al., 1999; Cochran et al., 2003). Major advances are being made in this area relating receptor subunit composition to behavioural relevance. For example, mice that overexpress GluN2B subunits show increased cognition and enhanced plasticity (Cui et al., 2011). While this level of detailed, molecular knowledge about receptors should lead to improved selectivity of drug treatments, it is equally important to understand the mechanisms leading to the activation or modulation of those receptors, including those involving the kynurenine pathway.
The tryptophan-kynurenine pathway
Decades of interest in the metabolic products of tryptophan have focused on 5HT (or serotonin). However, the synthesis of 5HT is now known to account for the metabolism of only 3% or less of non-protein tryptophan. The kynurenine pathway, in contrast, accounts for approximately 90% of tryptophan metabolism in most tissues (Stone and Darlington, 2002).
The kynurenine pathway (Figure 1) was identified in the early years of the 20th century as the catabolic source of one of the newly recognized vitamins – vitamin B3 (nicotinic acid, nicotinamide or niacin). It was regarded solely as a route for the endogenous production of the vitamin to compensate for any dietary deficiency. The first product of tryptophan oxidation by the haemoprotein enzymes indoleamine-2,3-dioxygenase (IDO, found in most tissues) and tryptophan-2,3-dioxygenase (TDO, found mainly in the liver) is kynurenine, a compound identified originally in studies of the chemical composition of canine urine, from which it takes its name. The breakthrough into cognitive neuroscience came when two of the major components of the pathway – quinolinic acid and kynurenic acid – were shown to act on NMDA receptors (NMDAR).
Figure 1.
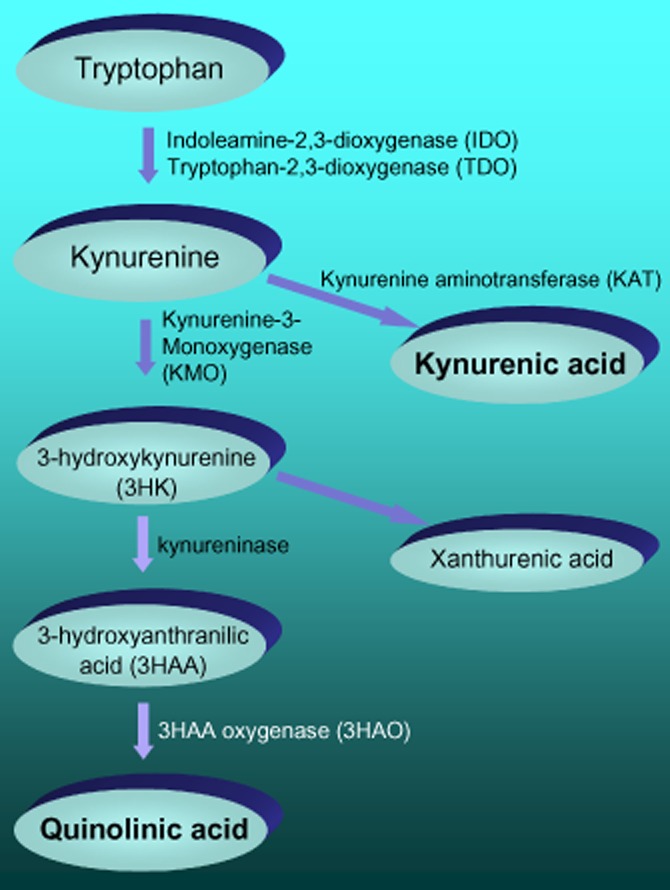
A summary of the major components of the kynurenine pathway for the oxidation of tryptophan.
Quinolinic acid
Quinolinic acid was originally found to excite neurones in the cerebral cortex of anaesthetized rats, an effect that was prevented by selective antagonists at NMDAR (Stone and Perkins, 1981). These receptors remain the most important sites of action of quinolinic acid, largely accounting for both its excitatory activity and its ability to produce axon-sparing excitotoxicity (Schwarcz et al., 1983).
There may be additional sites of action of quinolinic acid, although these have not yet been explored in detail. Thus it can generate, or promote the formation of, reactive oxygen species and has a generally proinflammatory profile within the immune system, increasing the expression and secretion of chemotactic molecules such as monocyte chemoattractant protein-1 (MCP-1) and regulated on activation, normal T cell expressed and secreted (RANTES) (Guillemin et al., 2003). These compounds can originate from several types of immune-competent cells peripherally but are also released by microglia in the CNS. Both proteins attract immune-activated macrophages, which secrete inflammatory cytokines including TNFα. In addition to modulating neuronal excitability, therefore, quinolinic acid can also act as an initiator and promoter of local inflammation within the CNS.
Kynurenic acid
Kynurenic acid is a product of tryptophan metabolism produced by the transamination of kynurenine by kynurenine aminotransferases (KAT) (Figure 1). Kynurenic acid is an antagonist at glutamate ionotropic receptors responding to amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), kainate or NMDA (Perkins and Stone, 1982), although it is most active at the latter. The antagonism of NMDA results from kynurenic acid's binding largely at the Gly-B site on the NMDAR complex (Stone and Darlington, 2002). Additional sites of action for kynurenic acid have been reported recently such as the aryl hydrocarbon receptor and the G-protein coupled receptor GPR35 (Wang et al., 2006; DiNatale et al., 2010; Stone et al., 2013), but their significance for brain function remains unclear.
Kynurenines and cognitive function
A direct examination of the effects of kynurenines on brain neurochemistry and behaviour in experimental animals and their roles in cognitive disorders in human patients has supported the view that this pathway plays a key role not only in several aspects of cognitive function but also in mediating the effects of environmental influences on cognition (Fig. 2).
Figure 2.
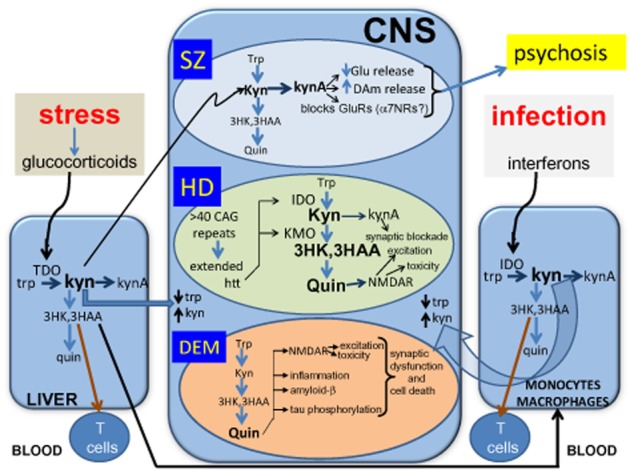
A summary of the involvement of kynurenines in three groups of CNS cognitive disorders: (A) schizophrenia and related conditions such as autism spectrum disorders (SZ), (B) Huntington's disease (HD) and (C) dementias such as HIV-Associated Neurocognitive Disorders (HAND), with probable relevance to conditions such as Alzheimer's disease. The diagram also illustrates the influences of stress on the kynurenine pathway via the induction of glucocorticoid secretion which induces TDO in the liver, and infections which cause the induction of IDO in several subsets of leucocytes via the action of interferons. The activation of IDO or TDO can produce localized depletion of tryptophan from proliferating cells while increasing the concentrations of kynurenine and its metabolites in tissues. Some of these metabolites such as kynurenic acid and quinolinic acid are active primarily on neuronal function, while 3HK and 3HAA have their main effects on T cell proliferation and cytokine secretion.
α7NRs, α7-nicotinic cholinoceptors; 3HAA, 3-hydroxyanthranilic acid; 3HK, 3-hydroxykynurenine; DAm, dopamine; Glu, glutamate; Htt, huntingtin; IDO, indoleamine-2,3-dioxygenase; Kyn, kynurenine; Kyna, kynurenic acid; Quin, quinolinic acid; TDO, tryptophan-2,3-dioxygenase; Trp, tryptophan.
The simplest of the kynurenine metabolites to manipulate experimentally is kynurenic acid since its concentrations in the brain can be increased substantially by the simple expedient of administering its precursor, kynurenine, which readily crosses the blood-brain barrier. Kynurenate levels can also be raised by inhibiting the enzyme kynurenine-3-monoxygenase (KMO; also known as kynurenine-3-hydroxylase; Figure 1) (Röver et al., 1997), which normally oxidises kynurenine to 3-hydroxykynurenine (3HK). Inhibition of KMO, therefore, increases the amount of kynurenine available for transamination to kynurenic acid.
Increasing cerebral kynurenate levels in this way produces behavioural and cognitive changes similar to those encountered in psychiatric conditions. Prominent among these changes are effects on sensory processing exemplified by changes in pre-pulse inhibition of auditory startle stimuli (Nilsson et al., 2006). This phenomenon has received a great deal of attention since, although it occurs in a number of psychiatric conditions, it is particularly prominent in schizophrenia and is reduced by treatment with antipsychotic drugs. It is usually considered to reflect sensorimotor gating and integration, properties that may underlie some of the confusional state that exists in early phases of schizophrenia. The ease with which endogenous levels of kynurenic acid can be increased to alter pre-pulse inhibition (Erhardt et al., 2004; Nilsson et al., 2006) favours the concept that a pathological change in the kynurenine pathway could be involved in psychiatric dysfunction.
Increased levels of kynurenic acid have also been linked with deficits of spatial working memory (Chess et al., 2007). A particularly fascinating aspect of this finding is that it occurs not only after the acute elevation of kynurenate in adult animals, but also in adults several months after raising kynurenate levels perinatally or during adolescence (Akagbosu et al., 2012; Pocivavsek et al., 2012). This latter observation presents an intriguing parallel with current views on the development of schizophrenia which suggest a neurodevelopmental origin established pre- or neonatally and leading to the emergence of behavioural disorder in later life. In addition to spatial learning, kynurenic acid levels modulate stimulus processing and conditioned responding, while later work discovered changes in contextual fear conditioning and context discrimination (Chess and Bucci, 2006). There is some selectivity in these actions; however, since cue-specific fear conditioning is not affected (Chess et al., 2009).
More recently, attention has moved to more sophisticated tests of cognitive performance. Increasing kynurenic acid levels by injections of kynurenine has been found to produce deficiencies in attentional set-shifting paradigms. While the treated and control animals tested were all able to acquire the initial, simple, compound discrimination and the moderately difficult intradimensional shift, animals with elevated CNS kynurenate exhibited deficits in acquiring and handling the extradimensional shift task (Alexander et al., 2012; Pocivavsek et al., 2012).
Although the artificial elevation of kynurenate levels is a valuable experimental approach to modelling a possible pathological state, it does not address the question of whether normal, physiological levels of kynurenate are involved in baseline cognitive function. This question has been addressed by lowering the levels of endogenous kynurenate. The deletion of the major KAT isoenzyme KAT-II results in a substantial (approximately 70%) decrease in the extracellular concentration of kynurenic acid in the rat CNS and this is accompanied by improved cognitive performance in a range of behavioural tasks including exploration, object recognition and passive avoidance learning (Potter et al., 2010). These effects are paralleled by alterations of neuronal excitability and plasticity demonstrated by electrophysiological recordings in the hippocampus. Importantly, the selectivity of the kynurenic acid change resulting from KAT-II deletion was confirmed by the fact that there were no associated changes in the levels of the NMDAR agonist quinolinic acid (Sapko et al., 2006). It is work of this kind that has led to the development of KAT inhibitor compounds for potential use as cognitive enhancers (Rossi et al., 2010; Dounay et al., 2012).
Cognitive disorders
Schizophrenia
One of the disorders characterized almost exclusively by cognitive dysfunction is schizophrenia. A widely accepted current view of this disorder is that it results from underactivation of glutamate receptors – the ‘hypo-glutamatergic’ hypothesis. This concept has expanded the earlier focus on dopamine function in which the fundamental problem was thought to be overactivity of dopaminergic neurons in the ventral tegmento-striatal (accumbens) projections, associated with metabolic hypofunction in the prefrontal cortex (PFC). It now seems likely that changes in glutamate neurotransmission represent the more biochemically proximal defect in schizophrenia, which can lead secondarily to altered dopamine release and/or receptor expression. Certainly, a primary role for glutamate is supported by the development of animal models showing cognitive deficits related to those in schizophrenia but induced by subchronic treatments with NMDAR antagonists such as ketamine, dizocilpine [MK-801] or phencyclidine (Jentsch and Roth, 1999; Newcomer et al., 1999; Cochran et al., 2003). The use of phencyclidine in particular reproduces cognitive abnormalities seen either in the early positive or later negative phases of schizophrenia, including dysfunctional sensorimotor and contextual integration or pre-pulse inhibition (Jentsch and Roth, 1999; Cochran et al., 2003; Phillips and Silverstein, 2003). These deficits are difficult to reproduce with older, dopamine-centred models (Lindsley et al., 2006).
The concentration of kynurenic acid is increased within brain tissue or CSF of patients with schizophrenia, a result that has been replicated by several groups in different cohorts of patients (Schwarcz et al., 2001; Erhardt et al., 2004; Nilsson et al., 2005; 2006). The concentrations of kynurenic acid were several-fold higher than in normal control subjects, especially in the PFC where, as noted above, the ‘hypo-frontality’ characterized by reduced metabolic function has become the focus of interest in studies of schizophrenia. As predicted on this hypothesis, treatment with neuroleptic drugs reduces kynurenic acid levels in rodents (Ceresoli-Borroni et al., 2006).
The reason for the elevation of kynurenic acid concentrations in schizophrenia is still unclear although it may be caused by reduced levels of the enzymes KMO and 3-hydroxy-anthranilic acid oxidase (3HAO) (Figure 1). The mRNA, protein levels and activity of these enzymes are reduced in post-mortem samples of PFC from schizophrenic patients, while activities of the first enzyme of the pathway (IDO), KAT and quinolinate phosphoribosyl transferase are normal (Sathyasaikumar et al., 2011; Wonodi et al., 2011). Experimentally, administration to rodents of the KMO blocking compound 3,4-dimethoxy-N-[4-(3-nitrophenyl)thiazol-2-yl]benzenesulphonamide (Ro61-8048; Röver et al., 1997) produces a clear increase in kynurenic acid concentrations, supporting the concept that reducing enzyme activity does increase kynurenine production (Cozzi et al., 1999; Clark et al., 2005).
Although there are few studies to date on the genetics of tryptophan catabolism in relation to schizophrenia, there is an association between a single nucleotide polymorphism of KMO (rs1053230) and levels of kynurenic acid in the CSF (Holtze et al., 2012). The amino acid change produced by this mutation was considered important for the interaction between enzyme and its substrate (kynurenine) and may be considered a proof-of-concept that a minor genetic alteration can lead to increased kynurenic acid production.
While most of these studies have been performed on CNS tissue or CSF, there are reports of lowered kynurenic acid concentrations in the blood of patients (Myint et al., 2011) or a social isolation-induced rodent model (Moeller et al., 2012). The uncertain relationship between brain and blood measurements places serious limitations on the value of the latter for interpretation of events with the CNS, but a resolution of the discrepant results might lie in the role of the immune system in schizophrenia. The literature on individual cytokine levels or gene polymorphisms in schizophrenia is confusing and often contradictory, but there seems to be a consensus that immune activation contributes to the symptomatology of the disorder, especially in acute exacerbations of the disease. Increased immune function, primarily in the blood, tends to induce the tissue expression and activity of both IDO and KMO, leading to higher levels of most kynurenine metabolites. Increased levels of kynurenine will pass into the brain and generate increased kynurenic acid which, as a charged, acidic molecule, will be trapped within the brain. This may be further enhanced by the presence of increased concentrations in the PFC of TNF-α (Paterson et al., 2006) and other proinflammatory compounds which activate IDO. The conversion of kynurenine to kynurenic acid will induce the diffusion and transport of more kynurenine from the periphery. The various apparently conflicting data may therefore be integrated in a model that takes these peripheral and central pharmacokinetic factors into account to explain raised kynurenine and kynurenic acid levels in the CNS, at the same time as reduced levels in blood (Fig. 2).
At the neurochemical level, increasing kynurenate levels in experimental animals in vivo increases the activity of dopaminergic neurons (Nilsson et al., 2006; Linderholm et al., 2007) and facilitates the dopamine release induced by amphetamine (Olsson et al., 2012). On the other hand, several studies which have examined directly the release and extracellular concentrations of dopamine have shown that kynurenic acid decreases dopamine levels in the striatum (Rassoulpour et al., 2005). These results are supported by the inhibition or deletion of KAT, which leads to reduced concentrations of kynurenate and which increases dopamine release in the striatum (Amori et al., 2009). Some of these changes in dopamine release may be indirectly caused by effects of kynurenate on glutamate levels which are decreased in the striatum, hippocampus or PFC by local applications of kynurenate or treatment with kynurenine (Carpenedo et al., 2001; Pocivavsek et al., 2011; 2012). Indeed, kynurenate appears to be a physiological modulator of glutamate and dopamine release, since release of both is increased when resting, endogenous levels of kynurenate are reduced by deleting KAT (Potter et al., 2010; Pocivavsek et al., 2011). The reduction in extracellular glutamate levels by kynurenic acid is accompanied by impaired performance of mice or rats in tests of cognitive function such as the water-maze test for visuospatial memory and passive avoidance paradigms, while inhibition of KAT and the resulting lowering of kynurenic acid levels produces increased glutamate release and improved performance in these tasks as well as object exploration, object recognition and spatial discrimination. These facilitated behaviours were associated with increased synaptic plasticity at the cellular level, reflected in greater long-term potentiation in the hippocampus in vitro (Pocivavsek et al., 2011) (Fig. 2).
Some of the data discussed here may be relevant to other conditions in which psychosis forms a major symptom. Thus, a proportion of patients infected with the human immunodeficiency virus (HIV) may develop severe psychotic symptoms resembling those seen in schizophrenia. In these patients, the symptom severity appears to relate to the levels of kynurenic acid in the CSF (Atlas et al., 2007; Kandanearatchi and Brew, 2012).
Dementia
Dementia appears in various guises such as Alzheimer's disease and vascular dementia or, as it is now believed, combinations of these two conditions. Early studies focused on the potential neurotoxic effects of quinolinic acid, based on the several hundred-fold increase in the levels of this compound in the CSF of patients with HIV-linked dementia (Heyes et al., 1991). This disorder, now often known as HIV-associated neurocognitive disorder (Kandanearatchi and Brew, 2012) is usually reversible as the infection is treated and quinolinic acid concentrations fall.
Symptoms are accompanied by invasion of the CNS by inflammatory cytokines which gain entry after internalization by peripheral monocytes and macrophages which enter the CNS before being transformed into microglia. Growing evidence suggests the existence of an inflammatory component in dementia in which quinolinic acid may be involved by means of its ability to induce the expression of chemokines such as MCP-1 and RANTES as noted above (Guillemin et al., 2003). These proinflammatory molecules are produced by immune-competent cells in the periphery and by microglia in the CNS – cells functionally related to, and developed from, macrophages. A major action of MCP-1 is to attract and activate macrophages which release more quinolinic acid and potent inflammatory cytokines including the powerfully proinflammatory TNFα. Quinolinic acid can, therefore, initiate and promote the development of local inflammation within the CNS, a situation thought to lead to brain damage subsequently (Fig. 2).
It has not proved possible to show any simple relationship between quinolinic acid and dementia in other conditions such as Alzheimer's disease, even though deposits of β-amyloid proteins in the brain are characteristic of Alzheimer's disease and links have been noted with activity along the kynurenine pathway. Activity of IDO and quinolinic acid concentrations are increased in the amyloid plaques and microfibrillary tangles (Guillemin et al., 2005; Bonda et al., 2010) and β-amyloid peptides can induce IDO and kynureninase activities (Guillemin et al., 2003; Walker et al., 2006). Quinolinic acid produces glial activation and cytokine release consistent with a role in the inflammatory and cytotoxic processes occurring in Alzheimer's disease (Ting et al., 2009) while proinflammatory cytokines potentiate the activation of IDO by β-amyloid fragments including the most toxic Aβ(1–42) (Yamada et al., 2009), establishing a damaging positive feedback cycle. Nevertheless, there may be a more direct relationship between dementia and kynurenic acid, since levels of this compound in the blood and brain, together with the activity of IDO, increase with age to the extent that they correlate well with cognitive decline and imminent mortality in patients (Pertovaara et al., 2006). The gradual rise in kynurenate levels probably produces a progressively greater blockade of NMDAR in the CNS causing a generalized disruption of synaptic transmission. Inhibitors of KAT (see below) might slow this cognitive decline.
Recent work suggests a role for quinolinic acid in the accumulation of tau proteins, abnormally hyperphosphorylated forms of which constitute the microfibrillary tangles which are frequently observed in Alzheimer's disease (Rahman et al., 2009).
Huntington's disease
Although this condition is often first diagnosed on the appearance of motor symptoms, these are preceded by cognitive abnormalities which can be revealed by appropriate testing. Patients with an increased likelihood of developing this inherited disorder show impaired performance on tasks such as attentional set shifting and semantic verbal fluency (Lawrence et al., 1998). Later work extended this profile to show deficits in pattern and spatial recognition memory, simultaneous and delayed matching-to-sample and aspects of visual working memory (Lawrence et al., 2000). Most of the disturbed cognitive functions are known to involve the striatum, consistent with the later emergence of motor symptoms caused by striatal neurodegeneration.
Early animal models of the disease involved the non-selective destruction of striatal neurons using excitotoxins such as kainic acid. Later, with the recognition that the agonist activity of quinolinic acid at NMDA receptors (Stone and Perkins, 1981) translated into neurotoxicity (Schwarcz et al., 1983), it soon became apparent that quinolinate generated a more selective animal model. Intrastriatal delivery of quinolinic acid produces a profile of neurochemical, electrophysiological and behavioural changes that closely mimic those seen in patients with Huntington's disease (Beal et al., 1991; Popoli et al., 1994; Shear et al., 1998; Schwarcz et al., 2009).
More recently developed models involve engineering the expression of the abnormal Huntington's disease protein huntingtin, the sequence of which is terminated by a series of glutamine residues, coded for by cytosine-adenine-guanine (CAG) repeats in the gene. Abnormal huntingtin can form aggregates that ultimately lead to neuronal death. The neuronal damage and behavioural sequelae seen in these animals appear to be mediated through activation of NMDA receptors (Heng et al., 2009). Levels of quinolinic acid and 3HK are increased in the neocortex and striatum and are also increased in human patients (Guidetti et al., 2006), along with activity of their interconverting enzyme 3HAO (Schwarcz et al., 1988; Pearson and Reynolds, 1992; Guidetti et al., 2000). The initial enzyme, IDO is also more active in patients in the more advanced stages of the disease (Stoy et al., 2005). The role of kynurenines may be strengthened by the finding that 3HK can enhance the toxic effects of quinolinic acid (Guidetti and Schwarcz, 1999). In the R6/2 mouse model of Huntington's disease, activity of KMO is also increased (Sathyasaikumar et al., 2010), results which would contribute to the increased conversion of tryptophan to 3HK and quinolinic acid (Fig. 2). Intriguingly, quinolinic acid itself can promote the production of huntingtin (Tatter et al., 1995).
This potential involvement of the kynurenine pathway has been strongly supported by the discovery of a correlation between levels of components of the pathway and the length of the triplet codon CAG repeat (Forrest et al., 2010). The codon can be repeated up to 35 times in normal individuals, but if it exceeds 40, most patients exhibit symptoms of the disorder. The correlation between codon repeat number and kynurenine pathway metabolites is one of very few that have been demonstrated between the genetic abnormality and a biochemical parameter. The relationship is strengthened by the fact that the pre-motor cognitive dysfunction also correlates with the CAG repeat length (Jason et al., 1997).
An important discovery was that the kynurenine : tryptophan ratio correlated with levels of the proinflammatory cytokine interleukin-23, as did levels of kynurenic acid and anthranilic acid (Forrest et al., 2010). Increased levels of the latter compound had been reported previously in several inflammatory conditions (Darlington et al., 2010). Correlations were also revealed between kynurenine metabolites and blood concentrations of the soluble human leukocyte antigen-G (sHLA-G), an anti-inflammatory molecule. This would be consistent with earlier data that tryptophan and several kynurenine metabolites can induce cell surface and secreted forms of sHLA-G in immune system dendritic cells, leading to their reduced ability to activate T cells. This concept in turn would add to the evidence that several kynurenine metabolites have critical functions within the immune system as well as the nervous system (Stone et al., 2013). Interestingly, since IFN-γ is one of the most potent inducers of IDO and sHLA-G can induce the production of IFN-γ from peripheral blood mononuclear cells, it is likely that sHLA-G produces an indirect activation of the kynurenine pathway. The mechanisms by which IDO and sHLA-G act – separately or in combination – to affect T cell function, and the extent to which they do so in a manner relevant to the motor or cognitive symptoms of Huntington's disease, remain unclear. Overall, there are strong arguments for overactivity of these compounds being involved in Huntington's disease, probably generating cognitive symptoms via the kynurenine pathway (Fig. 2). This proposal has been further strengthened by the discovery that expression of abnormal huntingtin in yeast cells can produce cell damage by increasing activity along the kynurenine pathway (Giorgini et al., 2005; Stone et al., 2012).
Immune function, kynurenines and cognition
The relationship between kynurenines and immune function may have a wide relevance to ‘sickness behaviour’ and the lasting effects of infection on motor and cognitive function in conditions such as cerebral malaria (Clark et al., 2005) and trypanosomiasis (Rodgers et al., 2009). Indeed, many features of what is often referred to as ‘sickness behaviour’ may be linked to kynurenine metabolism, including the anxiety and anhedonia or apathy that seems to be induced by activation of IDO (Salazar et al., 2012). The induction of IDO and KMO by infecting organisms is probably secondary to the increased expression of IFN-γ but may be the mechanism through which infection by viruses can produce cognitive dysfunction. The infection results in altered hippocampal neuron structure and impairs reversal learning in an adaptation of the water maze test of spatial memory (Jurgens et al., 2012).
Activation of the kynurenine pathway is also likely to account for the depression which follows viral or bacterial infections and treatment of patients with interferons (Dantzer et al., 2008; O'Connor et al., 2009; Raison et al., 2010).
Stress
Stress causes cognitive changes and alterations of emotionality (Davidson and McEwen, 2012) which may lead to mental illness (de Kloet et al., 2005). This may occur directly in adults, especially after experiencing chronic stress over many months or years (Marin et al., 2011) but can also appear in later life after the exposure to stress in early postnatal life, with the appearance of defective memory and executive functioning (Hedges and Woon, 2011; Pechtel and Pizzagalli, 2011). Indeed, there is good evidence that prenatal stress in the pregnant female can lead to the emergence of cognitive and emotional problems in the offspring in later life (Lupien et al., 2009).
The negative effect of stress on cognition may be linked to the activation of tryptophan metabolism by TDO (Kiank et al., 2010; Miura et al., 2011). Whereas IDO is rather non-selective in its metabolism of compounds with a basic indole structure, (including 5HT and melatonin) TDO is much more selective for tryptophan and is a high-capacity, low-affinity enzyme which maintains the blood levels of tryptophan around 60 μM. Hence, its importance is primarily to regulate the amounts of tryptophan to which immune system cells in the tissues or cells within the CNS are exposed. The role of TDO may be far more important than often realized since it is induced by glucocorticoids. These adrenal steroids are secreted in response to stress and their induction of TDO can potentially alter the blood levels of kynurenines to a much greater extent than IDO (Fig. 2). This, therefore, is another mechanism by which an environmental factor – stress induced by a wide variety of life situations – could modify brain levels of quinolinic acid and kynurenic acid and modulate neuronal activity leading to changes in behaviour and cognitive function, or a susceptibility to external influences arising subsequently.
Exposure to corticosterone can markedly affect NMDAR function (Tse et al., 2012) and may lead to severe and persisting clinical depression and other disorders (Muscatell et al., 2009). For example, repeated stress has adverse effects on temporal order recognition tests (involving the PFC), associated with reduced NMDAR and AMPA receptor function (Yuen et al., 2012). Whether cognitive function is enhanced or suppressed depends on the amount of steroid present and the relative activation of glucocorticoid and mineralocorticoid receptors (Popoli et al., 2012). Glucocorticoids may affect glutamate receptors particularly in the PFC where they are believed to be crucial in the altered metabolic activity there linked with schizophrenia (Marin et al., 2011).
Post-surgical cognitive recovery
One further example of a link between kynurenines and cognition has emerged from a study of cognitive function in patients after cardiac bypass or major thoracic surgery, which should probably be viewed as major stressors. It is well established that major surgery under general anaesthesia is followed by cognitive deficits that may take months or years to resolve. In a study employing six independent tests of cognitive function, highly significant correlations were established between the levels of kynurenine metabolites and the degree of cognitive impairment tested at three time points after thoracic surgery (Forrest et al., 2011). No comparable correlations were seen between cognition and the levels of proinflammatory cytokines. The extent of the correlation was greater than had been noted for any molecular marker of intellectual functioning in previous studies. A similar result was reported more recently in which, for patients recovering from a stroke, the level of kynurenine pathway activation was correlated inversely with cognitive recovery (Gold et al., 2011), a finding which also supports the previously noted correlations between kynurenine pathway activation and the post-stroke infarct size (Darlington et al., 2007).
Peripheral kynurenines and the CNS
Expression of the kynurenine pathway is not confined to the CNS. Many peripheral tissues express IDO, while the liver has a predominance of the unrelated and less substrate-selective enzyme TDO. Infection or inflammation can induce the expression and activity of IDO, largely through the action of interferon-γ, while TDO is induced by glucocorticoids and therefore responds to stressful situations. Both enzymes lower tryptophan levels and increase kynurenine concentrations, with the latter leading to increased generation of the more distal metabolites such as quinolinic acid and kynurenic acid. An important interface exists with the CNS since tryptophan, kynurenine and 3HK can cross the blood-brain barrier quite readily, partly a result of simple diffusion across the vascular membranes and partly the result of active transport via the large neutral amino acid transporter (Speciale et al., 1989; Speciale and Schwarcz, 1990; Fukui et al., 1991; Eastman et al., 1992). This leads to increased concentrations of those compounds as well as quinolinic acid in the brain (Jauch et al., 1993). On the other hand, kynurenic acid and 3-hydroxy-anthranilic acid (3HAA) do not cross into the CNS to an appreciable extent, but their concentrations are elevated markedly after systemic administration of kynurenine (Miller et al., 1992; Nozaki and Beal, 1992). Quinolinic acid does not normally cross the blood-brain barrier but, like many compounds, may do so when integrity of the barrier is compromised (Vezzani et al., 1989). These considerations mean that events largely limited to the periphery but which, via IDO or TDO, alter tryptophan/kynurenine ratios in the blood, can produce significant secondary changes in the amounts of kynurenine metabolites in the CNS (Saito et al., 1992), contributing no doubt to the effects of immune activity and stress as noted above.
Since increasing the concentration of kynurenic acid in the brain is neuroprotective, several groups have explored the possibility of using analogues or prodrugs of kynurenate for systemic administration. Nagy et al. (2011), for example, have reported on the ability of a derivative of kynurenic acid to cross the blood-brain barrier, leading to significant preservation of morphological and electrophysiological characteristics in the rodent hippocampus following a period of forebrain ischaemia (Gellert et al., 2011).
In addition to the role of endothelial transporters in the regulation of kynurenine concentrations in the CNS, the endothelial cells themselves may play an important role in determining the extent of tryptophan metabolism and thus the amounts of metabolites available for transport (Fig. 2). Kynurenine is taken up by vascular endothelial cells where it is converted to kynurenic acid via KAT (Wejksza et al., (2004). The rate of that conversion changes with local levels of acidity and oxygenation, leading to suggestions that it might play a modulatory role in vascular or neuronal function partly mediating tissue responses to local perfusion (Stazka et al., 2002; Wang et al., 2010).
The possible link between vascular function and neuromodulation (central or peripheral) is supported by the ability of glutamate to inhibit kynurenic acid generation. In addition, L-homocysteine or S-adenosyl-homocysteine can exert biphasic effects on kynurenic acid synthesis depending on concentration (Stazka et al., 2005). This has in turn led to proposals that endothelial kynurenines may contribute to arterial vessel relaxation and the control of blood pressure. The presence of infection or inflammation induces endothelial IDO which, in addition to decreasing the local tryptophan/kynurenine ratio, produces hypotension. The systemic administration of kynurenine decreases blood pressure in hypertensive rats. Conversely, inhibition of IDO increases blood pressure in mice. Both tryptophan and kynurenine dilate coronary arteries although the tryptophan effect results from its conversion to kynurenine in the endothelium. Overall, the expression of IDO in endothelial cells may contribute to the physiopathological regulation of vascular tone, and changes of vascular tone may influence neuronal activity via the associated alterations of kynurenine metabolism.
Furthermore, since homocysteine has been linked with the production of atherosclerosis, it has been suggested that this condition could be caused by the homocysteine-induced suppression of kynurenic acid production. Kynurenic acid itself appears to promote both the proliferation and migration of endothelial cells and the inhibition of these parameters by homocysteine could produce a degree of cellular stasis which might eventually form the basis of atherosclerotic disease. Certainly, increasing kynurenic acid concentrations prevents the toxic effects of homocysteine on endothelial cells (Wejksza et al., (2009) and should therefore protect against atherosclerosis. Quinolinic acid may also affect vascular cell function since local administration in the striatum is accompanied by extensive vascular remodelling (Ryu et al., 2009), although the extent to which this activity is shared by agonists at other glutamate receptors, or neurotoxins acting independently of glutamate receptors, is not clear.
The relevance of these various vascular considerations to the CNS is that tryptophan/kynurenine metabolism in endothelial cells may be an important contributor to the regulation of neurodegeneration and cognition. The induction of IDO in endothelial cells by inflammatory mediators including INF-γ, and the resulting changes of local tryptophan and kynurenine concentrations are likely to alter the levels of both compounds and their metabolites in the CNS, especially since there is evidence that the kynurenine generated is secreted preferentially from the basolateral pole of the endothelial cells, gaining direct access to the cerebral aspect of the blood-brain barrier (Owe-Young et al., 2008). This may be particularly important in conditions with a compromised blood-brain barrier. This relationship may be of special significance for understanding the cognitive and neurodegenerative effects of HIV infection in which there is strong evidence for a role of quinolinic acid neurotoxicity (Kandanearatchi and Brew, 2012). Local changes in tryptophan and kynurenine metabolite levels will also affect immune tolerance which may further exacerbate susceptibility of the CNS in some individuals (Owe-Young et al., 2008).
Kynurenines and brain development
If changes along the kynurenine pathway, whether induced by stress, infection, surgery or other factors, are involved in CNS abnormalities in the adult, the question arises as to whether changes in the pathway early in life, or even prenatally by conditions affecting the pregnant mother, could influence cognitive function or behaviour in the offspring. Factors such as stress or infection could predispose some individuals to develop behavioural abnormalities when exposed to subsequent life events which would have less or no effect on individuals not exposed to those factors. In all these situations, the activation of tryptophan metabolism along the kynurenine pathway induced by steroids, cytokines or other agents released by stress and infection may be the common factor mediating changes in glutamate receptor function.
Prenatal exposure to the viral-mimetic double-stranded RNA complex polyinosinic : polycytidylic acid can selectively alter the expression of a number of proteins involved in early neurogenesis, axonal guidance and synapse formation in the brains of the postnatal offspring examined 21 days after birth (Forrest et al., 2012). The changes observed included decreased expression of the NMDAR subunit GluN1, while subsequent work (unpublished) also indicates changes in the morphogenetic protein sonic hedgehog and α-synuclein levels, combined with an increased expression of tyrosine hydroxylase. Since these effects may involve the activation of the kynurenine pathway, studies were performed on the effects of inhibiting 3-hydroxy-kynurenine production using Ro61-8048.
When pregnant rats were treated prenatally with Ro61-8048, there was a substantial (more than 10-fold) increase in the brain levels of kynurenic acid together with decreased expression of GluN2A subunits and increased GluN2B in the embryo brains after only 5 h, with changes in sonic hedgehog protein after 24 h (Forrest et al., 2013). When the offspring were allowed to mature to 21 days of postnatal age, the brains exhibited increased expression of GluN2A, GluN2B and the associated postsynaptic density protein PSD-95. Levels of sonic hedgehog were now increased relative to vehicle-exposed animals, while increased expression of doublecortin and proliferating cell nuclear antigen suggested increased neurogenesis (Forrest et al., 2013). The overall amounts and ratio between the different glutamate receptor subunits is increasingly considered to be critical in synaptic function, plasticity and neurodegeneration (Hardingham and Bading, 2011). These molecular changes in early brain development were associated with enhanced neuronal excitability in the postnatal offspring and increased long-term potentiation, which is considered critical in some aspects of cognitive function (Forrest et al., 2013).
An important link between these various changes and behaviour is indicated by work showing that the administration of kynurenine to rats during adolescence can lead to impairments in social behaviour in adulthood while identical treatment to adults has no such effect (Trecartin and Bucci, 2011). In the related study referred to earlier (Pocivavsek et al., 2012), kynurenine treatment before and immediately after birth produced not only the expected increase in kynurenic acid levels but also produced significant cognitive dysfunction in adulthood, including abnormal performance in passive avoidance learning and in the water maze spatial learning task.
Nicotinic cholinoceptors
The focus of attention, in the relationship between kynurenines and cognition, continues to be on NMDAR but there is evidence that kynurenic acid may also have effects on α7-homomeric nicotinic receptors (α7NRs) for acetylcholine. Modifications in the expression and function of α7NRs can result in marked changes in some aspects of cognitive behaviour (Deiana et al., 2011; Graef et al., 2011). The septohippocampal projections include a substantial cholinergic component in which the release of acetylcholine can activate muscarinic and nicotinic receptors on the target neurones. A similar projection from the nucleus basalis to the neocortex seems to underlie aspects of learning as well as logical thought and reasoning. The α7NRs are involved in cognitive function and may be underactive in schizophrenia (Breese et al., 2000; Mexal et al., 2010), while their deletion has profoundly negative consequences for learning and memory (Hernandez et al., 2010). Conversely, selective agonists at α7NRs can suppress the positive and negative symptoms of schizophrenia, including aspects of cognitive function such as visual recognition, logic and rational thought (Mueller and Dursun, 2011). In the early stages of Alzheimer's disease, nicotinic cholinergic agonists or cholinesterase inhibitors have beneficial effects, restoring some degree of memory and reasoning in the disease and in animal models (Kihara et al., 2001; Ren et al., 2007).
The evidence for a role of kynurenic acid in modulating nicotinic receptor function, however, remains the subject of intense debate. The initial observations were reported using neuronal cultures in which depolarization was produced by agonists at α7NRs (Hilmas et al., 2001) and kynurenic acid appeared to be more potent an antagonist than at NMDARs. Subsequent work failed to confirm this greater potency in intact brain slices in which kynurenic acid blocked NMDARs and α7NRs with comparable potency (Stone, 2007). The differences between these conclusions may be related to the presence and access of endogenous glutamate and kynurenic acid itself to subsynaptic or extra-synaptic sites, which will be more accessible in cell culture than in slices (Ren et al., 2007). Subsequent studies have examined the phenomenon in more detail (Alkondon et al., 2004; 2011a,b) while others have failed to demonstrate any antagonism at α7NRs using concentrations of kynurenic acid which completely block responses to NMDA (Mok et al., 2009; Dobelis et al., 2012). In addition, a study of dopaminergic activation by kynurenic acid (discussed above) concluded that α7NRs were not involved, suggesting that the phenomenon was probably caused exclusively by blockade of NMDARs (Linderholm et al., 2007). Interestingly, the loss of kynurenic acid produced by the deletion of KAT-II was associated with increased sensitivity of α7NRs which was not caused by increased receptor expression (Alkondon et al., 2004). Overall, it is possible that the apparent effects of kynurenic acid on α7NRs may be secondary to the effects of kynurenic acid on NMDARs.
Drug development
It will be clear that there are several approaches to modulating activity along the kynurenine pathway that may have therapeutic potential in the treatment of cognitive disorders. One approach is the production of analogues or prodrugs of kynurenic acid, as described in an earlier section (Gellert et al., 2011; Nagy et al., 2011). In general, most medicinal chemistry has been focused on the inhibition of KMO or IDO. In neurodegenerative disorders such as Huntington's disease, the excitotoxic activity of quinolinic acid and the experimental evidence for the importance of this or other metabolites such as 3HK suggest that inhibition of KMO would be neuroprotective by reducing the generation of these toxins (Carpenedo et al., 1994; Pellicciari et al., 1994; Chiarugi et al., 1995; 1996). The continuing development of interest in kynurenine pathway inhibitors, especially for KMO (Cesura and Röver, 1999), is reflected in the growing number of patents being taken for novel series of compounds with acceptable levels of inhibitory activity. These include series of 2-aminobenzoyl compounds (Daily et al., 2010; Zisapel et al., 2012) (Figure 3A,B), pyrrole derivatives (Della Torre et al., 2001) (Figure 3C), a variety of 2-amino-4-phenyl-4-oxo-butanoic acid derivatives (Varasi et al., 2001) (Figure 3D) as well as a range of 5-(3-phenyl-3-oxo-propyl)-1H-tetrazole derivatives (Pevarello et al., 2000) (Figure 3E) and 4-phenyl-4oxo-2-butenoic acid derivatives (Giordani et al., 2000) (Figure 3F). Some of the compounds produced by these latter two groups showed IC50 values less than 1 μM against KMO, while three of the series synthesized by Varasi et al., (2001) had IC50s between 140 and 270nM. A different chemical avenue involved the development of cyclopropane-1-carboxylic acids (Benatti et al., 2005) (Figure 3G).
Figure 3.
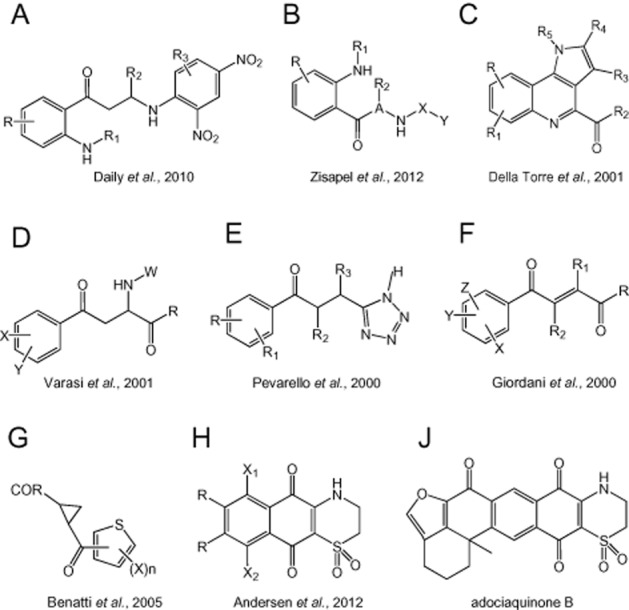
Structures of compounds described in patents for inhibitors of kynurenine-3-monoxygenase or indoleamine-2,3-dioxygenases targeted as CNS disorders.
Most of these patents specify a wide range of potential clinical applications, including neurodegenerative conditions such as Huntington's disease and stroke, while others have focused primarily on the role of kynurenine pathway involvement in disorders such as Huntington's disease (Muchowski et al., 2011) or on the cerebral side effects of metabolic disorders such as diabetes (Autier et al., 2010). On addition to patenting the concept of examining the kynurenine pathway as a route to the treatment of Huntington's disease (Muchowski and Giorgini, 2009), the group generated a series of compounds, several of which inhibited KMO by more than 50% at a concentration of 10 μM.
Several multicompound patents cover compounds designed to inhibit IDO (Andersen et al., 2012) (Figure 3H,J). Of a large series of compounds, the most potent was adociaquinone B (Figure 3J) with a Ki of 25nM against IDO. The inhibition of IDO may involve actions on both the CNS and immune systems and are more often being considered for their potential anti-cancer properties. IDO is a key player in immune surveillance and its control over infectious organisms and cancer cell removal. Although it is still considered possible that this role of IDO is mediated partly through the local depletion (starvation) of tryptophan concentrations resulting from its activation, it is increasingly thought that downstream metabolites of the kynurenine pathway, especially 3HK and 3HAA are involved, in view of their ability to alter the balance between proinflammatory and anti-inflammatory T cell populations such as Th1, Th2 and Th17. In fact, the potential impact of KMO inhibition on the immune system has not yet been explored in depth, so that it is not possible to state whether KMO inhibition would have any deleterious effects on immune function under basal or challenged conditions, to the extent that these would compromise their therapeutic utility.
In addition, KMO inhibition seems to have little effect on quinolinic acid concentrations, either because of compensatory changes in enzyme expression or activity, or because of the shift to an alternative pathway. The major suspect in the latter case would be the metabolism of kynurenine to anthranilic acid followed by the spontaneous oxidation of this compound to 3HAA and the normal conversion of the latter to quinolinic acid. Interestingly, there is growing evidence that this route is recruited in a range of inflammatory conditions in which the levels of anthranilic acid are increased several-fold above normal levels (Darlington et al., 2010).
KAT inhibition
A different therapeutic approach to the kynurenine pathway lies in the possibility of reducing kynurenic acid levels in order to decrease the overactivity in dopaminergic projections which characterizes psychotic disorders such as schizophrenia. Schizoid delusions and hallucinations can also follow the chronic use of psychedelic agents such as cannabis and may develop in the advanced phases of Parkinson's disease treated with dopamine-facilitating agents. Dopaminergic overactivity may be secondary to the hypoglutamatergic activity caused by increased kynurenic acid concentrations in these various situations.
The simplest practical approach to achieving reliably reduced levels of kynurenic acid in vivo would be to lower production by inhibiting KAT, since this not only has the desired effect on kynurenic acid production but seems to create little interference with the rest of the kynurenine metabolic pathway. Simple inhibitors such as L-cysteine sulphinate were described by Kocki et al. (2003), producing substantial inhibition at low micromolar concentrations. Several other relatively non-selective compounds have been shown to have inhibitory activity including the nitric oxide synthase inhibitor L-nitroarginine (Luchowski et al., 2001), 3-nitropropionic acid and 1-methyl-4-phenyl-pyridinium (Luchowski et al., 2002).
Several groups have subsequently produced more specific chemical structures with high selectivity and potency as KAT inhibitors. One of the first highly specific targeted compounds was (S)-4-(ethylsulfonyl)benzoylalanine (ESBA; Figure 4A) (Pellicciari et al., 2006) which showed high selectivity against rat KAT with little effect on any of the other enzymes of the kynurenine pathway. When administered to rats, ESBA reduced kynurenic acid levels in the brain and produced a corresponding increase in the release and extracellular concentrations of acetylcholine (Zmarowski et al., 2009), dopamine (Amori et al., 2009) and glutamate (Konradsson-Geuken et al., 2010). In addition to these neurochemical effects, the intracerebroventricular administration of ESBA to rats produced a significantly improved visuospatial memory performance in a water maze paradigm (Pocivavsek et al., 2011). Involvement of kynurenic acid in these neurochemical and behavioural effects of ESBA were prevented by the simultaneous administration of kynurenine or kynurenic acid itself. Unfortunately, later work indicated that ESBA was much less active as an inhibitor of the human KAT enzyme because of a higher molecular flexibility in the region of the substrate binding site (Casazza et al., 2011).
Figure 4.
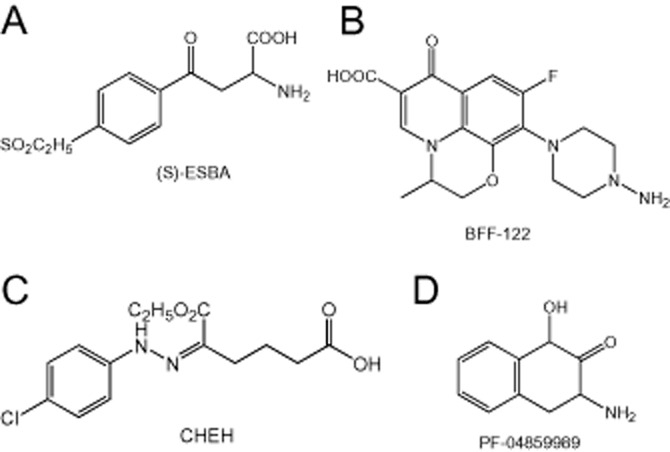
Structures of compounds described in patents for inhibitors of kynurenine aminotransferase.
A second compound developed as a more potent but still selective inhibitor was (S)-(-)-9-(4-aminopiperazin-1-yl)-8-fluoro-3-methyl- 6-oxo-2,3-dihydro-6H-1-oxa- 3a-azaphenalene-5-carboxylic acid (BFF-122; Figure 4B) (Rossi et al., 2010). This agent blocks KAT activity of in vitro brain slices (Alkondon et al., 2011a) and in vivo after direct, intrastriatal administration (Amori et al., 2009) but has not been examined in as much detail as ESBA, or in behavioural tests.
Akladios et al. (2012) reported that 6-ethoxy-6-oxo-5-(2-phenylhydrazono) hexanoic acid and 3-(2-carboxyethyl)-1H-indole- 2-carboxylic acid were promising compounds from which to derive novel inhibitors of human KAT-I. Of the 12 derivatives described, the most active was 5-(2-(4-chlorophenyl)-hydrazono)-6-ethoxy-6-oxohexanoic acid (CHEH; Figure 4C) which exhibited an IC50 of 19.8 μM. Even this level of activity is sometimes considered inadequate for the development of clinically useful drugs, especially when concentrations comparable with the IC50 must be achieved within the CNS without accompanying side effects that might be produced by the inevitably higher concentrations existing peripherally.
One route to compounds with significantly higher activity may be that pursued by Dounay et al. (2012) who generated the bicyclic compound PF-04859989 (Figure 4D) as a potent and selective inhibitor of human and rat KAT-II with an IC50 of approximately 20 nM. X-ray crystal structure and C-13 NMR studies of PF-04859989 bound to KAT-II reveal the formation of a covalent complex between the compound and pyridoxal phosphate, a key co-factor for KAT-II activity. The formation of this adduct effectively blocked activity of the enzyme in an irreversible fashion. A strong advantage of PF-04859989 over previous inhibitors is its ability to penetrate the CNS relatively readily. The same group has now extended the chemical family represented by PF-04859989 with a series of isosteric analogues, also active in the nanomolar range, which retain good penetration into the CNS after systemic administration (Henderson et al., 2013). Although no behavioural data have yet been reported using these compounds, they appear to have a promising, non-toxic profile which could lead to their further development.
Since kynurenic acid acts primarily at the Gly-B-binding site for glycine, acting partly in a competitive manner, the combined use of a KAT inhibitor with a glycine transport inhibitor to increase extracellular levels of glycine could represent an important synergistic approach yet to be tested experimentally.
A number of patents explore the molecular flexibility of blocking KAT using endogenous compounds as inhibitors with potential clinical utility. Some of these are targeted specifically at KAT, including a variety of naturally occurring aliphatic compounds (Guidetti et al., 2008) while others are intended as more general inhibitors of transaminases with the ability to include inhibition of KAT (Teichberg, 2008; 2010). Since most transaminases have limited selectivity for individual enzymes, the overall balance of inhibitory activity is probably similar with these two approaches.
Summary
The kynurenine pathway generates a series of neuroactive compounds, the most prominent of which can modulate the activity of neuronal pathways by altering the degree of activation (quinolinic acid) or blockade (kynurenic acid) of NMDARs. This review has highlighted some of the disorders for which there is strong evidence implicating the kynurenines in the behavioural and cognitive symptoms. With several enzymes along the route, the kynurenine pathway is eminently suitable for the development of pharmacological interventions to treat and, possibly, to prevent cognitive dysfunction in these and other CNS disorders.
Acknowledgments
The authors' own work referenced here was supported by the Medical Research Council, Biotechnology and Biological Sciences Research Council, The Wellcome Trust, Epsom Medical Research, The Peacock Trust and the Haddon Family Trust.
Conflict of interest
The authors declare that they have no conflicts of interest in the writing of this review.
References
- Akagbosu C, Evans GC, Gulick D, Suckow RF, Bucci DJ. Exposure to kynurenic acid during adolescence produces memory deficits in adulthood. Schizophr Bull. 2012;38:769–778. doi: 10.1093/schbul/sbq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akladios FN, Nadvi NA, Park J, Hanrahan JR, Kapoor V, Gorrell MD, et al. Design and synthesis of novel inhibitors of human kynurenine aminotransferase-I. Bioorg Med Chem Lett. 2012;22:1579–1581. doi: 10.1016/j.bmcl.2011.12.138. [DOI] [PubMed] [Google Scholar]
- Alexander KS, Wu H-Q, Schwarcz R, Bruno JP. Acute elevations of brain kynurenic acid impair cognitive flexibility: normalization by the alpha7 positive modulator galantamine. Psychopharmacology (Berl) 2012;220:627–637. doi: 10.1007/s00213-011-2539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EFR, Yu P, Arruda EZ, Almeida LEF, Guidetti P, et al. Targeted deletion of the kynurenine aminotransferase II gene reveals a critical role of endogenous kynurenic acid in the regulation of synaptic transmission via alpha 7 nicotinic receptors in the hippocampus. J Neurosci. 2004;24:4635–4648. doi: 10.1523/JNEUROSCI.5631-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EFR, Albuquerque EX. Endogenous activation of nAChRs and NMDA receptors contributes to the excitability of CA1 stratum radiatum interneurons in rat hippocampal slices: effects of kynurenic acid. Biochem Pharmacol. 2011a;82:842–851. doi: 10.1016/j.bcp.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EFR, Eisenberg HM, Kajii Y, Schwarcz R, Albuquerque EX. Age dependency of inhibition of alpha-7 nicotinic receptors and tonically active N-methyl-D-aspartate receptors by endogenously produced kynurenic acid in the brain. J Pharmacol Exp Ther. 2011b;337:572–582. doi: 10.1124/jpet.110.177386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amori L, Wu H-Q, Marinozzi M, Pellicciari R, Guidetti P, Schwarcz R. Specific inhibition of kynurenate synthesis enhances extracellular dopamine levels in the rodent striatum. Neuroscience. 2009;159:196–203. doi: 10.1016/j.neuroscience.2008.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen RJ, Pereira A, Huan X-H, Mauk G, Vottero E, Roberge M, et al. Patent US 7799776: indoleamine-2,3-dioxygenase (IDO) inhibitors. Alexandria, VA: US Patent and Trademark Offices; 2012. [Google Scholar]
- Atlas A, Gisslen M, Nordin C, Lindstrom L, Schwieler L. Acute psychotic symptoms in HIV-1 infected patients are associated with increased levels of kynurenic acid in cerebrospinal fluid. Brain Behav Immun. 2007;21:86–91. doi: 10.1016/j.bbi.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Autier V, De Vacqueur AA, Moinet G, Mariais D, Kargar C, Kergoat M. Patent US 7727977B2: kynurenine-3-hydroxylase inhibitors for the treatment of diabetes. Alexandria, VA: US Patent and Trademark Offices; 2010. [Google Scholar]
- Backman L, Lindenberger U, Li S-C, Nyberg L. Linking cognitive aging to alterations in dopamine neurotransmitter functioning: recent data and future avenues. Neurosci Biobehav Rev. 2010;34:670–677. doi: 10.1016/j.neubiorev.2009.12.008. [DOI] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ, Swartz KJ, Kowall NW. Chronic quinolinic acid lesions in rats closely resemble Huntington's disease. J Neurosci. 1991;11:1649–1659. doi: 10.1523/JNEUROSCI.11-06-01649.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatti L, Caccia C, Fariello R, Pellicciari R, Salvati P. Patent EP1565451: halothienoyl-cyclopropane-1-carboxylic acid derivatives. Munich, Germany: European Patent Office; 2005. [Google Scholar]
- Bonda DJ, Mailankot M, Stone JG, Garrett MR, Staniszewska M, Castellani RJ, et al. Indoleamine 2,3-dioxygenase and 3-hydroxy-kynurenine modifications are found in the neuropathology of Alzheimer's disease. Redox Rep. 2010;15:161–168. doi: 10.1179/174329210X12650506623645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese CR, Lee MJ, Adams CE, Sullivan B, Logel J, Gillen KM, et al. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23:351–364. doi: 10.1016/S0893-133X(00)00121-4. [DOI] [PubMed] [Google Scholar]
- Carpenedo R, Chiarugi A, Russi P, Lombardi G, Carla V, Pellicciari R, et al. Inhibitors of kynurenine hydroxylase and kynureninase increase cerebral formation of kynurenate and have sedative and anticonvulsant activities. Neuroscience. 1994;61:237–244. doi: 10.1016/0306-4522(94)90227-5. [DOI] [PubMed] [Google Scholar]
- Carpenedo R, Pittaluga A, Cozzi A, Attucci S, Galli A, Raiteri M, et al. Presynaptic kynurenate-sensitive receptors inhibit glutamate release. Eur J Neurosci. 2001;13:2141–2147. doi: 10.1046/j.0953-816x.2001.01592.x. [DOI] [PubMed] [Google Scholar]
- Casazza V, Rossi F, Rizzi M. Biochemical and structural investigations on kynurenine aminotransferase II: an example of conformation-driven species-specific Inhibition. Curr Top Med Chem. 2011;11:148–157. doi: 10.2174/156802611794863599. [DOI] [PubMed] [Google Scholar]
- Castellano C, Cestari V, Ciamei A. NMDA receptors and learning and memory processes. Curr Drug Targets. 2001;2:273–281. doi: 10.2174/1389450013348515. [DOI] [PubMed] [Google Scholar]
- Ceresoli-Borroni G, Rassoulpour A, Wu H-Q, Guidetti P, Schwarcz R. Chronic neuroleptic treatment reduces endogenous kynurenic acid levels in rat brain. J Neural Transm. 2006;113:1355–1365. doi: 10.1007/s00702-005-0432-z. [DOI] [PubMed] [Google Scholar]
- Cesura A, Röver S. Patent: US 5877193: use of N-(4-aryl-thiazol-2-yl)-sulfonamides. Alexandria, VA: US Patent and Trademark Offices; 1999. [Google Scholar]
- Chess AC, Bucci DJ. Increased concentration of cerebral kynurenic acid alters stimulus processing and conditioned responding. Behav Brain Res. 2006;170:326–332. doi: 10.1016/j.bbr.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Chess AC, Simoni MK, Alling TE, Bucci DJ. Elevations of endogenous kynurenic acid produce spatial working memory deficits. Schizophr Bull. 2007;33:797–804. doi: 10.1093/schbul/sbl033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess AC, Landers AM, Bucci DJ. L-kynurenine treatment alters contextual fear conditioning and context discrimination but not cue-specific fear conditioning. Behav Brain Res. 2009;201:325–331. doi: 10.1016/j.bbr.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Chiarugi A, Carpenedo R, Molina M, Mattoli L, Pellicciari R, Moroni F. Comparison of the neurochemical and behavioral-effects resulting from inhibition of kynurenine hydroxylase and/or kynureninase. J Neurochem. 1995;65:1176–1183. doi: 10.1046/j.1471-4159.1995.65031176.x. [DOI] [PubMed] [Google Scholar]
- Chiarugi A, Carpenedo R, Moroni F. Kynurenine disposition in blood and brain of mice: effects of selective inhibitors of kynurenine hydroxylase and of kynureninase. J Neurochem. 1996;67:692–698. doi: 10.1046/j.1471-4159.1996.67020692.x. [DOI] [PubMed] [Google Scholar]
- Clark CJ, Mackay GM, Smythe GA, Bustamante S, Stone TW, Phillips RS. Prolonged survival of a murine model of cerebral malaria by kynurenine pathway inhibition. Infect Immun. 2005;73:5249–5251. doi: 10.1128/IAI.73.8.5249-5251.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ. Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology. 2003;28:265–275. doi: 10.1038/sj.npp.1300031. [DOI] [PubMed] [Google Scholar]
- Cozzi R, Carpenedo R, Moroni F. Kynurenine hydroxylase inhibitors reduce ischaemic brain damage: studies with (m-nitrobenzoyl)alanine and 3,4-dimethoxy-[N-4-(nitrophenyl)thiazol-2-yl]-benzenesulfonamide (Ro 61-8048) in models of focal or global ischaemia. J Cereb Blood Flow Metab. 1999;19:771–777. doi: 10.1097/00004647-199907000-00007. [DOI] [PubMed] [Google Scholar]
- Cui Y, Jin J, Zhang X, Xu H, Yang LG, Du D, et al. Forebrain NR2B overexpression facilitating the prefrontal cortex long-term potentiation and enhancing working memory function in mice. PLoS ONE. 2011;6:e20312. doi: 10.1371/journal.pone.0020312. AR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily D, Laudon M, Zisapel N. Patent EP 1644316B1: 2-aminobenzoyl derivatives. Munich, Germany: European Patent Office; 2010. [Google Scholar]
- Dalley JW, Cardinal RN, Robbins TW. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci Biobehav Rev. 2004;28:771–784. doi: 10.1016/j.neubiorev.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–57. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlington LG, Mackay GM, Forrest CM, Stoy N, George C, Stone TW. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur J Neurosci. 2007;26:2211–2221. doi: 10.1111/j.1460-9568.2007.05838.x. [DOI] [PubMed] [Google Scholar]
- Darlington LG, Forrest CM, Mackay GM, Stoy N, Smith RA, Smith AJ, et al. On the biological significance of the 3-hydroxyanthranilic acid: anthranilic acid ratio. Int J Tryptophan Res. 2010;3:51–59. doi: 10.4137/ijtr.s4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson RJ, McEwen BS. Social influences on neuroplasticity: stress and interventions to promote well-being. Nat Neurosci. 2012;15:689–695. doi: 10.1038/nn.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiana S, Platt B, Riedel G. The cholinergic system and spatial learning. Behav Brain Res. 2011;221:389–411. doi: 10.1016/j.bbr.2010.11.036. [DOI] [PubMed] [Google Scholar]
- Della Torre A, Heidempergher F, Pevarello P, Speciale C, Varasi M. Patent EP 0922044: pyrrolo(3,2c) derivatives. Munich, Germany: European Patent Office; 2001. [Google Scholar]
- DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci. 2010;115:89–97. doi: 10.1093/toxsci/kfq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobelis P, Staley KJ, Cooper DC. Lack of modulation of nicotinic acetylcholine alpha-7 receptor currents by kynurenic acid in adult hippocampal interneurons. PLoS ONE. 2012;7:e41108. doi: 10.1371/journal.pone.0041108. AR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dounay AB, Anderson M, Bechle BM, Campbell BM, Claffey MM, Evdokimov A, et al. Discovery of brain-penetrant, irreversible kynurenine aminotransferase II inhibitors for schizophrenia. ACS Med Chem Lett. 2012;3:187–192. doi: 10.1021/ml200204m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman CL, Guilarte TR, Lever JR. Uptake of 3-hydroxykynurenine measured in rat-brain slices and in a neuronal cell-line. Brain Res. 1992;584:110–116. doi: 10.1016/0006-8993(92)90883-b. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Schwieler L, Emanuelsson C, Geyer M. Endogenous kynurenic acid disrupts prepulse inhibition. Biol Psychiatry. 2004;56:255–260. doi: 10.1016/j.biopsych.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Forrest CM, Mackay GM, Stoy N, Spiden SL, Taylor R, Stone TW, et al. Blood levels of kynurenines, interleukin-23 and sHLA-G at different stages of Huntington's disease. J Neurochem. 2010;112:112–122. doi: 10.1111/j.1471-4159.2009.06442.x. [DOI] [PubMed] [Google Scholar]
- Forrest CM, Mackay GM, Oxford L, Millar K, Darlington LG, Higgins MJ, et al. Kynurenine metabolism predicts cognitive function in patients following cardiac bypass and thoracic surgery. J Neurochem. 2011;119:136–152. doi: 10.1111/j.1471-4159.2011.07414.x. [DOI] [PubMed] [Google Scholar]
- Forrest CM, Khalil OS, Pisar M, Smith RA, Darlington LG, Stone TW. Prenatal activation of Toll like receptors-3 by administration of the viral mimetic poly(I:C) changes synaptic proteins, N-methyl-D-aspartate receptors and neurogenesis markers in offspring. Mol Brain. 2012;5:22. doi: 10.1186/1756-6606-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest CM, Khalil OS, Pisar M, Darlington LG, Stone TW. Prenatal inhibition of the tryptophan-kynurenine pathway alters synaptic plasticity and protein expression in the rat hippocampus. Brain Res. 2013;1504:1–15. doi: 10.1016/j.brainres.2013.01.031. [DOI] [PubMed] [Google Scholar]
- Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR. Blood-brain-barrier transport of kynurenines – implications for brain synthesis and metabolism. J Neurochem. 1991;56:2007–2017. doi: 10.1111/j.1471-4159.1991.tb03460.x. [DOI] [PubMed] [Google Scholar]
- Gellert L, Fuzik J, Goebloes A, Sarkoezi K, Marosi M, Kis Z, et al. Neuroprotection with a new kynurenic acid analog in the four-vessel occlusion model of ischemia. Eur J Pharmacol. 2011;667:182–187. doi: 10.1016/j.ejphar.2011.05.069. [DOI] [PubMed] [Google Scholar]
- Giordani A, Pevarello P, Speciale C, Varasi M. Patent US 6048896: 4-phenyl-4-oxo-2-butenoic acid derivatives with kynurenine-3-hydroxylase inhibiting activity. Alexandria, VA: US Patent and Trademark Offices; 2000. [Google Scholar]
- Giorgini F, Guidetti P, Nguyen QV, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine-3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005;37:526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold AB, Herrmann N, Swardfager W, Black SE, Aviv RI, Tennen G, et al. The relationship between indoleamine 2,3-dioxygenase activity and post-stroke cognitive impairment. J Neuroinflammation. 2011;8:17. doi: 10.1186/1742-2094-8-17. AR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef S, Schoenknecht P, Sabri O, Hegerl U. Cholinergic receptor subtypes and their role in cognition, emotion and vigilance control: a review of preclinical and clinical findings. Psychopharmacology (Berl) 2011;215:205–229. doi: 10.1007/s00213-010-2153-8. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Schwarcz R. 3-hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur J Neurosci. 1999;11:3857–3863. doi: 10.1046/j.1460-9568.1999.00806.x. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Reddy PH, Tagle DA, Schwarcz R. Early kynureninergic impairment in Huntington's disease and in a transgenic animal model. Neurosci Lett. 2000;283:233–235. doi: 10.1016/s0304-3940(00)00956-3. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, MacDonald ME, et al. Elevated brain 3-hydroxykynurenine and quinolinate in Huntington disease mice. Neurobiol Dis. 2006;23:190–197. doi: 10.1016/j.nbd.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Pellicciari R, Schwarcz R. Patent EP 1954665 and US 201110144064: inhibitors of kynurenine aminotransferase and uses therefore. Munich, Germany and Alexandria, VA: European Patent Office: US Patent and Trademark Offices; 2008. [Google Scholar]
- Guillemin GJ, Croitoru-Lamoury J, Dormont D, Armati PJ, Brew BJ. Quinolinic acid upregulates chemokine production and chemokine receptor expression in astrocytes. Glia. 2003;41:371–381. doi: 10.1002/glia.10175. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer's disease hippocampus. Neuropathol Appl Neurobiol. 2005;31:395–404. doi: 10.1111/j.1365-2990.2005.00655.x. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2011;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges DW, Woon FL. Early-life stress and cognitive outcome. Psychopharmacology (Berl) 2011;214:121–130. doi: 10.1007/s00213-010-2090-6. [DOI] [PubMed] [Google Scholar]
- Henderson JL, Sawant-Basak A, Tuttle JB, Dounay AB, McAllister LA, Pandit J, et al. Discovery of hydroxamate bioisosteres as KAT II inhibitors with improved oral bioavailability and pharmacokinetics. MedChemComm. 2013;4:125–129. [Google Scholar]
- Heng MY, Detloff PJ, Wang PL, Tsien JZ, Albin RL. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J Neurosci. 2009;29:3200–3205. doi: 10.1523/JNEUROSCI.5599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CM, Kayed R, Zheng H, Sweatt JD, Dineley KT. Loss of alpha-7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer's disease. J Neurosci. 2010;30:2442–2453. doi: 10.1523/JNEUROSCI.5038-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes MP, Brew BJ, Martin A, Price RW, Salazar AM, Sidtis JJ, et al. Quinolinic acid in cerebrospinal-fluid and serum in HIV-1 infection – relationship to clinical and neurological status. Ann Neurol. 1991;29:202–209. doi: 10.1002/ana.410290215. [DOI] [PubMed] [Google Scholar]
- Hilmas C, Pereira EFR, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolites kynurenic acid inhibits α7-nicotinic receptor activity and increases non-α7 nicotinic receptor expression. J Neurosci. 2001;21:7463–7473. doi: 10.1523/JNEUROSCI.21-19-07463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtze M, Saetre P, Engberg G, Schwieler L, Werge T, Andreassen OA, et al. Kynurenine 3-monooxygenase polymorphisms: relevance for kynurenic acid synthesis in patients with schizophrenia and healthy controls. J Psychiatry Neurosci. 2012;37:53–57. doi: 10.1503/jpn.100175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jason GW, Suchowersky O, Pajurkova EM, Graham L, Klimek ML, Garber AT, et al. Cognitive manifestations of Huntington's disease in relation to genetic structure and clinical onset. Arch Neurol. 1997;54:1081–1088. doi: 10.1001/archneur.1997.00550210019008. [DOI] [PubMed] [Google Scholar]
- Jauch DA, Sethy VH, Weick BG, Chase TN, Schwarcz R. Intravenous administration of l-kynurenine to rhesus-monkeys – effect on quinolinate and kynurenate levels in serum and cerebrospinal-fluid. Neuropharmacology. 1993;32:467–472. doi: 10.1016/0028-3908(93)90171-x. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–225. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Jurgens HA, Amancherla K, Johnson RW. Influenza infection induces neuroinflammation, alters hippocampal neuron morphology, and impairs cognition in adult mice. J Neurosci. 2012;32:3958–3968. doi: 10.1523/JNEUROSCI.6389-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisch R, Holt B, Petrovic P, De Martino B, Kloppel S, Buchel C, et al. The NMDA Agonist D-Cycloserine facilitates fear memory consolidation in humans. Cerebral Cortex. 2009;19:187–196. doi: 10.1093/cercor/bhn076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandanearatchi A, Brew BJ. The kynurenine pathway and quinolinic acid: pivotal roles in HIV associated neurocognitive disorders. FEBS J. 2012;279:1366–1374. doi: 10.1111/j.1742-4658.2012.08500.x. [DOI] [PubMed] [Google Scholar]
- Kiank C, Zeden J-P, Drude S, Domanska G, Fusch G, Otten W, et al. Psychological stress-induced, IDO1-dependent tryptophan catabolism: implications on immunosuppression in mice and humans. PLoS ONE. 2010;5:e11825. doi: 10.1371/journal.pone.0011825. AR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, et al. alpha-7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block a beta-amyloid-induced neurotoxicity. J Biol Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Kocki T, Luchowski P, Luchowska E, Wielosz M, Turski WA, Urbanska EM. L-cysteine sulphinate, endogenous sulphur-containing amino acid, inhibits rat brain kynurenic acid production via selective interference with kynurenine aminotransferase II. Neurosci Lett. 2003;346:97–100. doi: 10.1016/s0304-3940(03)00579-2. [DOI] [PubMed] [Google Scholar]
- Konradsson-Geuken A, Wu H-Q, Gash CR, Alexander KS, Campbell A, Sozeri Y, et al. Cortical kynurenic acid bi-directionally modulates prefrontal glutamate levels as assessed by microdialysis and rapid electrochemistry. Neuroscience. 2010;169:1848–1859. doi: 10.1016/j.neuroscience.2010.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence AD, Sahakian BJ, Robbins TW. Cognitive functions and corticostriatal circuits: insights from Huntington's disease. Trends Cogn Sci. 1998;2:379–388. doi: 10.1016/s1364-6613(98)01231-5. [DOI] [PubMed] [Google Scholar]
- Lawrence AD, Watkins LHA, Sahakian BJ, Hodges JR, Robbins TW. Visual object and visuospatial cognition in Huntington's disease: implications for information processing in corticostriatal circuits. Brain. 2000;123:1349–1364. doi: 10.1093/brain/123.7.1349. [DOI] [PubMed] [Google Scholar]
- Linderholm KR, Andersson A, Olsson S, Olsson E, Snodgrass R, Engberg G, et al. Activation of rat ventral tegmental area dopamine neurons by endogenous kynurenic acid: a pharmacological analysis. Neuropharmacology. 2007;53:918–924. doi: 10.1016/j.neuropharm.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Lindsley CW, Shipe WD, Wolkenberg SE, Theberge CR, Williams DL, Sur C, et al. Progress towards validating the NMDA receptor hypofunction hypothesis of schizophrenia. Curr Top Med Chem. 2006;6:771–785. doi: 10.2174/156802606777057599. [DOI] [PubMed] [Google Scholar]
- Luchowski P, Kocki T, Urbanska EM. N-G-nitro-L-arginine and its methyl ester inhibit brain synthesis of kynurenic acid possibly via nitric oxide-independent mechanism. Pol J Pharmacol. 2001;53:597–604. [PubMed] [Google Scholar]
- Luchowski P, Luchowska E, Turski WA, Urbanska EM. 1-methyl-4-phenylpyridinium and 3-nitropropionic acid diminish cortical synthesis of kynurenic acid via interference with kynurenine aminotransferases in rats. Neurosci Lett. 2002;330:49–52. doi: 10.1016/s0304-3940(02)00735-8. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- Marin M-F, Lord C, Andrews J, Juster R-P, Sindi S, Arsenault-Lapierre G, et al. Chronic stress, cognitive functioning and mental health. Neurobiol Learn Mem. 2011;96:583–595. doi: 10.1016/j.nlm.2011.02.016. [DOI] [PubMed] [Google Scholar]
- Mexal S, Berger R, Logel J, Ross RG, Freedman R, Leonard S. Differential regulation of alpha 7 nicotinic receptor gene (CHRNA7): expression in schizophrenic smokers. J Mol Neurosci. 2010;40:185–195. doi: 10.1007/s12031-009-9233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ, Agid Y, Bruene M, Bullmore ET, Carter CS, Clayton NS, et al. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov. 2012;11:141–168. doi: 10.1038/nrd3628. [DOI] [PubMed] [Google Scholar]
- Miller JM, Macgarvey U, Beal MF. The effect of peripheral loading with kynurenine and probenecid on extracellular striatal kynurenic acid concentrations. Neurosci Lett. 1992;146:115–118. doi: 10.1016/0304-3940(92)90186-b. [DOI] [PubMed] [Google Scholar]
- Minzenberg MJ, Carter CS. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology. 2008;33:1477–1502. doi: 10.1038/sj.npp.1301534. [DOI] [PubMed] [Google Scholar]
- Miura H, Ando Y, Noda Y, Isobe K, Ozaki N. Long-lasting effects of inescapable-predator stress on brain tryptophan metabolism and the behavior of juvenile mice. Stress. 2011;14:262–272. doi: 10.3109/10253890.2010.541539. [DOI] [PubMed] [Google Scholar]
- Moeller M, Du Preez JL, Emsley R, Harvey BH. Social isolation rearing in rats alters plasma tryptophan metabolism and is reversed by sub-chronic clozapine treatment. Neuropharmacology. 2012;62:2499–2506. doi: 10.1016/j.neuropharm.2012.02.021. [DOI] [PubMed] [Google Scholar]
- Mok MHS, Fricker AC, Weil A, Kew JNC. Electrophysiological characterisation of the actions of kynurenic acid at ligand-gated ion channels. Neuropharmacology. 2009;57:242–249. doi: 10.1016/j.neuropharm.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Giorgini F. Patents US 7618793B2: identifying agents for decreasing cellular toxicity associated with huntingtin toxicity. Alexandria, VA: US Patent and Trademark Offices; 2009. [Google Scholar]
- Muchowski PJ, Muchowski JM, Schwarcz R, Guidetti P. Patent US 8071631B2 and 7994338: small molecule inhibitors of kynurenine-3-monoxygenase. San Francisco, CA: The J D Gladstone Institute; 2011. [Google Scholar]
- Mueller N, Dursun SM. Schizophrenia genes, epigenetics and psychoneuroimmunology therapeutics: all make sense now. J Psychopharmacol. 2011;25:713–714. doi: 10.1177/0269881110364268. [DOI] [PubMed] [Google Scholar]
- Muscatell KA, Slavich GM, Monroe SM, Gotlib IH. Stressful life events, chronic difficulties, and the symptoms of clinical depression. J Nerv Ment Dis. 2009;197:154–160. doi: 10.1097/NMD.0b013e318199f77b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myint AM, Schwarz MJ, Verkerk R, Mueller HH, Zach J, Scharpe S, et al. Reversal of imbalance between kynurenic acid and 3-hydroxykynurenine by antipsychotics in medication-naive and medication-free schizophrenic patients. Brain Behav Immun. 2011;25:1576–1581. doi: 10.1016/j.bbi.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Nagy K, Plangar I, Tuka B, Gellert L, Varga D, Demeter I, et al. Synthesis and biological effects of some kynurenic acid analogs. Bioorg Med Chem. 2011;19:7590–7596. doi: 10.1016/j.bmc.2011.10.029. [DOI] [PubMed] [Google Scholar]
- Neill JC, Barnes S, Cook S, Grayson B, Idris NF, McLean SL, et al. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther. 2010;128:419–432. doi: 10.1016/j.pharmthera.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Newcomer JW, Krystal JH. NMDA receptor regulation of memory and behavior in humans. Hippocampus. 2001;11:529–542. doi: 10.1002/hipo.1069. [DOI] [PubMed] [Google Scholar]
- Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, et al. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–118. doi: 10.1016/S0893-133X(98)00067-0. [DOI] [PubMed] [Google Scholar]
- Nilsson LK, Linderholm KR, Engberg G, Paulson L, Blennow LLH, et al. Elevated levels of kynurenic acid in the cerebrospinal fluid of male patients with schizophrenia. Schizophr Res. 2005;80:315–322. doi: 10.1016/j.schres.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Nilsson LK, Linderholm KR, Erhardt S. Subchronic treatment with kynurenine and probenecid: effects on prepulse inhibition and firing of midbrain dopamine neurons. J Neural Transm. 2006;113:557–571. doi: 10.1007/s00702-005-0343-z. [DOI] [PubMed] [Google Scholar]
- Nozaki K, Beal MF. Neuroprotective effects of l-kynurenine on hypoxia ischemia and NMDA lesions in neonatal rats. J Cereb Blood Flow Metab. 1992;12:400–407. doi: 10.1038/jcbfm.1992.57. [DOI] [PubMed] [Google Scholar]
- O'Connor JC, Andre C, Wang Y, Lawson MA, Szegedi SS, Lestage J, et al. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to Bacillus Calmette-Guerin. J Neurosci. 2009;29:4200–4209. doi: 10.1523/JNEUROSCI.5032-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson SK, Larsson MK, Erhardt S. Subchronic elevation of brain kynurenic acid augments amphetamine-induced locomotor response in mice. J Neural Transm. 2012;119:155–163. doi: 10.1007/s00702-011-0706-6. [DOI] [PubMed] [Google Scholar]
- Onur O, Schlaepfer TE, Kukolja J, Bauer A, Jeung H, Patin A, et al. The N-Methyl-D-Aspartate receptor co-agonist d-cycloserine facilitates declarative learning and hippocampal activity in humans. Biol Psychiatry. 2010;67:1205–1211. doi: 10.1016/j.biopsych.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Owe-Young R, Webster NL, Mukhtar M, Pomerantz RJ, Smythe G, Walker D, et al. Kynurenine pathway metabolism in human blood-brain-barrier cells: implications for immune tolerance and neurotoxicity. J Neurochem. 2008;105:1346–1357. doi: 10.1111/j.1471-4159.2008.05241.x. [DOI] [PubMed] [Google Scholar]
- Paterson GJ, Ohashi Y, Reynolds GP, Pratt JA, Morris BJ. Selective increases in the cytokine, TNF-alpha, in the prefrontal cortex of PCP-treated rats and human schizophrenic subjects: influence of antipsychotic drugs. J Psychopharmacol. 2006;20:636–642. doi: 10.1177/0269881106062025. [DOI] [PubMed] [Google Scholar]
- Pearson SJ, Reynolds GP. Increased brain concentrations of a neurotoxin, 3-hydroxy-kynurenine, in Huntington's disease. Neurosci Lett. 1992;144:199–201. doi: 10.1016/0304-3940(92)90749-w. [DOI] [PubMed] [Google Scholar]
- Pechtel P, Pizzagalli DA. Effects of early life stress on cognitive and affective function: an integrated review of human literature. Psychopharmacology (Berl) 2011;214:55–70. doi: 10.1007/s00213-010-2009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicciari R, Natalini B, Costantino G, Mahmoud M, Mattoli L, Sadeghpour BM, et al. Modulation of the kynurenine pathway in search for new neuroprotective agents – synthesis and preliminary evaluation of (m-nitrobenzoyl)alanine, a potent inhibitor of kynurenine-3-hydroxylase. J Med Chem. 1994;37:647–655. doi: 10.1021/jm00031a015. [DOI] [PubMed] [Google Scholar]
- Pellicciari R, Rizzo RC, Costantino G, Marinozzi M, Amori L, Guidetti P, et al. Modulators of the kynurenine pathway of tryptophan metabolism: synthesis and preliminary biological evaluation of (S)-4-(ethylsulfonyl)benzoylaianine, a potent and selective kynurenine aminotransferase II (KAT II) inhibitor. ChemMedChem. 2006;1:528–531. doi: 10.1002/cmdc.200500095. [DOI] [PubMed] [Google Scholar]
- Perkins MN, Stone TW. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982;247:184–187. doi: 10.1016/0006-8993(82)91048-4. [DOI] [PubMed] [Google Scholar]
- Pertovaara M, Raitala A, Lehtimaki T, Karhunen PJ, Oja SS, Jylha M, et al. Indoleamine 2,3-dioxygenase activity in nonagenarians is markedly increased and predicts mortality. Mech Ageing Dev. 2006;127:497–499. doi: 10.1016/j.mad.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Pevarello P, Giordani A, Villa M, Speciale C, Varasi M. Patent US 6133302: 5-(3-phenyl-3-oxo-propyl)-1H-tetrazole derivatives. Alexandria, VA: US Patent and Trademark Offices; 2000. [Google Scholar]
- Phillips WA, Silverstein SM. Convergence of biological and psychological perspectives on cognitive coordination in schizophrenia. Behav Brain Sci. 2003;26:65–100. doi: 10.1017/s0140525x03000025. [DOI] [PubMed] [Google Scholar]
- Pocivavsek A, Wu H-Q, Potter MC, Elmer GI, Pellicciari R, Schwarcz R. Fluctuations in endogenous kynurenic acid control hippocampal glutamate and memory. Neuropsychopharmacology. 2011;36:2357–2367. doi: 10.1038/npp.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocivavsek A, Wu H-Q, Elmer GI, Bruno JP, Schwarcz R. Pre- and postnatal exposure to kynurenine causes cognitive deficits in adulthood. Eur J Neurosci. 2012;35:1605–1612. doi: 10.1111/j.1460-9568.2012.08064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoli P, Pezzola A, Domenici ME, Sagratella S, Diana G, Caporali MG, et al. Behavioural and electrophysiological correlates of the quinolinic acid rat model of Huntington's disease in rats. Brain Res Bull. 1994;35:329–335. doi: 10.1016/0361-9230(94)90109-0. [DOI] [PubMed] [Google Scholar]
- Potter MC, Elmer GI, Bergeron R, Albuquerque EX, Guidetti P, Wu H-Q, et al. Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behaviour. Neuropsychopharmacology. 2010;35:1734–1742. doi: 10.1038/npp.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, Kaka T, Cullen KM, Braidy N, Brew BJ, Guillemin GJ. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS ONE. 2009;4:e6344. doi: 10.1371/journal.pone.0006344. AR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry. 2010;15:393–403. doi: 10.1038/mp.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassoulpour A, Wu H-Q, Ferre S, Schwarcz R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J Neurochem. 2005;93:762–765. doi: 10.1111/j.1471-4159.2005.03134.x. [DOI] [PubMed] [Google Scholar]
- Ren K, Thinschmidt J, Liu J, Ai L, Papke RL, King MA, et al. Alpha-7 nicotinic receptor gene delivery into mouse hippocampal neurons leads to functional receptor expression, improved spatial memory-related performance, and tau hyperphosphorylation. Neuroscience. 2007;145:314–322. doi: 10.1016/j.neuroscience.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Robert PH, Benoit M. Neurochemistry of cognition: serotonergic and adrenergic mechanisms. Handb Clin Neurol. 2008;88:31–40. doi: 10.1016/S0072-9752(07)88002-X. DOI: 10.1016/S0072-9752(07)88002-X. [DOI] [PubMed] [Google Scholar]
- Rodgers J, Stone TW, Barratt MP, Bradley B, Kennedy PG. Kynurenine pathway inhibition reduces central nervous system inflammation in a model of human African trypanosomiasis. Brain. 2009;132:1259–1267. doi: 10.1093/brain/awp074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F, Valentina C, Garavaglia S, Sathyasaikumar KV, Schwarcz R, Kojima S, et al. Crystal structure-based selective targeting of the pyridoxal 5′-phosphate dependent enzyme kynurenine aminotransferase II for cognitive enhancement. J Med Chem. 2010;53:5684–5689. doi: 10.1021/jm100464k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röver S, Cesura AM, Hugenin P, Kettler R, Szente A. Synthesis and biochemical evaluation of N-(4-phenylthiazol-2-yl)benzenesulfonamides as high-affinity inhibitors of kynurenine 3-hydroxylase. J Med Chem. 1997;40:4378–4385. doi: 10.1021/jm970467t. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Jantaratnotai N, McLarnon JG. Thalidomide inhibition of vascular remodeling and inflammatory reactivity in the quinolinic acid-injected rat striatum. Neuroscience. 2009;163:601–608. doi: 10.1016/j.neuroscience.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Saito K, Markey SP, Heyes MP. Effects of immune activation on quinolinic acid and neuroactive kynurenines in the mouse. Neuroscience. 1992;51:25–39. doi: 10.1016/0306-4522(92)90467-g. [DOI] [PubMed] [Google Scholar]
- Salazar A, Gonzalez-Rivera BL, Redus L, Parrott JM, O'Connor JC. Indoleamine 2,3-dioxygenase mediates anhedonia and anxiety-like behaviors caused by peripheral lipopolysaccharide immune challenge. Horm Behav. 2012;62:202–209. doi: 10.1016/j.yhbeh.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapko MT, Guidetti P, Yu P, Tagle DA, Pellicciari R, Schwarcz R. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: implications for Huntington's disease. Exp Neurol. 2006;197:31–40. doi: 10.1016/j.expneurol.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Sarter M, Bruno JP, Parikh V. Abnormal neurotransmitter release underlying behavioral and cognitive disorders: toward concepts of dynamic and function-specific dysregulation. Neuropsychopharmacology. 2007;32:1452–1461. doi: 10.1038/sj.npp.1301285. [DOI] [PubMed] [Google Scholar]
- Sathyasaikumar KV, Stachowski EK, Amori L, Guidetti P, Muchowski PJ, Schwarcz R. Dysfunctional kynurenine pathway metabolism in the R6/2 mouse model of Huntington's disease. J Neurochem. 2010;113:1416–1425. doi: 10.1111/j.1471-4159.2010.06675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyasaikumar KV, Stachowski EK, Wonodi I, Roberts RC, Rassoulpour A, McMahon RP, et al. Impaired kynurenine pathway metabolism in the prefrontal cortex of individuals with schizophrenia. Schizophr Bull. 2011;37:1147–1156. doi: 10.1093/schbul/sbq112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Whetsell WO, Jr, Mangano RM. Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science. 1983;219:316–318. doi: 10.1126/science.6849138. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Okuno E, White RJ, Bird ED, Jnr WWO. 3-Hydroxyanthranilate oxygenase activity is increased in the brains of Huntington disease victims. Proc Natl Acad Sci USA. 1988;85:4079–4081. doi: 10.1073/pnas.85.11.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Rassoulpour A, Wu H-Q, Medoff D, Tamminga CA, Roberts RC. Increased cortical kynurenate content in schizophrenia. Biol Psychiatry. 2001;50:521–530. doi: 10.1016/s0006-3223(01)01078-2. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Guidetti P, Sathyasaikumar KV, Muchowski PJ. Of mice, rats and men: revisiting the quinolinic acid hypothesis of Huntington's disease. Prog Neurobiol. 2009;90:230–245. doi: 10.1016/j.pneurobio.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shear DA, Dong J, Gundy CD, Haik-Creguer KL, Dunbar GL. Comparison of intrastriatal injections of quinolinic acid and 3-nitropropionic acid for use in animal models of Huntington's disease. Prog Neuropsychopharmacol Biol Psychiatry. 1998;22:1217–1240. doi: 10.1016/s0278-5846(98)00070-0. [DOI] [PubMed] [Google Scholar]
- Speciale C, Schwarcz R. Uptake of kynurenine into rat-brain slices. J Neurochem. 1990;54:156–163. doi: 10.1111/j.1471-4159.1990.tb13296.x. [DOI] [PubMed] [Google Scholar]
- Speciale C, Hares K, Schwarcz R, Brookes N. High-affinity uptake of l-kynurenine by a Na+-independent transporter of neutral amino-acids in astrocytes. J Neurosci. 1989;9:2066–2072. doi: 10.1523/JNEUROSCI.09-06-02066.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stazka J, Luchowski P, Wielosz M, Kleinrok Z, Urbanska EM. Endothelium-dependent production and liberation of kynurenic acid by rat aortic rings exposed to L-kynurenine. Eur J Pharmacol. 2002;448:133–137. doi: 10.1016/s0014-2999(02)01943-x. [DOI] [PubMed] [Google Scholar]
- Stazka J, Luchowski P, Urbanska EM. Homocysteine, a risk factor for atherosclerosis, biphasically changes the endothelial production of kynurenic acid. Eur J Pharmacol. 2005;517:217–223. doi: 10.1016/j.ejphar.2005.04.048. [DOI] [PubMed] [Google Scholar]
- Stone TW. Kynurenic acid blocks nicotinic synaptic transmission to hippocampal interneurons in young rats. Eur J Neurosci. 2007;25:2656–2665. doi: 10.1111/j.1460-9568.2007.05540.x. [DOI] [PubMed] [Google Scholar]
- Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov. 2002;1:609–620. doi: 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- Stone TW, Perkins MN. Quinolinic acid: a potent endogenous excitant at amino acid receptors in the CNS. Eur J Pharmacol. 1981;72:411–412. doi: 10.1016/0014-2999(81)90587-2. [DOI] [PubMed] [Google Scholar]
- Stone TW, Forrest CM, Stoy N, Darlington LG. Involvement of kynurenines in Huntington's disease and stroke-induced brain damage. J Neural Transm. 2012;119:261–274. doi: 10.1007/s00702-011-0676-8. [DOI] [PubMed] [Google Scholar]
- Stone TW, Stoy N, Darlington LG. An expanding range of targets for kynurenic acid. Trends Pharmacol Sci. 2013;34:136–143. doi: 10.1016/j.tips.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Stoy N, Mackay GM, Forrest CM, Christofides J, Egerton M, Stone TW, et al. Tryptophan metabolism and oxidative stress in patients with Huntington's disease. J Neurochem. 2005;93:611–623. doi: 10.1111/j.1471-4159.2005.03070.x. [DOI] [PubMed] [Google Scholar]
- Tadaiesky MT, Dombrowski PA, Figueiredo CP, Cargnin-Ferreira E, Da Cunha C, Takahashi RN. Emotional, cognitive and neurochemical alterations in a premotor stage model of Parkinson's disease. Neuroscience. 2008;156:830–840. doi: 10.1016/j.neuroscience.2008.08.035. [DOI] [PubMed] [Google Scholar]
- Tatter SB, Galpern WR, Hoogeveen AT, Isacson O. Effects of striatal excitotoxicity on Huntington-like immunoreactivity. Neuroreport. 1995;6:1125–1129. doi: 10.1097/00001756-199505300-00013. [DOI] [PubMed] [Google Scholar]
- Teichberg VI. Patent US 7404951: Method and Composition for Protecting Neuronal Tissue from Damage Induced by Elevated Glutamate Levels. Alexandria, VA: US Patent and Trademark Offices; 2008. [Google Scholar]
- Teichberg VI. Patent US 7767204: Method and Composition for Protecting Neuronal Tissue from Damage Induced by Elevated Glutamate Levels. Alexandria, VA: US Patent and Trademark Offices; 2010. [Google Scholar]
- Ting KK, Brew BJ, Guillemin GJ. Effect of quinolinic acid on human astrocytes morphology and functions: implications in Alzheimer's disease. J Neuroinflammation. 2009;6:36. doi: 10.1186/1742-2094-6-36. AR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trecartin KV, Bucci DJ. Administration of kynurenine during adolescence, but not during adulthood, impairs social behavior in rats. Schizophr Res. 2011;133:156–158. doi: 10.1016/j.schres.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse YC, Bagot RC, Wong TP. Dynamic regulation of NMDAR function in the adult brain by the stress hormone corticosterone. Front Cell Neurosci. 2012;6:9. doi: 10.3389/fncel.2012.00009. AR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varasi M, Giordani A, Cini M, Speciale C, Bianchetti A. Patent US 6207709B1: N-substituted-2-amino-4-phenyl-4-oxo-butanoic acid compounds having kynurenine-3-hydroxy base inhibitory activity. Alexandria, VA: US Patent and Trademark Offices; 2001. [Google Scholar]
- Vezzani A, Stasi MA, Wu HQ, Castiglioni M, Weckermann B, Samanin R. Studies on the potential neurotoxic and convulsant effects of increased blood-levels of quinolinic acid in rats with altered blood-brain-barrier permeability. Exp Neurol. 1989;106:90–98. doi: 10.1016/0014-4886(89)90149-0. [DOI] [PubMed] [Google Scholar]
- Walker DG, Link J, Lue L-F, Dalsing-Hernandez JE, Boyes BE. Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol. 2006;79:596–610. doi: 10.1189/jlb.0705377. [DOI] [PubMed] [Google Scholar]
- Wang J, Simonavicius N, Wu X, Swaminath G, Reagen J, Tian H, et al. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem. 2006;281:22021–22028. doi: 10.1074/jbc.M603503200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu H, McKenzie G, Witting PK, Stasch J-P HM, et al. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat Med. 2010;16:279–285. doi: 10.1038/nm.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wejksza K, Rzeski W, Parada-Turska J, Zdzisinska B, Rejdak R, Kocki T, et al. Kynurenic acid production in cultured bovine aortic endothelial cells. Homocysteine is a potent inhibitor. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:300–304. doi: 10.1007/s00210-004-0872-2. [DOI] [PubMed] [Google Scholar]
- Wejksza K, Rzeski W, Turski WA. Kynurenic acid protects against the homocysteine-induced impairment of endothelial cells. Pharmacol Res. 2009;61:751–756. doi: 10.1016/s1734-1140(09)70130-6. [DOI] [PubMed] [Google Scholar]
- Wonodi I, Stine OC, Sathyasaikumar KV, Roberts RC, Mitchell BD, Hong LE, et al. Downregulated kynurenine 3-monooxygenase gene expression and enzyme activity in schizophrenia and genetic association with schizophrenia endophenotypes. Arch Gen Psychiatry. 2011;68:665–674. doi: 10.1001/archgenpsychiatry.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Yan J, Zhou P, Li J, Gao H, Xia Y, et al. Neurotransmitter receptors and cognitive dysfunction in Alzheimer's disease and Parkinson's disease. Prog Neurobiol. 2012;97:1–13. doi: 10.1016/j.pneurobio.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A, Akimoto H, Kagawa S, Guillemin GJ, Takikawa O. Proinflammatory cytokine interferon-gamma increases induction of indoleamine 2,3-dioxygenase in monocytic cells primed with amyloid beta peptide 1-42: implications for the pathogenesis of Alzheimer's disease. J Neurochem. 2009;110:791–800. doi: 10.1111/j.1471-4159.2009.06175.x. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Wei J, Liu W, Zhong P, Li Xiangning YZ. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. 2012;73:962–977. doi: 10.1016/j.neuron.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisapel N, Laudon M, Daily D. Patent US 8309767B2: 2-aminobenzoyl Derivatives. Alexandria, VA: US Patent and Trademark Offices; 2012. [Google Scholar]
- Zmarowski A, Wu H-Q, Brooks JM, Potter MC, Pellicciari R, Schwarcz R, et al. Astrocyte-derived kynurenic acid modulates basal and evoked cortical acetylcholine release. Eur J Neurosci. 2009;29:529–538. doi: 10.1111/j.1460-9568.2008.06594.x. [DOI] [PubMed] [Google Scholar]