

Abstract
Background and Purpose
The 5-HT3 receptor is a ligand-gated ion channel that is modulated allosterically by various compounds including colchicine, alcohols and volatile anaesthetics. However the positive allosteric modulators (PAMs) identified to date have low affinity, which hinders investigation because of non-selective effects at pharmacologically active concentrations. The present study identifies 5-chloroindole (Cl-indole) as a potent PAM of the 5-HT3 receptor.
Experimental Approach
5-HT3 receptor function was assessed by the increase in intracellular calcium and single-cell electrophysiological recordings in HEK293 cells stably expressing the h5-HT3A receptor and also the mouse native 5-HT3 receptor that increases neuronal contraction of bladder smooth muscle.
Key Results
Cl-indole (1–100 μM) potentiated agonist (5-HT) and particularly partial agonist [(S)-zacopride, DDP733, RR210, quipazine, dopamine, 2-methyl-5-HT, SR57227A, meta chlorophenyl biguanide] induced h5-HT3A receptor-mediated responses. This effect of Cl-indole was also apparent at the mouse native 5-HT3 receptor. Radioligand-binding studies identified that Cl-indole induced a small (∼twofold) increase in the apparent affinity of 5-HT for the h5-HT3A receptor, whereas there was no effect upon the affinity of the antagonist, tropisetron. Cl-indole was able to reactivate desensitized 5-HT3 receptors. In contrast to its effect on the 5-HT3 receptor, Cl-indole did not alter human nicotinic α7 receptor responses.
Conclusions and Implications
The present study identifies Cl-indole as a relatively potent and selective PAM of the 5-HT3 receptor; such compounds will aid investigation of the molecular basis for allosteric modulation of the 5-HT3 receptor and may assist the discovery of novel therapeutic drugs targeting this receptor.
Linked Articles
Recent reviews on allosteric modulation can be found at:
Kenakin, T (2013). New concepts in pharmacological efficacy at 7TM receptors: IUPHAR Review 2. British Journal of Pharmacology 168: 554–575. doi: 10.1111/j.1476-5381.2012.02223.x
Roche D, Gil D and Giraldo J (2013). Mechanistic analysis of the function of agonists and allosteric modulators: reconciling two-state and operational models. British Journal of Pharmacology 169: 1189–1202. doi: 10.1111/bph.12231
Keywords: 5-HT3 receptor, ligand-gated ion channel, allosteric modulation
Introduction
The 5-HT3 receptor mediates fast synaptic neurotransmission in neurons of both the central and peripheral nervous systems (Barnes et al., 2009; Walstab et al., 2010). In addition, the receptor is expressed by a number of other cell types including several subsets of immune cells (e.g. Fiebich et al., 2004; Rinaldi et al., 2010). Various antagonists of the 5-HT3 receptor offer benefit to patients experiencing nausea and vomiting, most commonly those arising from aggressive anti-cancer chemo- and radiation-therapy, or following recovery from general anaesthesia (post-operative nausea and vomiting). In addition, 5-HT3 receptor antagonists such as alosetron offer symptomatic relief to patients with irritable bowel syndrome, although rare instances of severe constipation and ischaemic colitis have limited use of these medications (Barnes et al., 2009; Walstab et al., 2010).
At the molecular level, the 5-HT3 receptor belongs to the Cys-loop ligand-gated ion channel (LGIC) superfamily that also includes nicotinic acetylcholine, GABAA, strychnine-sensitive glycine receptors and a much less studied Zn2+-activated cation channel (Barnes et al., 2009). In common with other superfamily members, the functional receptor is a pentameric complex in which the second transmembrane domain of each subunit lines the transmembrane pore and contributes significantly to the integral ion channel (Boess et al., 1995; Barnes et al., 2009). However, other regions such as the membrane-associated stretch within the C-terminal end of the large second intracellular loop (Kelley et al., 2003; Peters et al., 2005) and the lining of the extracellular domain (Livesey et al., 2011) make important contributions to the permeation pathway.
Five human genes have been identified to date that encode the subunits 5-HT3A-E (Barnes et al., 2009; Walstab et al., 2010; Alexander et al., 2011). Atypical for the Cys-loop superfamily, the 5-HT3A subunit assembles efficiently as a functional homomeric receptor complex that possesses many of the pharmacological and biophysical characteristics of some native 5-HT3 receptors, particularly those expressed by certain neuroblastoma cell lines (Lambert et al., 1989; Gill et al., 1995). However, not all biophysical characteristics of other native 5-HT3 receptors, such as those of rodent superior cervical ganglion and hippocampal neurones (Yang et al., 1992; Hussy et al., 1994; Jones and Surprenant, 1994) are mimicked by homomeric 5-HT3A receptors (Fletcher and Barnes, 1998) and incorporation of the 5-HT3B subunit would appear to account for this biophysical diversity (Davies et al., 1999; Dubin et al., 1999; Peters et al., 2005).
The orthosteric 5-HT recognition site is located at subunit interfaces within the extracellular domain (Barnes et al., 2009). In common with other Cys-loop superfamily members, the 5-HT3 receptor possesses topologically distinct allosteric sites that appear to recognize a diverse array of compounds. These include various alcohols and indole derivatives and colchicine (Parker et al., 1996; Hu et al., 2006; Hu and Lovinger, 2008; Walstab et al., 2010; Davies, 2011). The therapeutic potential of allosteric receptor ligands is increasingly recognized in pharmaceutical research and development due to their ability to modulate receptor function yet retain spatial and temporal control (e.g. Williams et al., 2011).
The best-characterized positive allosteric modulator (PAM), 5-hydroxyindole, has diverse effects on 5HT3 receptors. Studies show a mix of allosteric and orthosteric effects (Kooyman et al., 1994; Hu and Lovinger, 2008). Indeed, one study failed to show any effect, suggesting that 5-hydroxyindole is a PAM at rodent receptors, with no activity at their human counterpart (Grønlien et al., 2010). Furthermore, 5-hydroxindole lacks selectivity having a well-described allosteric action on nicotinic α7 receptors. The relatively low affinity (mM) and poor selectivity hampers the utility of the currently characterized PAMs as tools to study their pharmacological impact. In the present study, we report the identification of the 5-substituted indole, 5-chloroindole (Cl-indole; Moretti et al., 1996; Supporting Information Figure S1), as a selective allosteric modulator of the 5-HT3 receptor that displays micromolar potency.
Materials and methods
Cell culture
HEK293 cells stably expressing the human 5-HT3A subunit (HEKh5-HT3A cells; Brady et al., 2001) were grown in DMEM, supplemented with 10% (v/v) FBS, 1% (v/v) penicillin/streptomycin (10 000 U·mL−1 penicillin and 10 mg·mL−1 streptomycin) and G418 (250 μg·mL−1) and maintained at 37°C, 5% CO2, 95% air at 95% relative humidity. Approximately 24 h prior to intracellular calcium assays, cells were seeded directly into poly-D-lysine coated, black-walled, clear bottomed, 96-well plates (Costar, Bio-Rad Laboratories Ltd., Hemel Hempstead, UK) at a density of 1 × 105 cells per well. HEK293 cells stably expressing the human α7 nicotinic receptor and the human ric3 protein (HEKα7ric3; G. Grafton et al., unpubl.) were grown in DMEM supplemented with 10% (v/v) FBS, 10 000 U·mL−1 penicillin, 10 mg mL−1 streptomycin, 250 μg·mL−1 G418 and 250 μg·mL−1 hygromycin B and were maintained as per HEKh5-HT3A cells.
Intracellular Ca2+ measurements
Cells were washed with 1 × HBSS (Invitrogen, Paisley, UK) and incubated with fluo-4 acetoxymethyl (AM) ester (2.5 μM; Molecular Probes, Paisley, UK) for 60 min at room temperature. Cells were then washed in HBSS and incubated for a further 30 min (room temperature) prior to assay. Changes in intracellular calcium were measured using a FlexStation (Molecular Devices, Sunnyvale, CA, USA) with fluorescence levels assessed every 3 s. Buffer, or Cl-indole, was added after 20 s, agonist/partial agonist (or antagonist tested for potential emergent agonist activity) was added after 80 s and recordings continued for at least 4 min. In antagonism studies, antagonists were pre-incubated for half an hour prior to analysis.
Radioligand binding
Radioligand-binding assays were performed similar to our previous studies (e.g. Monk et al., 2004). Briefly, HEKh5-HT3A cells were homogenized (Polytron, Fisher Scientific, Loughborough, UK) in Tris/Krebs buffer (in mM; Tris 50.0, NaCl 118.0, KCl 4.75, KH2PO4 1.2, MgSO4 1.2, CaCl2 2.5, NaHCO3 25.0, glucose 11.0, final pH 7.4 adjusted with NaOH) and washed twice by centrifugation (48 000× g; 4°C) before re-suspension in Tris/Krebs buffer. Radioligand-binding assays were performed in triplicate; glass tubes contained 500 μL of competing drug, or vehicle (Tris/Krebs buffer), and 100 μL of [3H]-granisetron (∼1 nM for competition studies, or a range of concentrations between 0.1 and 15 nM for saturation studies; ∼3 TBqmmol−1; NEN Life Science Products, PerkinElmer, Seer Green, UK). An aliquot (100 μL) of the cell homogenate was added to initiate binding, which was allowed to proceed at 37°C for 60 min before termination by rapid filtration and washing under vacuum through Whatman GF/B filters, followed by assay of the radioactivity remaining on the filters.
Single-cell electrophysiology
Macroscopic currents were recorded in the whole-cell recording mode of the patch-clamp technique from HEKh5-HT3A cells cultured on coverslips using infrared DIC (inverted Olympus FV1000 confocal microscope; Olympus Keymed, Southend on Sea, UK). Cells were superfused at ∼4 mL·min−1 with an extracellular solution (in mM; NaCl 140, KCl 2.8, CaCl2 1.0, glucose 10, HEPES 10, pH 7.4 adjusted with NaOH). Patch electrodes were pulled from borosilicate glass (o.d. 1.2 mm, i.d. 0.69 mm; Harvard Apparatus, Edenbridge, UK) using a P-97 puller (Sutter, Novato, CA, USA) and filled with intracellular solution consisting of (in mM) 135 CsCl, 2 MgCl2, 10 HEPES, 1 EGTA, 2 Mg-ATP and 0.3 Na-GTP; pH adjusted to 7.3 with KOH (osmolarity ∼285 mOsm). Patch electrodes typically had open tip resistances of 4–7 MΩ. Membrane currents were recorded using an NPI SEC-10 L amplifier (Scientifica, Harpenden, UK), low-pass Bessel filtered at 1 kHz (NL-125; Digitimer Ltd., Welwyn Garden City, UK) and digitized at 10 kHz by a Power 1401 (CED Ltd., Cambridge, UK). Experiments were performed at room temperature with the cells voltage-clamped at −60 mV. Stimulation and data acquisition were controlled using Signal software (version 3; CED).
Agonist-evoked currents were elicited by either pressure ejection (20-kPa; Picospritzer II; General Valve, Fairfield, NJ, USA) of agonist (5-HT 1.0–10 μM, or DDP733 100 nM) from modified patch pipettes placed ∼30 μm from the recorded cell, or by bath application of 5-HT (1.0 μM).
Neuronal contraction of mouse bladder
Male BALB/c albino mice, aged 8–12 weeks, were killed with a rising CO2 concentration followed by cervical fracture. All experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act 1986 and European Communities Council Directive 86/09/EEC. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). The urinary bladder was removed and placed in physiological saline (in mM; NaCl 118.4, NaHCO3 25.0, NaH2PO4 1.13, CaCl2 1.8, KCl 4.7, MgCl2 1.3, glucose 11.1 pH and [O2] were regulated by continuously bubbling the solution reservoir with 95% O2 and 5% CO2). Other organs were harvested for experimental use in other projects. Connective tissue surrounding the bladder was first removed before the ventral wall of the bladder was opened longitudinally from the bladder neck to the apex of the dome with the urothelium left intact. Tissue strips (6–8 mm long, 1–2 mm wide) were cut along the craniocaudal axis of the dorsal surface for organ bath studies. Each bladder strip was mounted in a 500 μL organ bath and connected to an isometric transducer (HLT050/D; ADinstrument, Chalgrove, UK) under an initial tension of 9.8 mN and allowed to equilibrate (i.e. to accommodate under tension) for at least 60 min. Electrical field stimulation was delivered every minute by a Grass S48 stimulator (Grass Instruments, Quincy, MA, USA) with a 0.1 ms pulse width, at 90 V with a 500 ms train duration at a train frequency of 10 Hz. Contraction data were digitized using a Powerlab/4SP data acquisition system using Chart v.4.2.3 software (ADInstruments) and data stored on a Macintosh computer.
Drugs were dissolved to the required concentrations in gassed physiological saline solution, then administered to the strips via continuous perfusion using a peristaltic pump. For antagonism studies, ondansetron was allowed a minimum equilibration time of 15 min prior to electrical stimulation and the subsequent application of agonist, or Cl-indole.
Data analysis
Concentration response and radioligand-binding data were analysed by computer-assisted iterative curve fitting according to a three parameter logistic equation (Barnes et al., 1992). Electrophysiological data were analysed using Signal3 and Origin 8 (Silverdale Scientific, Stoke Mandeville, UK). Decay times of the 5-HT3 currents were expressed as the time taken to decay to 50% of the maximum response. Where relevant, statistical differences were identified using a t-test (GraphPad Software, La Jolla, CA, USA).
Drugs
Cl-indole, dopamine, 5-HT, quipazine and PNU 282 987 (N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide) were from Sigma-Aldrich (Poole, UK). Alosetron, DDP733 (MKC-733, pumosetrag), palonosetron, ramosetron and RR210 [5-chloro-7-methyl-2-(4-methyl-1,4-diazepan-1-yl)benzo(d)oxazole] were from AMRI Inc. (Albany, NY, USA; a generous gift from Dr Dave Manning). 2-Methyl-5-HT, meta chlorophenyl biguanide (mCPBG), SR57227A [4-amino-(6-chloro-2-pyridyl)-1 piperidine hydrochloride], tropisetron and PNU 120 596 were from Tocris (Bristol, UK). BRL46470 {endo-N-[8-methyl-8-azabicyclo(3,2,l)oct-3y1]-2, 3-dihydro-3, 3-dimethyl-indole-l-carboxamide} was from SmithKline Beecham (Harlow, UK; a generous gift from Dr Tom Blackburn). Ondansetron was from GSK (Stevenage, UK). (S)-Zacopride was from Delalande (Paris, France; a generous gift from Dr Jean-Claude Levy).
Results
5-HT3 receptor agonists increase intracellular calcium in HEKh5-HT3A cells
The endogenous full agonist, 5-HT (10 nM–10 μM), or a range of structurally diverse partial 5-HT3 receptor agonists [(S)-zacopride, DDP733, RR210, quipazine, dopamine, 2-methyl-5-HT, SR57227A, mCPBG], evoked concentration-dependent increases in intracellular calcium in HEKh5-HT3A cells (Figure 1; Supporting Information Figures S2 and S3). Maximal responses evoked by the partial agonists ranged from inconsistent responses barely above baseline [∼3% (S)-zacopride] to 89% (mCPBG) of the maximal response to 5-HT (Figure 1; Supporting Information Figures S2 and S3). It was notable that in the continued presence of 5-HT, or partial agonists with relatively high intrinsic activity, particularly at the higher concentrations investigated, there was tachyphylaxis of the intracellular calcium response (e.g. Figure 1).
Figure 1.

Concentration-dependent ability of 5-HT (A) and DDP733 (B) to increase intracellular calcium in HEK293 cells expressing the h5-HT3A receptor and blockade of such responses by prior incubation with the selective 5-HT3 receptor antagonist, ondansetron (500 nM). Data representative of at least five independent experiments.
Untransfected HEK 293 cells did not respond to either 5-HT (10 μM), or DDP733 (1 μM), although these cells responded to the muscarinic acetylcholine receptor agonist, carbachol (1 mM; data not shown).
Potentiation by Cl-indole of the h5-HT3A receptor-mediated increase in intracellular calcium in HEKh5-HT3A cells
Application of Cl-indole (1–100 μM) potentiated 5-HT responses in a concentration-dependent manner (Figure 2A and B). Cl-indole produced a small potentiation of the peak Ca2+ signal and slowed the decay of the Ca2+ signal in the response to a maximally effective concentration of 5-HT (3 μM). The effects of Cl-indole on 5–HT-induced Ca2+ increases were more pronounced with sub-maximal concentrations of 5-HT (Figure 2). Cl-indole alone did not increase intracellular calcium in HEKh5-HT3A cells.
Figure 2.

Concentration-dependent ability of Cl-indole to potentiate responses to 5-HT (A; 0.3 μM, B; 3.0 μM) and DDP733 (C; 100 nM) to increase intracellular calcium in HEK293 cells expressing the h5-HT3A receptor (data representative from 3 to 6 experiments). In A–C, drugs were added where indicated by the horizontal bar. D; Cl-indole concentration–responses fitted to a three-parameter logistic equation (100 nM DDP – triangles, 0.3 μM 5-HT – squares and 3.0 μM 5-HT – circles and inset).
Cl-indole also induced a concentration-dependent increase in the maximal responses evoked by a range of 5-HT3 receptor partial agonists (Figure 2C and D and Supporting Information Figures S2 and S3A).
Increases in intracellular calcium in HEKh5-HT3A cells evoked by 5-HT and the partial agonists in the absence and presence of Cl-indole (10–100 μM) were completely blocked by prior application of the selective antagonists, ondansetron (500 nM; Supporting Information Figure S3B – data for 10 μM Cl-indole not shown) and granisetron (500 nM; data not shown). In contrast to the 5-HT3 receptor agonists, the selective 5-HT3 receptor antagonists alosetron, BRL46470, ondansetron, ramosetron, and palonosetron (all individually at 3 μM) did not evoke increases in intracellular calcium in HEKh5-HT3A cells in either the absence, or presence, of Cl-indole (10–100 μM; Supporting Information Figure S4).
Potentiation by Cl-indole of agonist-evoked currents recorded under voltage-clamp from HEKh5-HT3A cells
At a holding potential of −60 mV, a brief application of 5-HT (10 μM; 100 ms) to HEKh5-HT3A cells elicited a transient inward current (560 ± 130 pA, n = 6; Figure 3A). The rising phase was best fitted by a single exponential function with a mean time constant of 0.9 ± 0.1 s and the current decayed slowly back to baseline (t50 2.1 ± 0.4 s). Repeated applications (5 min interval) of 5-HT significantly reduced the amplitude of the inward current, such that by the third application the peak amplitude was approximately one-third of the initial response (Figure 3A). In contrast, 5-HT3 receptor currents evoked by a 10 ms application of 5-HT (10 μM) at the same frequency were not reduced by repeated stimulation (Figure 3A) and hence this protocol was employed for further investigations.
Figure 3.

A: ability of repeated application of 5-HT (10 μM) for 100 ms (i) and 10 ms (ii) to evoke responses from voltage-clamped HEK293 cells expressing the h5-HT3A receptor. Traces are representative of at least three independent experiments, numbers represent the traces corresponding to the application number of the 5-HT. Data in histograms represent mean ± SEM, n = 5–6. B: ability of 5-HT (10 μM applied for 10 ms) in the absence (control) and presence of Cl-indole (10 μM, grey trace) to evoke responses from voltage-clamped HEK293 cells expressing the h5-HT3A receptor (i) with histograms displaying impact upon decay time (ii), amplitude (iii) and rise time (iv); the right hand column in each of these histograms represents results after wash-out of Cl-indole. *P < 0.05, **P < 0.01.
Superfusion of Cl-indole (10 μM) did not alter the holding current of 5-HT3A receptor expressing HEK 293 cells, nor did the compound alter the amplitude (control 411 ± 107 pA, Cl-indole 399 ± 106 pA; n = 6; P = 0.86) or rise time (control 76 ± 16 ms, Cl-indole 110 ± 40 ms; n = 6; P = 0.31) of 5-HT (10 μM)-induced currents (Figure 3B). However, Cl-indole significantly increased the decay time for 5-HT3 receptor-mediated currents (control t50 = 2.8 ± 0.7 s, Cl-indole t50 = 12.2 ± 2.5 s; n = 6; P = 0.005); the effect of Cl-indole on decay rate was partially reversible after washout (t50 = 6.3 ± 1.0 s; P = 0.04; Figure 3B).
The selective 5-HT3 receptor antagonist, ondansetron (100 nM), completely blocked the activation of 5-HT3 receptors by 5-HT (10 μM) in both the absence and presence of Cl-indole (10 μM). The effect of ondansetron was reversible upon washout with subsequent application of 5-HT evoking inward current, which was modulated by Cl-indole (Figure 4A).
Figure 4.
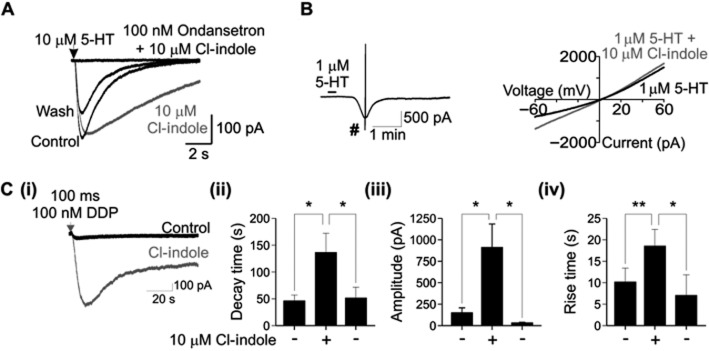
Ability of Cl-indole (10 μM) to potentiate 5-HT3 receptor agonist responses. A: The presence of the selective 5-HT3 receptor antagonist, ondansetron (100 nM), completely prevented a response to 5-HT (10 μM applied for 10 ms) in the presence of Cl-indole (10 μM). The effect of ondansetron was partly reversible by wash-out [subsequent response to 5-HT (10 μM applied for 10 ms; wash)]. Traces are representative of at least three independent experiments. B: I–V plots arising in the presence of 5-HT (1.0 μM) or 5-HT (1.0 μM) plus Cl-indole (10 μM) determined around the peak response (indicated #). Drugs were bath applied for 15 s. Traces are representative of at least three independent experiments. C: DDP733 (100 nM) in the absence and presence of Cl-indole (10 μM); recordings from voltage-clamped HEK293 cells expressing the h5-HT3A receptor (i) with histograms displaying impact upon decay time (ii), amplitude (iii) and rise time (iv); the right-hand column in each of these histograms represents results after wash-out of Cl-indole. Traces are representative of at least three independent experiments. Data in histograms represent mean ± SEM, n = 5–6. *P < 0.05, **P < 0.01.
To assess whether Cl-indole affected the ionic permeability of the h5-HT3A receptor, the I–V relationship of the macroscopic current response to 5-HT was constructed by a voltage ramp (−100 to +60 mV) recorded at the peak of the current response to bath-applied 5-HT (1.0 μM; 15 s). The reversal potential (E5-HT) in the presence and absence of Cl-indole (10 μM) was −2.2 ± 3.1 and −0.5 ± 2.9 mV, respectively n = 6, P = 0.74) (Figure 4B), suggesting that Cl-indole does not grossly affect the ionic permeability of h5-HT3A receptors. The peak 5-HT current recorded at −60 mV was significantly increased by Cl-indole (Control 265.8 ± 127.8 pA; Cl-indole 1430.8 ± 362.9 pA; n = 5, P < 0.05)
Application of the 5-HT3 receptor partial agonist, DDP733 (100 nM; 100 ms), evoked a relatively small inward current (149 ± 58 pA, n = 6; Figure 4C) which had a slow rise time (10.2 ± 3.2 s) and slow decay time (46.5 ± 10.5 s). Superfusion of Cl-indole (10 μM), significantly increased the amplitude of DDP733-induced currents (912 ± 272 pA; P < 0.05; Figure 4C) and slowed the rise time (18.6 ± 3.9 s; P < 0.005). Cl-indole also prolonged the decay rate of DDP733-induced currents (137 ± 35 s; P < 0.01; Figure 4C). The effect of Cl-indole upon DDP733-evoked amplitude, rise time and decay rate was fully reversed by washout of Cl-indole (33.1 ± 6.5 pA, 11.6 ± 3.9 s, 52 ± 20 s, respectively).
Modulation of neuronally mediated contraction of mouse bladder
To investigate the ability of Cl-indole to modulate native 5-HT3 receptors, the 5-HT3 receptor-mediated increase in bladder smooth muscle ‘twitch’ was investigated (Chetty et al., 2007). Under control conditions, electrical field stimulation induced a tetrodotoxin (100 nM)-sensitive contraction of the muscle strips. Using a sub-maximal stimulus protocol (six pulses at 10 Hz), consistent contraction amplitudes were obtained when the stimuli were delivered 1 min apart. The selective 5-HT3 receptor agonist, mCPBG (3.0 μM), transiently increased the amplitude of contraction, reaching a peak increase of 20 ± 4% (n = 4 strips; P < 0.05), which subsequently declined in the continued presence of mCPBG (30 μM; Figure 5). Cl-indole (30 μM) alone had no effect on the amplitude of contraction (6 ± 3%; n = 4; P = 0.46; Figure 5), but subsequent addition of mCPBG (3.0 μM) greatly increased the peak amplitude by 54 ± 3%, an effect that was maintained in the continued presence of mCPBG (30 μM; Figure 5).
Figure 5.

Cl-indole augments the mCPBG-induced potentiation of the neurogenic contractile response in mouse bladder strips. (A) Sample traces from different bladder strips showing that Cl-indole (30 μM) augments the mCPBG-induced potentiation of contractile response in mouse bladder strips. (Ai) mCPBG (3.0 μM) alone only subtly increases the amplitude of contractile force generated following field stimulation. (Aii) In the presence of Cl-indole (30 μM), mCPBG increases to a greater relative extent the contractile force. For each panel, the black line indicates the neurogenic contraction in the absence of mCPBG, and the grey line indicates the subsequent response in the presence of mCPBG in the same bladder strip. (Ai) and (Aii) show the response of different bladder strips from the same mouse and measured in parallel; the mean amplitudes did not vary significantly amongst the two test groups, although different muscle strips (even from the same animals) showed a range of typical contractile forces. (Bi) Field stimulation of mouse bladder strips (open bars) is subtly augmented by mCPBG (3.0 μM), an effect that shows tachyphylaxis (i.e. the plateau contractile amplitude in the continued presence of mCPBG is lower than the peak response). This effect is reversible upon removing mCPBG (washout). (Bii, Ci) In the presence of Cl-indole (30 μM), the peak response to mCPBG (3.0 μM) was potentiated and the tachyphylaxis was prevented (closed bars). The effect of Cl-indole was only partially reversed on wash. Cl-indole alone had no significant effect on contraction. (Cii) Responses to mCPBG (3.0 μM) in the presence of Cl-indole were blocked by the 5-HT3 receptor antagonist ondansetron (open bar). *P < 0.05 compared with vehicle, #P < 0.05 compared with mCPBG response in the absence of Cl-indole.
In the presence of the 5-HT3 receptor antagonist ondansetron (500 nM) and subsequent application of Cl-indole (30 μM), mCPBG (3.0 μM) did not significantly increase the amplitude of contraction (8 ± 7%; n = 6; P = 0.30; Figure 5Cii, open bars): in the matching set of bladder strips, Cl-indole (30 μM) + mCPBG (3.0 μM) in the absence of ondansetron increased the peak amplitude by 31 ± 10% (n = 5; P < 0.05; Figure 5Cii filled bars).
Effect of Cl-indole upon ligand affinity for the h5-HT3A receptor
Cl-indole (10 μM) had no effect upon saturation data arising from [3H]granisetron (0.1–10 nM) binding the h5-HT3A receptor expressed by HEKh5-HT3A cells (Bmax = 1130 ± 146 and 1027 ± 66 fmol·mg−1 protein, pKd = 8.85 ± 0.20 and 8.77 ± 0.16 calculated in the absence and presence of Cl-indole, respectively [mean ± SEM, n = 3], P > 0.05; non-specific binding defined by ondansetron, 10 μM). In competition studies with 5-HT and the antagonist, tropisetron, Cl-indole (10 μM) increased the apparent affinity of 5-HT for the [3H]granisetron specific binding site in HEKh5-HT3A cell homogenates (P < 0.05), but did not modify the affinity of tropisetron (P > 0.05; Figure 6 and Table 1).
Figure 6.

Impact of Cl-indole (10 μM; filled symbols) on the ability of 5-HT (circles) and tropisetron (squares) to compete for specific [3H]granisetron binding to HEK293 cells expressing the h5-HT3A receptor. Data represent mean from four independent experiments.
Table 1.
Impact of Cl-indole (10 μM) upon the affinity (pKi) of 5-HT and tropisetron (ICS 205–930; (1R,5S)-8-methyl-8-azabicyclo[3.2.1]octan-3-yl 1methyl-indole-3-carboxylate) and Hill coefficient for the h5-HT3A receptor radiolabelled with [3H]granisetron
| pKi | Hill coefficient | |
|---|---|---|
| 5-HT | 6.31 ± 0.03 | 1.59 ± 0.21 |
| 5-HT + Cl-indole | 6.57 ± 0.03* | 1.44 ± 0.13 |
| Tropisetron | 8.50 ± 0.10 | 1.00 ± 0.03 |
| Tropisetron + Cl-indole | 8.39 ± 0.06 | 0.99 ± 0.04 |
Data represents mean ± SEM from four independent experiments. Effect of Cl-indole
P < 0.05.
Further characterization of Cl-indole as a PAM
To further characterize the effect of Cl-indole, we examined whether it could re-activate desensitized 5-HT3 receptors. Prolonged bath application of 5-HT (10 μM) produced a large transient current (2.52 ± 0.75 nA; n = 4) that decayed rapidly to 8.9 ± 4.0% of the peak response. In the continued presence of 5-HT, pressure ejection of Cl-indole (10 μM) elicited an inward current (0.97 ± 0.54 pA; Figure 7).
Figure 7.

Characterization of Cl-indole. Prolonged bath application of 5-HT (10 μM) induced an inward current through 5-HT3A receptors which rapidly and completely desensitized. Subsequent picospritzer application of 10 μM Cl-indole (1 s, 20 psi; denoted by arrow) revealed a smaller inward current.
Previously described PAMs of the 5-HT3 receptor suffer from a lack of potency and specificity; for example, 5-hydroxyindole, is a PAM of both 5-HT3 (van Hooft et al., 1997) and nicotinic α7 receptors (Zwart et al., 2002). To establish the specificity of the modulation by Cl-indole, we tested its effect on nicotinic α7 receptors stably co-expressed with ric3 in HEK-293 cells (G. Grafton et al., unpubl.). Application of the selective nicotinic α7 agonist, PNU 282 987 (1 μM), induced a small inward current 38.4 ± 14.0 pA (n = 5; Figure 8). While nicotinic α7 currents were potentiated by the PAM, PNU 120 596 (control, 43.1 ± 12.5 pA; PNU 120 596, 1332.2 ± 296.3 pA; n = 5 P < 0.05), Cl-indole did not modulate the nicotinic α7 current (27.8 ± 5.8 pA, P = 0.34), suggesting that Cl-indole is a selective PAM of 5-HT3 receptors.
Figure 8.

Cl-indole does not act as a positive allosteric modulator of nicotinic α7 channels. (Ai) Pressure ejection of the nicotinic α7 agonist, PNU 282 987 (1 μM, 3 s, 20 psi; PNU agonist), evoked an inward current in an α7/Ric3 cell line (black line) that was not modulated by Cl-indole (grey trace). (Aii) mean data (±SEM) showed no significant alteration of nicotinic α7 currents by Cl-indole (n = 5). (B) Nicotinic α7 receptor currents evoked by the nicotinic α7 agonist, PNU 282 987, were significantly potentiated by the nicotinic α7 PAM, PNU 120 596 (PNU PAM). Where relevant, allosteric modulators (Cl-indole or PNU 120 596) were bath applied for 10 min prior to application of agonist and were present for the remainder of the recording.
Discussion
The present study demonstrates that Cl-indole is a relatively potent and selective PAM of the h5-HT3A receptor, with this action appearing to extend to the mouse native 5-HT3 receptor. Our studies initially measured the 5-HT3 receptor-mediated increase in intracellular calcium, which demonstrated that HEKh5-HT3A cells possessed a functional h5-HT3A receptor based on receptor pharmacology with a range of selective and non-selective 5-HT3 receptor ligands. The pharmacological profile included differential maximal responses by a variety of partial agonists, the majority with recognized sub-maximal intrinsic activities. Further evidence supporting a selective modulation of intracellular calcium via the 5-HT3A receptor came from studies performed in parallel where 5-HT3 receptor agonists failed to evoke responses in native HEK293 cells.
When applied alone to HEKh5-HT3A cells, Cl-indole did not cause an increase in intracellular calcium yet, in a concentration-dependent manner, potentiated the response to a range of agonists for the 5-HT3 receptor. It is noteworthy that the efficacy of Cl-indole was strongly agonist-dependent. Thus, at maximally effective concentrations of the agonists, Cl-indole promoted only a small percentage increase in the maximal effect of the full agonist 5-HT, yet responses by agonists with lower intrinsic activity for the 5-HT3 receptor, were potentiated to a much greater degree. Thus in the presence of Cl-indole, all of the partial 5-HT3 receptor agonists tested in the present study ((S)-zacopride, DDP733, dopamine, mCPBG, 2-methyl-5-HT, quipazine, RR 210, SR57727A; Richardson et al., 1985; Neijt et al., 1986; Kilpatrick et al., 1990; Bachy et al., 1993; Downie et al., 1995; Yamazaki et al., 1996; Evangelista, 2007) evoked considerably larger responses.
It is now appreciated that 5-HT3 receptor antagonists can have contrasting characteristics. For example, palonosetron binds with positive cooperativity (Rojas et al., 2008) as do 5-HT3 receptor agonists, including most with low intrinsic activity (e.g. Barnes et al., 1992). In addition, palonosetron evokes some receptor internalization that is less apparent with ondansetron and granisetron (Rojas et al., 2010). Another atypical 5-HT3 receptor antagonist, BRL46470, has an enhanced binding capacity to recombinant 5-HT3A receptors (Steward et al., 1995). However, in the present study both palonosetron and BRL46470 behaved as other ‘classical’ 5-HT3 receptor antagonists (alosetron, ondansetron, ramosetron) by not displaying agonist activity even in the presence of a maximal concentration of Cl-indole as assessed by measurements of intracellular calcium in HEKh5-HT3A cells. This directly contrasts with the ability of Cl-indole to elicit robust 5-HT3 receptor mediated responses from the low intrinsic activity partial agonists in the same experimental paradigm [e.g. (S)-zacopride, which has previously been designated an antagonist (Smith et al., 1988)]. Such findings suggest that the atypical characteristics of the 5-HT3 receptor ligands, palonosetron and BRL46470, are not attributable to a low level of intrinsic activity for the h5-HT3A receptor. In contrast, the atypical characteristics of (S)-zacopride in comparison with other selective 5-HT3 receptor antagonists (e.g. Barnes et al., 1990) may be attributed to a partial agonist action upon the 5-HT3 receptor.
Patch clamp recordings using HEKh5-HT3A cells demonstrated that Cl-indole potentiated h5-HT3A receptor mediated responses, particularly the maximal responses evoked by the partial agonist DDP733 in comparison to 5-HT. This effect of Cl-indole was not associated with change in the driving force for the 5-HT3 receptor mediated current as the reversal potential (E5-HT) was unchanged in the presence of Cl-indole.
To determine whether the action of Cl-indole translated from recombinant to native 5-HT3 receptors, we investigated the effect of the compound upon the previously described 5-HT3 receptor mediated increase in the neurogenic contraction of the mouse urinary bladder (Chetty et al., 2007). Due to the presence of 5-HT receptors other than those of the 5-HT3 receptor subtype in this preparation (Chetty et al., 2007), a just-maximal concentration of the selective 5-HT3 receptor agonist, mCPBG, which behaves as a full agonist in this preparation, was employed (Chetty et al., 2007). While mCPBG alone caused a small increase in the amplitude of nerve-stimulation evoked contraction, this effect showed marked tachyphylaxis consistent with 5-HT3 receptor desensitization. Similar to the results obtained with recombinant 5-HT3 receptors, Cl-indole potentiated mCPBG increases in the neurogenic contraction of the mouse urinary bladder and prevented the tachyphylaxis. Responses to mCPBG in the absence or presence of Cl-indole were prevented by the inclusion of ondansetron (500 nM). Together with the data from HEKh5-HT3A cells, these observations suggest that Cl-indole acts similarly on native and heterologously expressed 5-HT3 receptors. In contrast to reports that 5-hydroxyindole is a PAM at rodent 5-HT3 receptors but not at human receptors (Grønlien et al., 2010), Cl-indole evoked equivalent responses in both species.
5-substituted indoles, such as 5-hydroxyindole, have been long recognized as PAMs of 5-HT3 receptors. However several reports indicate that 5-hydroxyindole has both orthosteric and allosteric binding activities (Kooyman et al., 1994; Hu and Lovinger, 2008) suggesting at least two binding sites, one of which may overlap the orthosteric site. Furthermore, 5-hydroxyindole has allosteric effects on the nicotinic α7 receptor (Bertrand and Gopalakrishnan, 2007) and one report suggests that 5-hydroxyindole is a PAM at rodent but not human receptors (Grønlien et al., 2010). These studies cast considerable doubt on 5-hydroxyindole's selectivity and its mixed mode of action renders its use as a pharmacological tool problematical. In the present study we demonstrated, that unlike 5-hydroxyindole, Cl-indole is not an allosteric modulator of nicotinic α7 receptors highlighting the selectivity of Cl-indole over 5-hydroxyindole. Although closely related structures, Cl-indole exhibits a higher potency and selectivity than 5-hydroxyindole, making it a more attractive pharmacological tool for the exploration of the allosteric modulation of the 5-HT3 receptor.
In conclusion, the present study has identified Cl-indole as a relatively potent and selective PAM of the 5-HT3 receptor. Availability of such compounds will aid investigation of the molecular basis for allosteric modulation of the 5-HT3 receptor to help the discovery of novel therapeutic drugs targeting this receptor.
Acknowledgments
Funding from the University of Birmingham (NMB) is gratefully acknowledged.
Glossary
- AM
acetoxymethyl
- Cl-indole
5-chloroindole
- E5-HT
reversal potential for 5-HT-evoked current
- HEKh5-HT3A
HEK293 cells stably expressing the human 5-HT3A subunit
- LGIC
ligand-gated ion channel
- mCPBG
meta chlorophenyl biguanide
- PAMs
positive allosteric modulators
Conflict of interest
Authors declare that they have not any conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Structure of Cl-indole.
Figure S2 Concentration-dependent ability of various 5-HT3 receptor agonists to induce increases in intracellular calcium in HEK293 cells expressing the h5-HT3A receptor in the absence and presence of Cl-indole (10 μM). Data represent mean ± SEM, n = 3–6.
Figure S3 Ability of Cl-indole (10, 30 and 100 μM) to potentiate the action of various 5-HT3 receptor agonists (A) to increase intracellular calcium in HEK293 cells expressing the h5-HT3A receptor and the blockade of the response by the selective 5-HT3 receptor antagonist, ondansetron (500 nM) (B) 5-HT (3.0 μM), (S)-zacopride (1.0 μM), DDP 733 (1.0 μM), RR210 (1.0 μM), quipazine (30 nM), dopamine (300 μM), SR57227A (1.0 μM), mCPBG (1.0 μM) and 2-methyl-5-HT (10 μM). Data represent mean % response compared with 5-HT ± SEM (n = 3–15). Effect of Cl-indole *P < 0.05, **P < 0.01, ***P < 0.001; antagonism by ondansetron P < 0.01 in all instances except compared with (S)-zacopride alone.
Figure S4 Failure of Cl-indole (10, 30 and 100 μM) to induce responses from various 5-HT3 receptor antagonists assessed by intracellular calcium in HEK293 cells expressing the h5-HT3A receptor. 5-HT, full agonist for comparison, and the antagonists; ramosetron, palonosetron, BRL46470, ondansetron, alosetron and tropisetron (all 3.0 μM). Data represent mean % response compared with 5-HT ± SEM (n = 3–8).
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition (2009) Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachy A, Héaulme M, Giudice A, Michaud JC, Lefevre IA, Souilhac J, et al. SR 57227A: a potent and selective agonist at central and peripheral 5-HT3 receptors in vitro and in vivo. Eur J Pharmacol. 1993;237:299–309. doi: 10.1016/0014-2999(93)90282-m. [DOI] [PubMed] [Google Scholar]
- Barnes JM, Barnes NM, Costall B, Domeney AM, Kelly ME, Naylor RJ. The differential activities of (R+) and (S-)-Zacopride as 5-HT3 receptor antagonists. Pharmacol Biochem Behav. 1990;37:717–727. doi: 10.1016/0091-3057(90)90554-u. [DOI] [PubMed] [Google Scholar]
- Barnes JM, Barnes NM, Costall B, Jagger SM, Naylor RJ, Robertson DW, et al. Agonist interactions with 5-HT3 receptor recognition sites in the rat entorhinal cortex labelled by structurally diverse radioligands. Br J Pharmacol. 1992;105:500–504. doi: 10.1111/j.1476-5381.1992.tb14283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes NM, Hales TG, Lummis SC, Peters JA. The 5-HT3 receptor – the relationship between structure and function. Neuropharmacology. 2009;56:273–284. doi: 10.1016/j.neuropharm.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M. Allosteric modulation of nicotinic acethylcholine receptors. Biochem Pharmacol. 2007;74:1155–1163. doi: 10.1016/j.bcp.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Boess FG, Beroukhim R, Martin IL. Ultrastructure of the 5-hydroxytryptamine3 receptor. J Neurochem. 1995;64:1401–1405. doi: 10.1046/j.1471-4159.1995.64031401.x. [DOI] [PubMed] [Google Scholar]
- Brady CA, Stanford IM, Ali I, Lin L, Williams JM, Dubin AE, et al. Pharmacological comparison of human homomeric 5-HT3A receptors versus heteromeric 5-HT3A/3B receptors. Neuropharmacology. 2001;41:282–284. doi: 10.1016/s0028-3908(01)00074-0. [DOI] [PubMed] [Google Scholar]
- Chetty N, Coupar IM, Chess-Williams R, Kerr KP. Demonstration of 5-HT3 receptor function and expression in the mouse bladder. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:359–368. doi: 10.1007/s00210-007-0173-7. [DOI] [PubMed] [Google Scholar]
- Davies PA. Allosteric moduation of the 5-HT3 receptor. Curr Opin Pharmacol. 2011;11:75–80. doi: 10.1016/j.coph.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PA, Pistis M, Hanna MC, Peters JA, Lambert JJ, Hales TG, et al. The 5-HT3B subunit is a major determinant of serotonin-receptor function. Nature. 1999;397:359–363. doi: 10.1038/16941. [DOI] [PubMed] [Google Scholar]
- Downie DL, Hope AG, Belelli D, Lambert JJ, Peters JA, Bentley KR, et al. The interaction of trichloroethanol with murine recombinant 5-HT3 receptors. Br J Pharmacol. 1995;114:1641–1651. doi: 10.1111/j.1476-5381.1995.tb14952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin AE, Huvar R, D'Andrea MR, Pyati J, Zhu JY, Joy KC, et al. The pharmacological and functional characteristics of the serotonin 5-HT3A receptor are specifically modified by a 5-HT3B receptor subunit. J Biol Chem. 1999;274:30799–30810. doi: 10.1074/jbc.274.43.30799. [DOI] [PubMed] [Google Scholar]
- Evangelista S. Drug evaluation: Pumosetrag for the treatment of irritable bowel syndrome and gastroesophageal reflux disease. Curr Opin Investig Drugs. 2007;8:416–422. [PubMed] [Google Scholar]
- Fiebich BL, Akundi RS, Seidel M, Geyer V, Haus U, Müller W, et al. Expression of 5-HT3A receptors in cells of the immune system. Scand J Rheumatol Suppl. 2004;119:9–11. [PubMed] [Google Scholar]
- Fletcher S, Barnes NM. Desperately seeking subunits: are native 5-HT3 receptors really homomeric complexes? Trends Pharmacol Sci. 1998;19:212–215. doi: 10.1016/s0165-6147(98)01210-3. [DOI] [PubMed] [Google Scholar]
- Gill CH, Peters JA, Lambert JJ. An electrophysiological investigation of the properties of a murine recombinant 5-HT3 receptor stably expressed in HEK 293 cells. Br J Pharmacol. 1995;114:1211–1221. doi: 10.1111/j.1476-5381.1995.tb13335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grønlien JH, Ween H, Thorin-Hagene K, Cassar S, Li J, Briggs CA, et al. Importance of M2-M3 loop in governing properties of genistein at the α7 nicotinic acetylcholine receptor inferred from α7/5-HT3A chimera. Eur J Pharmacol. 2010;647:37–47. doi: 10.1016/j.ejphar.2010.08.027. [DOI] [PubMed] [Google Scholar]
- van Hooft JA, van der Haar E, Vijverberg HPM. Allosteric potentiation of the 5-HT3 receptor-mediated ion current in N1E-115 neuroblastoma cells by 5-hydroxyindole and analogues. Neuropharm. 1997;36:649–653. doi: 10.1016/s0028-3908(97)00045-2. [DOI] [PubMed] [Google Scholar]
- Hu XQ, Lovinger DM. The L293 residue in transmembrane domain 2 of the 5-HT3A receptor is a molecular determinant of allosteric modulation by 5-hydroxyindole. Neuropharmacology. 2008;54:1153–1165. doi: 10.1016/j.neuropharm.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XQ, Hayrapetyan V, Gadhiya JJ, Rhubottom HE, Lovinger DM, Machu TK. Mutations of L293 in transmembrane two of the mouse 5-hydroxytryptamine3A receptor alter gating and alcohol modulatory actions. Br J Pharmacol. 2006;148:88–101. doi: 10.1038/sj.bjp.0706685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussy N, Lukas W, Jones KA. Functional properties of a cloned 5-hydroxytryptamine ionotropic receptor subunit: comparison with native mouse receptors. J Physiol. 1994;481:311–323. doi: 10.1113/jphysiol.1994.sp020441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Surprenant A. Single channel properties of the 5-HT3 subtype of serotonin receptor in primary cultures of rodent hippocampus. Neurosci Lett. 1994;174:133–136. doi: 10.1016/0304-3940(94)90004-3. [DOI] [PubMed] [Google Scholar]
- Kelley SP, Dunlop JI, Kirkness EF, Lambert JJ, Peters JA. A cytoplasmic region determines single-channel conductance in 5-HT3 receptors. Nature. 2003;424:321–324. doi: 10.1038/nature01788. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick GJ, Butler A, Burridge J, Oxford AW. 1-(m-chlorophenyl)-biguanide, a potent high affinity 5-HT3 receptor agonist. Eur J Pharmacol. 1990;182:193–197. doi: 10.1016/0014-2999(90)90513-6. [DOI] [PubMed] [Google Scholar]
- Kooyman AR, van Hooft JA, Vanderheijden PML, Vijverberg HPM. Competitive and non-competitve effects of 5-hydroxyindole on 5-HT3 receptors in N1E-115 neuroblastoma cells. Br J Pharmacol. 1994;112:541–546. doi: 10.1111/j.1476-5381.1994.tb13107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JJ, Peters JA, Hales TG, Dempster J. The properties of 5-HT3 receptors in clonal cell lines studied by patch-clamp techniques. Br J Pharmacol. 1989;97:27–40. doi: 10.1111/j.1476-5381.1989.tb11920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey MR, Cooper MA, Lambert JJ, Peters JA. Rings of charge within the extracellular vestibule influence ion permeation of the 5-HT3A receptor. J Biol Chem. 2011;286:16008–16017. doi: 10.1074/jbc.M111.219618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk SA, Williams JM, Hope AG, Barnes NM. Identification and importance of N-glycosylation of the human 5-hydroxytryptamine3A receptor subunit. Biochem Pharmacol. 2004;68:1787–1796. doi: 10.1016/j.bcp.2004.06.034. [DOI] [PubMed] [Google Scholar]
- Moretti G, Quartarone G, Tassan A, Zingales A. 5-Amino- and 5-chloro-indole as mild steel corrosion inhibitors in 1N sulphuric acid. Electrochim Acta. 1996;41:1971–1980. [Google Scholar]
- Neijt HC, Vijverberg HPM, van den Bercken J. The dopamine response in mouse neuroblastoma cells is mediated by serotonin 5-HT3 receptors. Eur J Pharmacol. 1986;127:217–274. doi: 10.1016/0014-2999(86)90374-2. [DOI] [PubMed] [Google Scholar]
- Parker RM, Bentley KR, Barnes NM. Allosteric modulation of 5-HT3 receptors: focus on alcohols and anaesthetic agents. Trends Pharmacol Sci. 1996;17:95–99. doi: 10.1016/0165-6147(96)10003-1. [DOI] [PubMed] [Google Scholar]
- Peters JA, Hales TG, Lambert JJ. Molecular determinants of single-channel conductance and ion selectivity in the Cys-loop family: insights from the 5-HT3 receptor. Trends Pharmacol Sci. 2005;26:587–594. doi: 10.1016/j.tips.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Richardson BP, Engel G, Donatsch P, Stadler PA. Identification of serotonin M-receptor subtypes and their specific blockade by a new class of drugs. Nature. 1985;316:126–131. doi: 10.1038/316126a0. [DOI] [PubMed] [Google Scholar]
- Rinaldi A, Chiaravalli AM, Mian M, Zucca E, Tibiletti MG, Capella C, et al. Serotonin receptor 3A expression in normal and neoplastic B cells. Pathobiology. 2010;77:129–135. doi: 10.1159/000292646. [DOI] [PubMed] [Google Scholar]
- Rojas C, Stathis M, Thomas AG, Massuda EB, Alt J, Zhang J, et al. Palonosetron exhibits unique molecular interactions with the 5-HT3 receptor. Anesth Analg. 2008;107:469–478. doi: 10.1213/ane.0b013e318172fa74. [DOI] [PubMed] [Google Scholar]
- Rojas C, Thomas AG, Alt J, Stathis M, Zhang J, Rubenstein EB, et al. Palonosetron triggers 5-HT3 receptor internalization and causes prolonged inhibition of receptor function. Eur J Pharmacol. 2010;626:193–199. doi: 10.1016/j.ejphar.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Smith WW, Sancilio LF, Owera-Atepo JB, Naylor RJ, Lambert L. Zacopride, a potent 5-HT3 antagonist. J Pharm Pharmacol. 1988;40:301–302. doi: 10.1111/j.2042-7158.1988.tb05253.x. [DOI] [PubMed] [Google Scholar]
- Steward LJ, Ge J, Bentley KR, Barber PC, Hope AG, Lambert JJ, et al. Evidence that the atypical 5-HT3 receptor ligand, [3H]-BRL46470, labels additional 5-HT3 binding sites compared to [3H]-granisetron. Br J Pharmacol. 1995;116:1781–1788. doi: 10.1111/j.1476-5381.1995.tb16663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walstab J, Rappold G, Niesler B. 5-HT3 receptors: Role in disease and target of drugs. Pharmacol Ther. 2010;128:146–169. doi: 10.1016/j.pharmthera.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Williams DK, Wang J, Tabe A. Positive allosteric modulators as an approach to nicotinic acetylcholine-targeted therapeutics: advantages and limitations. Biochem Pharmacol. 2011;82:915–930. doi: 10.1016/j.bcp.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Yano T, Tabe A. Species differences in the intrinsic activity of MKC-733 for 5-HT3 receptor. Jpn J Pharmacol. 1996;71:62P. [Google Scholar]
- Yang J, Mathie A, Hille B. 5-HT3 receptor channels in dissociated rat superior cervical ganglion neurons. J Physiol. 1992;448:237–256. doi: 10.1113/jphysiol.1992.sp019039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, De Filippi G, Broad LM, McPhie GI, Pearson KH, Baldwinson T, et al. 5-hydroxyindole potentiates human alpha 7 nicotinic receptor mediated responses and enhances acethylcholine-induced glutamate release in cerebellar slices. Neuropharm. 2002;43:374–384. doi: 10.1016/s0028-3908(02)00094-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.