

Abstract
Background and Purpose
Dietary indole derivatives, indole-3-carbinol (I3C) and diindolylmethane (DIM), possess anti-cancer properties and exhibit the characteristics of aryl hydrocarbon receptor (AhR) ligands. Because AhR activation has recently been shown to regulate T cell differentiation, we tested the hypothesis that I3C and DIM may mediate anti-inflammatory properties by promoting regulatory T cell (T-regs) differentiation while inhibiting Th17 cells.
Experimental Approach
We investigated the therapeutic efficacy of I3C and DIM against experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis (MS). The efficacy was evaluated based on clinical scores of paralysis, histopathology, serum cytokines and infiltration of T cells in the CNS. We next studied the mechanism of induction of T cells against myelin oligodendrocyte glycoprotein (MOG35–55) peptide, both in vivo and in vitro, specifically investigating the differentiation of T-regs and Th17 cells, and determined if indoles were acting through AhR.
Key Results
Pretreatment of EAE mice with I3C or DIM completely prevented the clinical symptoms and cellular infiltration into the CNS. Also, post-treatment of EAE with I3C or DIM proved highly effective in curtailing the overall severity of the disease. In addition, I3C or DIM promoted the generation of T-regs, while down-regulating the induction of MOG-specific Th17 cells. The regulation of FoxP3 induction and suppression of Th17 cells by indoles in vivo and in vitro were found to be AhR-dependent.
Conclusions and Implications
Together, our studies demonstrate for the first time that I3C and DIM may serve as novel therapeutics to suppress neuroinflammation seen during MS through activation of AhR.
Keywords: aryl hydrocarbon receptor (AhR), diindolylmethane (DIM), experimental autoimmune encephalomyelitis (EAE), indole-3-carbinol (I3C), multiple sclerosis (MS), regulatory T cell (T-regs), T helper 17 cell (Th17)
Introduction
Multiple sclerosis (MS) is a debilitating autoimmune disease that affects approximately 400 000 people a year in the US, and more than 2.1 million people worldwide. Women appear to be two–three times more prone to being diagnosed with the disease than men. Patients suffering from MS typically develop partial to complete paralysis as well as impaired cognitive function. While the exact trigger of MS remains unclear, it is known that the disease involves autoreactive T cells that infiltrate into the CNS and target myelinated cells. These autoreactive T cells create an inflammatory microenvironment that destroys the CNS, leading to disruption in neuronal signalling and eventually a loss of motor function (Imam et al., 2007).
Experimental autoimmune encephalomyelitis (EAE) is a murine model often used to study the development of MS and test possible treatment options because it mimics many of the disease patterns. During EAE development and MS relapse, there is a bias towards CD4+ T-helper 1 (Th1) and pathogenic T-helper type 17 (Th17) cell differentiation (Stromnes and Goverman, 2006; Imam et al., 2007). Furthermore, mice deficient in either T-bet or STAT4, transcription factors needed for Th1 differentiation, have been shown to be resistant to EAE (Bettelli et al., 2004). Recently, Th17 cells have also been reported to contribute to the pathogenesis of EAE when adoptively transferred into naïve mice (Aggarwal et al., 2003; Langrish et al., 2005). In another study, these IL23p19-deficient mice displayed a significant reduction in IL-17-producing T cells in the CNS, which correlated with lower disease scores compared to wild-type EAE mice (Harrington et al., 2005; Park et al., 2005). The role of IL-17 in MS patients was also confirmed when IL-17 levels obtained from myelin basic protein-stimulated peripheral blood cells obtained from MS patients or controls correlated with the active lesions as seen with MRI (Hedegaard et al., 2008). In addition, the increased levels of pro-inflammatory cytokines, such as IFNγ, TNFα, IL-6 and IL-17, significantly drive disease progression, while simultaneously suppressing anti-inflammatory T-helper 2 (Th2) and regulatory T cell (T-regs) subsets (Bettelli et al., 2006). Studies have shown the presence of T-regs can be critical in preventing generalized multiorgan autoimmunity (Kohm et al., 2002; Reddy et al., 2004; Sakaguchi, 2005; Kim et al., 2007). In addition, MS patients have been reported to possess an impairment in T-reg suppressor function (Viglietta et al., 2004; Haas et al., 2005), thus contributing to the development of the disease.
The aryl hydrocarbon receptor (AhR) has been shown recently to be involved in cellular development and differentiation. Activation of AhR can regulate the development of the nervous system, metabolism and stress responses to hypoxia (Esser et al., 2009). AhR has also been shown to be expressed in many immune cells including lymphocytes (T and B cells), macrophages, dendritic cells (DCs), granulocytes and natural killer cells (Lawrence and Kerkvliet, 2007). Based on studies involving various AhR ligands, AhR has been postulated as an important regulator by controlling the balance between T-regs and pathogenic Th17 cells (Kimura et al., 2008; Quintana et al., 2008; Veldhoen et al., 2008).
Dietary indoles, such as 3,3′-diindolylmethane (DIM) and its precursor, indole-3-carbinol (I3C), found in high amounts in cruciferous vegetables, have been demonstrated to exert beneficial effects against cancer. Epidemiological studies have reported that people with high dietary intake of cruciferous vegetables have a lower risk of developing cancer (Verhoeven et al., 1996). Similarly, a metabolite of tryptophan known as indirubin, an active ingredient of the Chinese medicine Danggui Longhui Wan, has been demonstrated to exert therapeutic effects against breast cancer and chronic myelocytic leukemia (Xiao et al., 2002; Nicolaou et al., 2008). Both I3C and DIM, as well as indirubin, have also been reported to interact with AhR based on their chemical structure (Adachi et al., 2001; Stresser et al., 1995; Renwick et al., 1999) and abilities to induce CYP1A1 and CYP1A2 (De Kruif et al., 1991; Shertzer and Senft, 2000; Adachi et al., 2001; Guengerich et al., 2004; Nishiumi et al., 2012). While the anti-cancer effects of these indole derivatives have been extensively studied, their ability to modulate the immune cells and their response during disease in an AhR-dependent manner has not been well established.
To investigate the potential therapeutic role of dietary AhR ligands, we tested the efficacy of indirubin, I3C and DIM in the treatment of EAE. We hypothesized that these compounds may activate AhR and thereby promote the differentiation of CD4+ T cell subsets into T-regs while suppressing Th17. In the current study, we demonstrate that pretreatment with I3C or DIM prevents development of EAE. Moreover, I3C and DIM were also effective in reducing the clinical symptoms of EAE when administered after disease onset. The mechanism involves promoting the generation of T-regs, while simultaneously down-regulating the differentiation of Th17 cells following activating AhR.
Material and methods
Mice
Female C57BL/6 mice (6–8 weeks old) were purchased from the National Cancer Institute.
Ethics statement
All animals were housed at the AAALAC-accredited University of South Carolina, School of Medicine (Columbia, SC). All animal procedures were performed according to NIH guidelines under protocols approved by the Institutional Animal Care and Use Committee of the University of South Carolina. All studies involving animals are in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Reagents and monoclonal antibodies (mAbs)
The reagents used in this study were purchased as described below: Indirubin-3′-oxime (indirubin), I3C and DIM, AhR antagonist CH223191 (CH), concanavalin A (ConA), RBC lysis buffer, and corn oil, from Sigma-Aldrich (St. Louis, MO, USA); Percoll from GE Healthcare Life Sciences (Pittsburgh, PA, USA); RPMI 1640, L-glutamine, HEPES, PBS and FBS, from VWR (West Chester, PA, USA); Myelin oligodendrocyte glycoprotein (MOG35–55) peptide, H-MEVGWYRSPFSRVVHLYRNGK-OH, from PolyPeptide Laboratories San Diego (San Diego, CA, USA); TUNEL kits from Roche (Indianapolis, IN, USA); RNeasy Mini kit from QIAGEN (Valencia, CA, USA); MasterAmp PCR premix F from Epicentre (Madison, WI, USA); iScript cDNA synthesis kit and iTaq DNA Polymerase from Bio-Rad (Hercules, CA, USA).
The following mAbs were purchased from Biolegend (San Diego, CA, USA): Fluorescein isothiocyanate (FITC)-conjugated anti-mouse CD4 (L3T4) mAb (GK1.5; rat IgG2b); Phycoerythrin (PE) conjugated anti-mouse CD3e (145-2C11; hamster IgG); Allophycocyanin (APC) anti-mouse CD8 (Ly-2) (53-6.7; rat IgG2a), Pychoerthrin-Cy5.5 (PE-Cy5.5)-conjugated anti-mouse CD45.2 (104; mouse IgG2a) and affinity purified anti-mouse CD16/32 (93; rat IgGa2a). The following mAbs were purchased from eBioscience (San Diego, CA, USA): PE-conjugated anti-mouse IL-17A (eBio17B7; rat IgG2a) and PE-conjugated anti-mouse/rat FoxP3 (FJK-16 s; IgG2a).
In addition, FoxP3 staining kit was purchased from Biologend. For intracellular cytokine staining, BD Cytofix/CytoPerm Fixation/Permeabilization Solution Kit was purchased from BD Biosciences (San Jose, CA, USA). All ELISA kits used in cytokine analysis were purchased from Biolegend and used according to the manufacturer's manual.
Effects of I3C and DIM on EAE in mice
EAE was induced in female C57BL/6 mice (6–8 weeks old) as described previously (Singh et al., 2007; Zhou et al., 2010; Guan et al., 2011). Briefly, via subcutaneous immunization with 100 μL of 150 μg myelin oligodendrocyte glycoprotein (MOG35–55) peptide emulsified in complete Freund's adjuvant (Difco, Detroit, MI, USA) containing 4 mg·mL−1 killed Mycobacterium tuberculosis (strain H37Ra; Difco). Following immunization, 200 ng of pertussis toxin (List Labs) was injected i.p. into mice on Day 0, followed by a 400 ng pertussis toxin i.p. injection on Day 2. For pretreatment studies, mice were administered 50 mg·kg−1 indirubin, 20 mg·kg−1 I3C or DIM or vehicle (2% DMSO + corn oil). Injections were performed i.p. one day prior to the induction of EAE and continued every other day throughout the remainder of the study. For post-treatment studies, EAE was induced in naïve mice and 10 days later, these mice were completely randomized into groups that received treatment with vehicle, I3C (40 mg·kg−1) or DIM (40 mg·kg−1). This treatment was continued every day throughout the remainder of the study. Clinical scores (0, no symptoms; 1, limp tail; 2, partial paralysis of hind limbs; 3, complete paralysis of hind limbs or partial hind and front limb paralysis; 4, tetraparalysis; 5, moribund; 6, death) were recorded on a daily basis. The mean score was calculated for each group every day.
Histological analysis of cell infiltration
Spinal cords from naive, EAE + vehicle, EAE + I3C or EAE + DIM mice were collected 15 days after immunization. Spinal cord tissue was fixed with 10% formalin and paraffin blocks were prepared. Microtome sections (10 μm) were generated, and tissue sections were stained with haematoxylin and eosin (H&E).
Isolation of immune cells and flow cytometry
EAE-induced mice were given vehicle, I3C or DIM as indicated earlier. On day 15, blood was collected and serum was isolated for cytokine/chemokine analysis. Spleen and inguinal lymph nodes were excised prior to perfusion. Mice were then perfused with 10 mL heparinized PBS, and whole brain tissue was isolated. Tissues were homogenized separately into a single-cell suspension and subjected to red blood cell lysis. Mononuclear cells from whole brain homogenates were isolated using 33% Percoll. Cells were counted and stained with fluorescently tagged mAbs as indicated. Total cell number was calculated using the following equation: Total cells bearing a specific maker = Percentage of cells with the marker as analyzed by flow cytometry x absolute number of cells/100.
Cell culture
Cultures of T cells were maintained in complete RPMI 1640 media supplemented with 10% heat inactivated FBS, 10 mM L-glutamine, 10 mM HEPES, 50 μM β- mercaptoethanol and 100 μg·mL−1 penicillin/streptomycin at 37°C and 5% CO2.
Effects of I3C and DIM on MOG35–55 stimulated T cells from EAE mice
Splenocytes from naive, EAE + vehicle, EAE + I3C and EAE + DIM mice were isolated 15 days after immunization and cultured in a 24-well plate in the presence of 30 μg·mL−1 MOG35–55 for 3 days. Supernatants were collected for cytokine analysis. Prior to harvest, splenocytes were stimulated with phorbol myristate acetage (PMA) (50 ng·mL−1) and ionomyocin (1 μg·mL−1) and Golgi-plug for 4 h. Cells were harvested and processed for CD4 and IL-17 staining using BD Cytofix/CytoPerm Fixation/Permeabilization Solution Kit. Cells were analysed using a flow cytometer.
Determination of cytokines/chemokines
Serum was collected at day 15 from naïve, EAE + vehicle, EAE + I3C and EAE + DIM mice. Cell culture supernatants were collected from in vitro studies as described above. Cytokines/chemokines present in the serum or cell culture supernatants were determined using individual ELISA kits for TNF-α (Biolegend), IL-6 (Biolegend) and IL-17 (BD Bioscience). ELISAs were conducted according to the protocol of the companies (Biolegend).
Determination of regulatory T cell (T-reg) induction in vivo
Spleen and inguinal lymph node (LN) were isolated from vehicle, I3C (40 mg·kg−1, i.p.) or DIM (40 mg·kg−1, i.p.) injected mice after 4 days of daily treatment. Tissues were processed, and cells were stained for CD4 and FoxP3 using the Biolegend FoxP3 Fix/Perm kit.
Detection of Foxp3 expression by real time (RT)-PCR
Total RNA from spleen and inguinal LN were isolated from mice injected with vehicle, I3C (40 mg·kg−1, i.p.) or DIM (40 mg·kg−1, i.p.) after 4 days daily treatment using the RNeasy minikit. RNA quality was measured and quantified using a spectrophotometer. First-strand cDNA synthesis was performed using iScript according to the manufacturer's protocol in a 20-μL reaction mix containing 1 μg total RNA. After first-strand synthesis, 1 μL was used as a template for PCR amplification. To detect the expression of Foxp3, forward (5′-GGG GAA GCC ATG GCA ATA GTT-3′) and reverse (5′-TGA AGT AGG CGA ACA TGC GAG TAA-3′) primers specific to mouse Foxp3 were used. PCR was performed for 40 cycles using the following conditions: 30 s at 95°C (denaturing temperature), 40 s at 58.0°C (annealing temperature) and 60 s at 72°C (extension temperature), with a final incubation at 72°C for 10 min. The FoxP3 PCR products were normalized against 18S PCR products, which were used as an internal control. The 18S PCR products were generated based on forward (5′-GCC CGA GCC GCC TGG ATA-3′) and reverse (5′-CCG GCG GGT CAT GGG AAT AAC-3′) primers. After electrophoresis on 1.5% agarose gel, PCR products were visualized using a Bio-Rad image analysis system (Bio-Rad Laboratories).
Determination of cytokine production ex vivo
Spleen and inguinal LN were isolated from mice treated with vehicle, I3C (40 mg·kg−1, i.p.) or DIM (40 mg·kg−1, i.p.) after 4 days daily treatment. Tissues were processed, and cells from each respective group were plated at 1 × 106 cells/well in a 96-well plate. Cells were cultured as indicated for the required duration. Cells were stimulated with PMA (50 ng·mL−1) and ionomyocin (1 μg·mL−1) for 4 h. Supernatants were collected for cytokine analysis by ELISA.
Determination of Th17 differentiation ex vivo
Splenocytes were isolated from mice injected with vehicle, I3C (40 mg·kg−1, i.p.) or DIM (40 mg·kg−1, i.p.) after 4 days daily treatment. Tissues were processed, and cells from each respective group were plated at 1 × 106 cells/well in a 96-well plate. Cells were cultured as indicated for the required duration. Cells were stimulated with PMA (50 ng·mL−1), calcium ionomyocin (1 μg·mL−1) and Golgi-Plug for 4 h. Cells were processed and stained for CD4 and IL-17 using the BD Cytofix/CytoPerm Fixation/Permeabilization Solution Kit.
Effect of indoles on T-reg/Th17 differentiation in vitro
Naïve splenocytes were processed and plated at 1 × 106 cells/well in a 96-well plate. Cells were activated with ConA (2.5 μg·mL−1) in the absence or presence of pertussis toxin (PTX) (5 μg·mL−1) for 24 h as indicated. AhR antagonist CH (50 μM) was added to cultures 2 h prior to I3C or DIM (100 μM) treatment. For Th17 staining, cells were stimulated with PMA (50 ng·mL−1), ionomyocin (1 μg·mL−1) and Golgi-Plug for 4 h prior to harvest.
Effect of indoles on cytokine production in vitro
Naïve splenocytes were processed and plated at 1 × 106 cells/well in a 96-well plate. Cells were activated with ConA (2.5 μg·mL−1) in the absence or presence of PTX (5 μg·mL−1) for 24 h, or plate-bound anti-CD3 in the presence of PTX (5 μg·mL−1) for 4 days. I3C or DIM (100 μM) were added simultaneously to cells cultured in the same conditions. Cells were stimulated with PMA (50 ng·mL−1) and ionomyocin (1 μg·mL−1) for 4 h. Supernatants were collected for cytokine analysis by ELISA.
Role of AhR in I3C- and DIM-mediated amelioration of EAE in mice
EAE was generated in mice using MOG35–55 peptide, as described for pretreatment studies above. We used CH, as an AhR antagonist. One day earlier, all the mice were pretreated with vehicle, I3C (20 mg·kg−1 body weight), DIM (20 mg·kg−1 body weight) or CH, the AhR-specific antagonist (5 mg·kg−1 body weight), or CH + I3C or CH + DIM. From day 2 onwards, the mice were treated with vehicle, I3C, DIM, CH, CH + I3C, CH + DIM on alternate days. The mice were monitored for EAE symptoms as described above. On day 16, mice were sacrificed and inguinal LN and brain were collected, single cell suspensions were prepared. Mononuclear cells from brain tissue of the various groups were isolated, and the cells from both LN and brain were stained for the presence of T-regs (CD4-FITC and FoxP3-PE) and Th17 cells (CD4-FITC and IL-17-PE) in LN and brain.
Statistics
For EAE clinical studies, we used groups of 10 mice. Clinical scores were evaluated using the Mann–Whitney test. All cytokine and in vitro assays were performed in triplicate. The means ± SEM are shown for experiments that are applicable. The statistical differences within experiments were calculated using anova, while differences between groups were analysed with the Bonferrroni post hoc test. A P-value of ≤0.05 was considered to be statistically significant. The data that are shown in this paper represent at least three independent experiments.
Results
I3C/DIM treatment mitigates the clinical symptoms of EAE development
First, we investigated the effects of indirubin, I3C and DIM administered before (pretreatment) or after (post-treatment) immunization with MOG, on the clinical course of EAE. For pretreatment studies, the indole derivatives were administered at a dose of 20 mg·kg−1 i.p., 1 day prior to the induction of EAE and continued every other day until Day 25 (Figure 1A). Clinical scores revealed that indirubin treatment only marginally delayed the onset of EAE. Remarkably, pretreatment with either I3C or DIM completely prevented disease development (Figure 1B). Histological data taken from the spinal cord at the peak of the disease, Day 15, correlated with the observed clinical scores. Spinal cords taken from vehicle-treated EAE mice displayed cellular infiltration, particularly within the myelinated regions. The accumulation of these inflammatory cells was accompanied by immense tissue damage (Figure 1C). EAE mice pretreated with indirubin also showed dramatic levels of infiltration and altered tissue morphology compared to naïve histology (data not shown). Pretreatment with I3C or DIM markedly reduced inflammatory cells in the spinal cord. Furthermore, I3C or DIM pretreatment appeared to preserve the normal architecture of the myelinated regions within the spinal cord, resembling controls (Figure 1C).
Figure 1.

Effect of treatment with I3C or DIM on the development of EAE in C57BL/6 mice. (A) Timeline schematic of studies involving EAE mice pretreated with vehicle, indirubin, I3C or DIM. Treatment was administered at time points indicated in red arrows. (B) Clinical scores (n = 10 per group) for EAE mice pretreated with vehicle, indirubin, I3C or DIM. (****, P < 0.0001; EAE + I3C or EAE + DIM vs. EAE + vehicle, Mann–Whitney test) (C) Representative images of spinal cord histopathology stained with H&E (10×) at Day 15 for pretreatment studies. Arrows indicate regions of cellular infiltration. Architecture is noted as white matter (WM) and grey matter (GM). (D) Timeline schematic of studies involving EAE mice post-treated with vehicle, I3C or DIM. Treatment was administered at time points indicated in red arrows. (E) Clinical scores (n = 10 per group) for EAE mice post-treated with vehicle, I3C or DIM. (**, P < 0.01; EAE + I3C or EAE + DIM vs. EAE + vehicle, Mann–Whitney test) (F) Representative images of spinal cord histopathology stained with H&E (10×) at Day 15 for post-treatment studies. Architecture is noted as white matter (WM) and grey matter (GM).
Based on the marked effectiveness of I3C and DIM pretreatment, we continued our investigation with only these two indole derivatives. Next, we decided to delay the administration of treatment until 10 days after EAE induction (post-treatment) (Figure 1D), which is a clinically relevant model for treating patients with MS. Because there are no biomarkers of susceptibility to MS, we felt that post-treatment would provide significant translational impact in treating patients diagnosed with the disease. Preliminary experiments using a post-treatment dose of 20 mg·kg−1 I3C or DIM (i.p., daily) demonstrated mild protection against EAE development (data not shown). Interestingly, when the dose was increased to 40 mg·kg−1 (i.p., daily), clinical symptoms were not only delayed, but the overall severity was also substantially diminished in both I3C and DIM post-treated mice compared to vehicle-treated EAE mice (Figure 1E). On Day 15, spinal cord histopathology once again confirmed that vehicle-treated EAE mice possessed marked inflammation and tissue damage. Remarkably, post-treatment with I3C or DIM limited the number of infiltrating cells and maintained spinal cord tissue integrity (Figure 1F).
Treatment of I3C or DIM decreases pro-inflammatory cytokine levels in serum of EAE mice
Serum taken from EAE mice at the peak of disease displayed elevated levels of TNFα, IL-17 and IL-6 compared to naïve mice, confirming the existence of a disease promoting inflammatory environment (Figure 2, Supporting Information Figure S1). In contrast, pretreatment of EAE mice with I3C or DIM caused dramatic decrease in the levels of these inflammatory cytokines (Figure 2A, B, Supporting Information Figure S1). In the post-treatment groups, we did not observe any changes in TNFα levels (Figure 2C) while only I3C but not DIM caused a drop in IL-17 (Figure 2D). Overall, these data correlated with the clinical scores and possibly explained why pretreatment was more effective in preventing EAE than post-treatment. We also found that I3C and DIM treatment led to increased production of IL-10 compared to vehicle treatment (data not shown).
Figure 2.
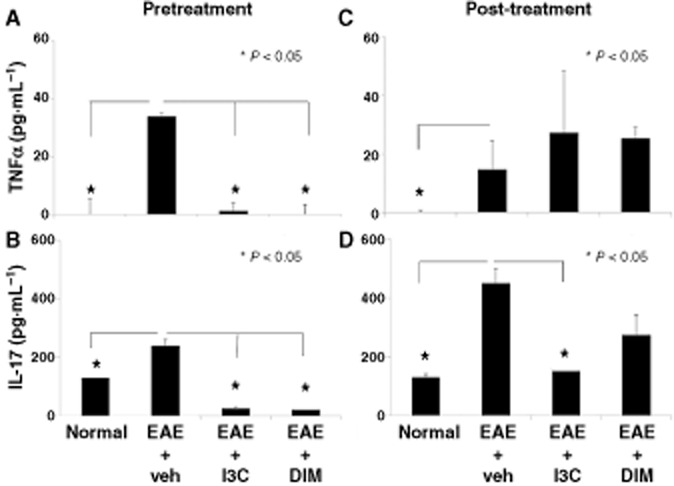
Treatment with I3C or DIM decreases pro-inflammatory cytokines in serum of EAE mice. Serum was collected on Day 15 from normal mice or EAE mice pre- or post-treated with either vehicle, I3C or DIM. Pro-inflammatory cytokines TNFα and IL-17, respectively, were analysed by ELISA for both pretreatment (A, B) and post-treatment (C, D) studies. Data presented as mean ± SEM of three pooled samples (n = 3 per sample). The statistical differences within experiments were calculated using anova, while differences between groups were analysed with the Bonferrroni post hoc test. In all panels, there were statistically significant differences between normal versus EAE + vehicle groups as indicated by an asterisk. Also, significant differences between EAE + vehicle versus EAE+I3C or EAE+DIM were indicated with an asterisk.
Post-treatment with I3C or DIM reduces T cell infiltration within brain tissue of EAE mice
Because there are no biomarkers that predict the susceptibility to MS, and to further address the clinical relevance of I3C's and DIM's therapeutic potential in patients diagnosed with MS, we decided to focus our studies on this post-treatment model to evaluate the mechanisms (Figure 1D). Mononuclear cells were isolated on Day 15 from brain tissue of naïve or EAE mice treated with vehicle, I3C or DIM. Cells were processed and assessed for their profile of various T cell subsets. Vehicle-treated EAE mice showed a significant spike in the percentage and total number of CD3+ T cells when compared to naïve mice, thereby indicating infiltration of CNS with T cells (Figure 3A, C). While both CD4+ and CD8+ T cell subsets increased in vehicle-treated EAE mice compared to naïve mice, the majority of the infiltrating cells were CD4+. Post-treatment with I3C and DIM resulted in moderate reduction in the proportion CD3+ T cells, particularly in CD4+ and not CD8+ (Figure 3A, B). This suggests that ratio of infiltrating cellular populations after the isolation of mononuclear cells from diseased mice remain the same even with treatment. However, when total cell numbers were enumerated, I3C and DIM caused dramatic decreases in both CD3+ and CD4+ T cells, similar to levels seen in naïve animals, indicating these dietary indoles may be eliciting their protective effects in the periphery by limiting immune cell trafficking to the CNS (Figure 3C).
Figure 3.

Post-treatment with indoles reduces T cell infiltration within brain tissue of EAE mice. Mononuclear cells were isolated from brain tissue on Day 15 from EAE mice given vehicle, I3C or DIM. The cells were stained with various mAbs, analysed using flow cytometry and representative histograms depicted. (A) Single stained CD3 T cells; (B) double-stained for CD4 and CD8. (C) By calculating the percentage of T cell subsets determined by Flow cytometer multiplied by absolute number of cells found in the brain and divided by 100, gave the total cell number for CD3+, CD4+ and CD8+ T cells, which were then plotted (**P < 0.01; normal vs. EAE+vehicle; EAE+I3C or EAE+DIM vs. EAE+vehicle). Data presented as mean ± SEM of three pooled samples (n = 3 per sample).
Post-treatment with I3C and DIM leads to induction of regulatory T cells, while reciprocally reducing Th17 populations
At the peak of the disease, EAE + vehicle treated mice, in their inguinal LN which was the draining LN from the site of MOG immunization, showed similar proportions of T-regs but increased percentage of Th17 cells when compared to naïve mice. Interestingly, post-treatment of EAE mice with I3C or DIM resulted in an increased proportion of CD4+/FoxP3+ T-regs and decreased percentage of Th17 cells, compared to vehicle-treated EAE mice (Figure 4A, B). Furthermore, when we enumerated the total number of T-regs and Th17 cells in the inguinal LN of EAE mice, we found that DIM but not I3C caused a significant increase in T-regs, while both I3C and DIM caused a significant decrease in Th17 cells (Figure 4C). Thus, considering both the percentage and total cell numbers, the overall trend was that the indoles caused an increase in T-regs while decreasing the Th17 cells.
Figure 4.

Post-treatment with indoles leads to T-reg induction, while reciprocally reducing Th17 cells, in vivo. Cells were isolated on Day 15 from inguinal lymph nodes of EAE mice post-treated with vehicle, I3C or DIM. Representative flow cytometry plots show cells gated on CD4 to examine (A) CD4+/FoxP3+ T-regs or (B) CD4+/IL-17+ Th17 populations. Data are representative of three independent experiments. Panel C shows total number of T-regs and Th17 cells calculated as described in Figure 3C. (*P < 0.005; EAE + vehicle vs. normal, I3C or DIM; #P < 0.05 normal vs. EAE + I3C; ##P < 0.005; normal vs. EAE + vehicle, EAE + I3C or EAE + DIM).
To further corroborate the impact of I3C and DIM on the effector Th17 cell differentiation, we cultured splenocytes from naïve and EAE mice treated with vehicle, I3C or DIM, in vitro in the presence of MOG35–55 peptide for 3 days. Such cultures from EAE mice displayed elevated proportion and numbers of Th17 cells compared to naïve, suggesting the generation and expansion of Th17 cells in response to MOG peptide (Figure 5A, B). However, cells from I3C or DIM treated EAE mice exhibited a significant reduction in the proportion and numbers of Th17 cells.
Figure 5.

Post-treatment with indoles decreases MOG35–55 activated Th17 cell generation ex vivo. Splenocytes were isolated on Day 15 from normal or EAE mice post-treated with vehicle, I3C or DIM. Cells were cultured in the presence of 30 ug·mL−1 MOG for 3 days and stained for CD4 and IL-17. (A) Representative flow cytometric plots show cells gated on CD4 to examine CD4+/IL-17+ Th17 populations. (B) Based on the proportions of Th17+ cells and absolute number of cells isolated from the cultures, total cell number of Th17 cells were calculated and plotted (*P < 0.05; normal, EAE + I3C or EAE + DIM vs. EAE + vehicle). Data presented as mean ± SEM (n = 3 per sample).
Administration of I3C or DIM promotes the generation of T-regs in naïve mice
While we studied the effect of indoles on inducible T-regs to MOG-immunization, we next determined if I3C or DIM could modulate natural T-regs found in naïve animals. To this end, we administered I3C or DIM to naïve mice at 40 mg·kg−1. After 4 consecutive days of treatment, spleen and inguinal LN were isolated to investigate the induction of T-regs. Flow cytometric analysis revealed that mice exposed to indole treatment demonstrated an increased proportion of CD4+ T cells that co-expressed FoxP3 in both spleen and inguinal LN compared to untreated naïve mice (Figure 6A). When we enumerated the total numbers of T-regs, DIM but not I3C caused a significant increase (Figure 6B). To confirm if the T cells expressed increased levels of FoxP3, we performed RT-PCR and found that naïve mice treated with I3C or DIM showed increased FoxP3 mRNA expression in spleen and inguinal LN compared to untreated mice (Figure 6C). Together, these data suggest that naïve mice exposed to indoles do exhibit an increased proportion of T-regs and higher expression of FoxP3, which may explain, at least in part, why pretreatment with indoles protects mice from developing EAE (Figure 1).
Figure 6.

Indole treatment leads to generation of T-regs in vivo. Spleen and inguinal LN were isolated from normal mice treated with vehicle, I3C (40 mg·kg−1) or DIM (40 mg·kg−1) daily for 4 days. (A) CD4+ gated T cells were examined for CD4+/FoxP3+ T-regs using flow cytometry. (B) Based on the proportions of T-regs and absolute number of cells isolated from these organs, total cell number of CD4+/FoxP3+ T-regs were calculated and plotted (*P < 0.05 I3C or DIM vs. naïve). Data presented as mean ± SEM (n = 3 per sample). (C) Expression of FoxP3 in spleen and inguinal LN was measured by RT-PCR, using 18S as an internal control. Splenocytes were isolated from naïve, I3C (40 mg·kg−1) or DIM (40 mg·kg−1) after 4 days daily treatment.
Exposure to I3C or DIM suppresses Th17 differentiation
We noted that pretreatment with indoles completely suppressed EAE development (Figure 1). We therefore determined if exposure to indoles in naïve animals inhibits Th17 differentiation upon T cell activation. To this end, we cultured splenocytes from vehicle, I3C or DIM treated naïve mice with ConA (2.5 μg·mL−1) in the absence or presence of PTX (5 μg·mL−1) for 24 h. We used PTX because it helps promote Th17 cell differentiation as previously shown by us (Singh et al., 2011). As expected, vehicle treated splenocytes showed a significant proportion of Th17 cells after ConA-activation, which was further enhanced in the presence of PTX. Splenocytes that had been exposed to I3C or DIM maintained a low proportion of Th17 cells even after ConA+PTX stimulation (Figure 7A). We also tested the ability of splenocytes from vehicle or indole treated mice cultured with ConA with or without PTX (Figure 7B) or with anti-CD3 with or without PTX (Figure 7C) to produce IL-17. In most culture conditions, splenocytes from indole-treated mice produced significantly less amounts of IL-17 (Figure 7B, C). We were also curious to see if I3C or DIM could directly impact Th17 differentiation when these indoles are added directly to culture. In these studies, we cultured naïve splenocytes with ConA + PTX for 24 h (Figure 7D) or anti-CD3+PTX for 4 days (Figure 7E) to promote Th17 differentiation. At the same time, cells were exposed in vitro to vehicle, I3C (100 μM) or DIM (100 μM). Vehicle treated cells expressed significant levels of IL-17 in culture supernatants. Cultures treated with I3C showed a substantial decrease in IL-17 production, while DIM suppressed IL-17 levels to non-detectable levels (Figure 7D, E). Together, these studies demonstrated that pretreatment with indoles reduced Th17 differentiation and IL-17 production even when T cells are favoured to differentiate into Th17 cells.
Figure 7.

Indole treatment in naïve mice leads to decreased Th17 generation upon activation ex vivo. (A) Splenocytes were isolated from naïve mice treated with vehicle, I3C (40 mg·kg−1) or DIM (40 mg·kg−1) daily for 4 days. Cells were activated with ConA (2.5 μg·mL−1) in the absence or presence of PTX (5 μg·mL−1) for 24 h. Representative plots for CD4+ gated cells examined for IL-17 expression using flow cytometry. (B) Splenocytes activated with ConA (2.5 μg·mL−1) in the absence or presence of PTX (5 μg·mL−1) for 24 h or (C) plate-bound anti-CD3 in the absence or presence of PTX (5 μg·mL−1) for 4 days. The supernatants were analysed by ELISA for IL-17(*P < 0.05, **P < 0.01 I3C or DIM vs. vehicle or PTX as indicated). (D) Indole treatment reduces IL-17 production under Th17 polarizing conditions in vitro. Naïve splenocytes were cultured under Th17 polarizing conditions such as with ConA (2.5 μg·mL−1) and PTX (5 μg·mL−1) in the absence or presence of I3C (100 μM) or DIM (100 μM) for 24 h (*P < 0.05 vs. PTX) or (E) cells were cultured with plate-bound anti-CD3 and PTX (5 μg·mL−1) in the absence or presence of I3C (100 μM) or DIM (100 μM) for 4 days (*P < 0.05 vs. PTX). Supernatants were analysed for IL-17 by ELISA. Data are representative of three independent experiments.
Role of AhR in I3C- and DIM-mediated differentiation of T-regs and Th17cells in vitro
To investigate if I3C and DIM mediated their effects on T-reg cell differentiation through AhR in vitro, we used a selective AhR antagonist, CH. To this end, naïve splenocytes were activated with ConA (2.5 μg·mL−1) in the absence or presence of I3C (100 μM) or DIM (100 μM) for 24 h in the absence or presence of AhR antagonist CH (50 μM). The data indicated that CH reversed the ability of I3C and DIM to promote the differentiation of FoxP3+ T-regs (Figure 8A). Similar experiments performed using Th17 polarizing conditions revealed inconclusive results because addition of CH to vehicle control itself caused marked inhibition of Th17 differentiation (from 23.1% to 6.6%), suggesting possible involvement of endogenous ligands for AhR in Th17 differentiation (Figure 8B). These data suggested that indoles may indirectly suppress Th17 differentiation by inducing T-regs which in turn suppress Th17 differentiation.
Figure 8.

Role of AhR in I3C- and DIM-mediated differentiation of T-regs and Th17cells in vitro. Naïve splenocytes were activated with ConA (2.5 μg·mL−1) in the absence and presence of I3C (100 μM) or DIM (100 μM) for 24 h. AhR antagonist CH223191 (CH)(50 μM) was added 2 h prior to the addition of indoles. Cells were gated on CD4 and analysed by flow cytometry for CD4+/FoxP3+ or CD4+/IL-17+.
Role of AhR in I3C- and DIM-mediated amelioration of EAE in mice and differentiation of T-regs and Th17cells in lymph nodes and brain
To investigate if I3C- or DIM-mediated amelioration of EAE in mice resulted from AhR activation, we used a novel AhR antagonist (CH) in vivo and tested if this would reverse the protection afforded by the indoles as well as reverse the induction of T-regs and decreased proportion of Th17 cells. To this end, EAE was induced in groups of 10 mice using MOG35–55 peptide as described in Figure 1A. One day earlier, all the mice were pretreated with vehicle, I3C (20 mg·kg−1 body weight), DIM (20 mg·kg−1 body weight) or CH, the AhR-specific antagonist (5 mg·kg−1 body weight), or CH + I3C or CH + DIM. From day 2 onwards, the mice were treated with vehicle, I3C, DIM, CH, CH + I3C, CH + DIM on alternate days. The mice were monitored for EAE symptoms. As shown in Figure 9A, both I3C and DIM profoundly suppressed EAE symptoms, when compared to mice that received vehicle treatment. However, treatment of these mice with CH (CH + I3C or CH + DIM) reversed the protective effects of I3C and DIM on EAE and such mice showed heightened clinical disease. These data suggested that I3C and DIM were mediating the protective effect against EAE through AhR activation.
Figure 9.
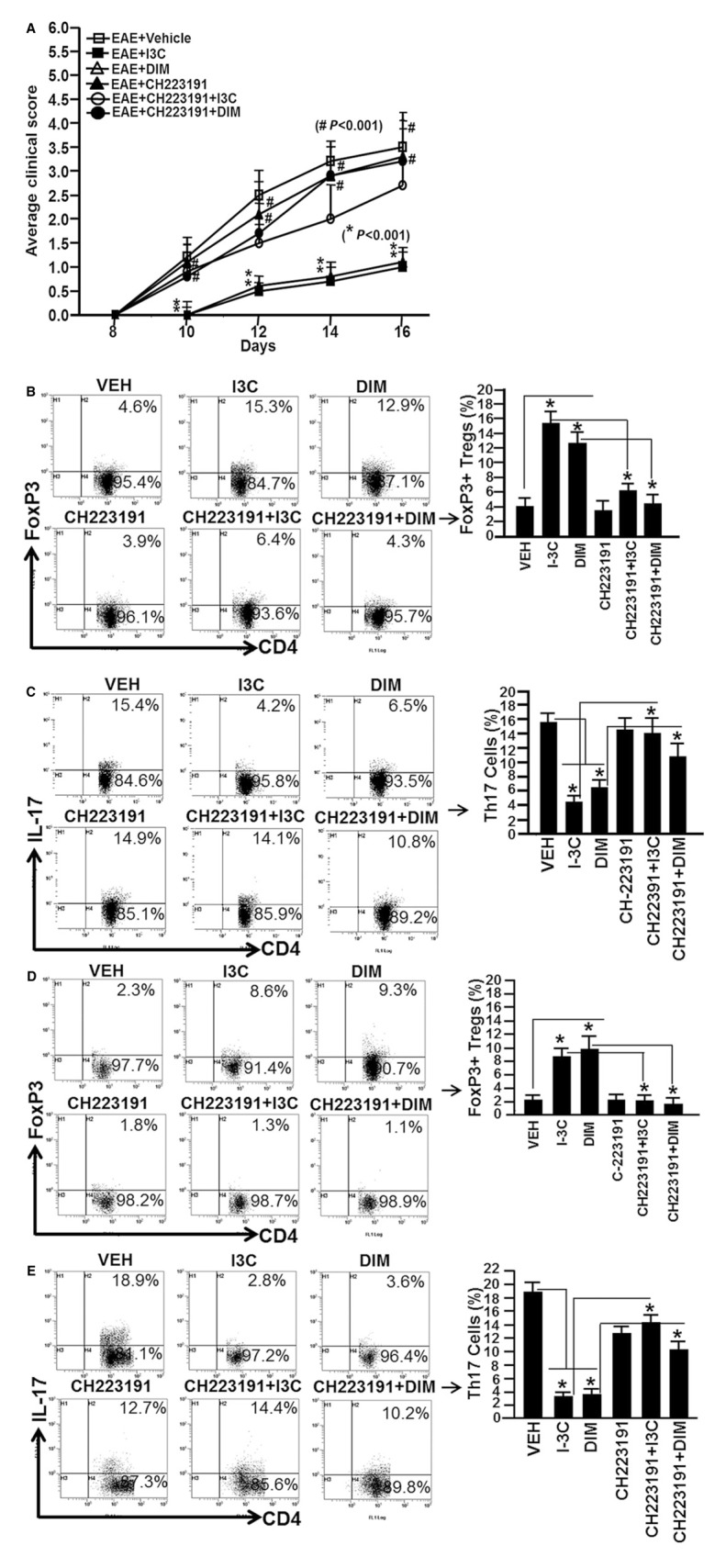
Role of AhR in I3C- or DIM-mediated amelioration of EAE in mice. (A) Clinical scores (n = 10 mice per group) for EAE mice pretreated with vehicle or I3C, or DIM or CH223191 or CH223191 + I3C or CH223191 + DIM. Symbols (#) represent significant (P < 0.001) reversal of EAE symptoms in mice with EAE that received CH223191 prior to I3C (CH223191 + I3C) or DIM (CH223191 + DIM) treatment, when compared to mice with EAE that received either I3C or DIM treatment. Symbol (*) indicates P < 0.001 when EAE + I3C or EAE + DIM were compared to EAE + vehicle. (B, C). In vivo generation of T-regs (CD4+/FoxP3+; B) and Th17 (CD4+/IL-17+; C) cells in inguinal LN in the presence of I3C or DIM (D-E). In vivo generation of T-regs (CD4+/FoxP3+; D) and Th17 (CD4+/IL-17+; E) cells in the brain of mice that received I3C or DIM treatments. The effect of I3C or DIM was significantly reversed (P < 0.05) in mice that received CH223191 treatment prior to I3C or DIM treatments. The data represent one of three independent experiments. Asterisk (*) represent significant (P < 0.05) effect of I3C or DIM on generation of T-regs and Th17 cells, when compared to vehicle in vivo.
On day 16, we examined the generation of T-regs and Th17 cells in the draining inguinal LN and brains of mice with EAE and treated with various regimens as described above. Upon analysis of T-regs in both LN and brain, we observed significant induction of T-regs (CD4+/FoxP3+) in mice that received either I3C or DIM treatment, when compared to vehicle treated group (Figure 9B, D). However, administration of CH reversed the effect of both I3C and DIM (Figure 9B, D). Upon examination of Th17 (CD4+/IL-17+) cells in LN and brain, we noted significantly less induction of Th17 cells in both LN and brain of mice with EAE that received either I3C or DIM treatment (Figure 9C, E). However, in the presence of CH, the effect of I3C and DIM was reversed as there was significantly higher induction of Th17 cells in both LN and brain (Figure 9C, E). These data together demonstrated that I3C and DIM-mediated effects on T-reg and Th17 generation were triggered through AhR.
Discussion
AhR was first discovered and well characterized as a transcription factor responsible for the activation of genes encoding a number of xenobiotic metabolizing enzymes and mediate the toxicity of chemicals such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Interestingly, recent studies indicated that AhR activation plays diverse roles in cellular functions, including the regulation of the immune system (Singh et al., 2007; 2011; 2008). Recent studies suggested that AhR activation may play a critical role in the regulation of T cell differentiation, more specifically the differentiation of T-regs and Th17 cells (Kimura et al., 2008; Quintana et al., 2008; Veldhoen et al., 2008). One possible mechanism may be due to the fact that in addition to expressing AhR, the FoxP3 gene of T-regs contains dioxin response elements. Thus, upon ligand binding to AhR, these cells can become transcriptionally activated and generate factors that promote T-reg differentiation (Quintana et al., 2008). Recent studies from our laboratory have shown that TCDD can protect against the development of acute dextran sodium sulphate-induced acute colitis via epigenetic mechanisms (Singh et al., 2011). These actions were found to occur through AhR-dependent induction of T-regs, which inhibited Th17 differentiation and inflammation in these mice. Further analysis revealed that TCDD treatment decreased demethylation of CpG islands of Foxp3 and increased methylation of IL-17 promoters within mesenteric lymph node or lamina propria cells during colitis.
Interestingly, two recent studies published using TCDD and 6-formylindolo(3,2-b)carbazole (FICZ) provided contradictory results on the role of AhR in the regulation of T-regs and Th17 cells and consequently, the clinical outcome of EAE (Quintana et al., 2008; Veldhoen et al., 2008). In one study, activation of AhR by FICZ, promoted Th17 differentiation and accelerated the onset and increased pathology in wild-type mice, but not in AhR-deficient mice (Veldhoen et al., 2008). Thus, the authors concluded that AhR activation promotes Th17 development. In contrast, in the second study, using TCDD as an AhR ligand, AhR activation induced T-regs and thereby suppressed clinical EAE (Quintana et al., 2008). However, in this study, treatment with FICZ decreased T-reg development, boosted Th17 cell differentiation, and enhanced the clinical scores of EAE. Such studies suggest that AhR ligands may exhibit differences in their ability to activate AhR and promote T cell differentiation into Th17 or T-regs. However, the role of dietary AhR ligands in regulating the immune response and T cell differentiation is intriguing and not well studied. Thus, our studies on indoles on T cell differentiation and regulation of EAE are novel and highly significant.
In this study, we demonstrate for the first time how dietary AhR ligands I3C and DIM can alleviate the development of EAE through the reciprocal induction of T-regs and Th17 cells. In pharmacokinetic studies using animals, I3C and DIM have been found to possess a 24 h half-life, which can be extended up to greater than 48 h after 1 week of continuous administration (Stresser et al., 1995). In addition, I3C and DIM have been stated as being relatively non-toxic, and can be taken by individuals at dosage ranging from 200 to 800 mg daily, resulting in tissue concentrations over 1 mM (Ho et al., 1998). Considering this, our in vitro studies used significantly lesser concentrations. Moreover, based on the calculated human equivalent dose, the doses of dietary indoles given to mice during our studies fell within the tolerable range available as dietary supplements.
During our investigation, we found mice pretreated with I3C or DIM were resistant to developing EAE compare to vehicle control mice. Meanwhile, pretreatment with indirubin offered no therapeutic benefit against the disease progression. Moreover, when treatment was delayed 10 days after EAE induction, I3C and DIM were still able to reduce the severity and onset of clinical symptoms, which was confirmed with histopathology and flow cytomteric analysis of the CNS. Our studies suggested that pretreatment of I3C and DIM promotes the expansion of natural T-regs, while inhibiting the differentiation of Th17 cells upon the onset of disease, suggesting a possible mechanism for mediating protection. During post-treatment, however, Th17 cells have already been generated, and the enrichment of the T-reg population at 20 mg·kg−1 may not be as effective to counter the development of disease. As a result, a higher dose of I3C or DIM was needed to enhance the production of T-regs and their suppressive function. Further studies in naïve mice confirmed I3C or DIM alone were sufficient to induce T-regs in the absence of disease-mediated factors. These data further indicate the ability of I3C and DIM to generate T-regs and promote an immunosuppressive environment in order to prevent or alleviate disease symptoms.
Additionally, we wanted to confirm the central role of AhR in the protective effects I3C or DIM observed during EAE. To that end, we used AhR antagonist rather than AhR KO mice because these mice have intrinsic differences in susceptibility to EAE which could confound the results. We used AhR-specific antagonist (CH) and not compounds such as alpha-naphthoflavone or 3′-methoxy-4′-nitroflavone, because the latter have also been reported to act as AhR partial agonist (Santostefano et al., 1993; Zhao et al., 2010; Choi et al., 2012). CH, on the other hand, was identified as an AhR antagonist using a chemical library and past studies have shown that it is a potent, pure AhR antagonist, which does not act as an AhR agonist (Zhao et al., 2010). Also, CH has been shown to inhibit TCDD-mediated nuclear translocation and DNA binding of AhR, as well as TCDD-induced luciferase activity and activation of CYP1A1 (Kim et al., 2006).
In our studies, we demonstrated that I3C and DIM in vivo and in vitro promoted the induction of T-regs through interaction with the AhR, indicating that these dietary indoles can modulate immune cells and their responses by acting as AhR ligands. At this time, we have not yet evaluated whether I3C and DIM cause AhR activation and affect signalling in DCs, which has been shown to promote the differentiation of T-regs (Hauben et al., 2008; Quintana et al., 2010). In addition, when we tried to use the AhR antagonist CH to block indole-mediated suppression of Th17 differentiation in vivo, we were able to reverse the of effects I3C- and DIM-mediated decrease in Th17 cell differentiation. In vitro, we also noted that the antagonist itself was capable of inhibiting Th17 differentiation. These results also raise the possible role played by endogenous AhR ligands and the fact that AhR is needed for the generation of Th17 cells, which correlates with the previous findings (Quintana et al., 2008; Veldhoen et al., 2009). Furthermore, our results also indicate that indoles may indirectly suppress Th17 differentiation by inducing T-regs which in turn may inhibit Th17 cells. Altogether, these data suggest I3C and DIM appear to be more effective as a pretreatment regimen compared to post-treatment due to their pre-emptive ability to generate T-regs, which can suppress T cell differentiation into disease-promoting Th17cells. Meanwhile, I3C and DIM were observed to still able to promote T-regs when administered as a post-treatment. However, the therapeutic extent of post-treatment appeared to be limited due to length of time mice received treatment and the fact these dietary indoles may hinder de novo Th17 differentiation, but may not have been able to inhibit effector cells that developed prior to administration. An additional protective effect of indole-induced T-regs could be due to the production of IL-10, which has been shown to be influenced by AhR and plays an important role in regulating the development and progression of EAE (Apetoh et al., 2010; Gandhi et al., 2010; Wu et al., 2011).
In summary, our studies indicate for the first time that dietary supplementation of I3C or DIM may prevent inflammation and that such indoles can also be used to alleviate the clinical symptoms and destruction of the CNS that are typically associated with EAE development. The mechanism seems to primarily involve the induction of T-regs and suppression of Th17 cells via activation of AhR.
Glossary
- DC
dendritic cells
- DIM
3,3′-diindolylmethane
- EAE
experimental autoimmune encephalomyelitis
- I3C
indole-3-carbinol
- MS
multiple sclerosis
- Th17
T-helper type 17
- T-regs
regulatory T cells
Conflicts of interest
The authors declare no competing interests exist.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Treatment with I3C or DIM decreases IL-6 levels in serum of EAE mice. Serum was collected on Day 15 from normal mice or EAE mice pre-treated with vehicle, I3C, or DIM. Pro-inflammatory cytokine IL-6 was analyzed by ELISA for pretreatment studies. Data presented as mean ± SEM of three pooled samples (n = 3 per sample). The statistical differences within experiments were calculated using anova, while differences between groups were analysed with the Bonferroni post hoc test. (*P < 0.05; EAE + vehicle vs. normal, EAE + I3C or EAE + DIM).
References
- Adachi J, Mori Y, Matsui S, Takigami H, Fujino J, Kitagawa H, et al. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J Biol Chem. 2001;276:31475–31478. doi: 10.1074/jbc.C100238200. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Ghilardi N, Xie MH, De Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol. 2010;11:854–861. doi: 10.1038/ni.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Choi EY, Lee H, Dingle RW, Kim KB, Swanson HI. Development of novel CH223191-based antagonists of the aryl hydrocarbon receptor. Mol Pharmacol. 2012;81:3–11. doi: 10.1124/mol.111.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kruif CA, Marsman JW, Venekamp JC, Falke HE, Noordhoek J, Blaauboer BJ, et al. Structure elucidation of acid reaction products of indole-3-carbinol: detection in vivo and enzyme induction in vitro. Chem Biol Interact. 1991;80:303–315. doi: 10.1016/0009-2797(91)90090-t. [DOI] [PubMed] [Google Scholar]
- Esser C, Rannug A, Stockringer B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009;30:447–454. doi: 10.1016/j.it.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol. 2010;11:846–853. doi: 10.1038/ni.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H, Nagarkatti PS, Nagarkatti M. CD44 Reciprocally regulates the differentiation of encephalitogenic Th1/Th17 and Th2/regulatory T cells through epigenetic modulation involving DNA methylation of cytokine gene promoters, thereby controlling the development of experimental autoimmune encephalomyelitis. J Immunol. 2011;186:6955–6964. doi: 10.4049/jimmunol.1004043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich F, Martin MV, McCormick WA, Nguyen LP, Glover E, Bradfield CA. Aryl hydrocarbon receptor response to indigoids in vitro and in vivo. Arch Biochem Biophys. 2004;423:309–316. doi: 10.1016/j.abb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Haas J, Hug A, Viehöver A, Fritzsching B, Falk CS, Filser A, et al. Reduced suppressive effect of CD4+CD25 high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. 2005;35:3343–3352. doi: 10.1002/eji.200526065. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Hauben E, Gregori S, Draghici E, Migliavacca B, Olivieri S, Woisetschläger M, et al. Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell-mediated effects on regulatory T cells. Blood. 2008;112:1214–1222. doi: 10.1182/blood-2007-08-109843. [DOI] [PubMed] [Google Scholar]
- Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. 2008;125:161–169. doi: 10.1111/j.1365-2567.2008.02837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho GH, Luo XW, Ji CY, Foo SC, Ng EH. Urinary 2/16 alpha-hydroxyestrone ration: correlation with serum insulin-like growth factor binding protein-3 and a potential biomarker of breast cancer risk. Ann Acad Med Singapore. 1998;27:294–299. [PubMed] [Google Scholar]
- Imam SA, Guyton MK, Haque A, Vandenbark A, Tyor WR, Ray SK, et al. Increased calpain correlates with Th1 cytokine profile in PBMCs from MS patients. J Neuroimmunol. 2007;190:139–145. doi: 10.1016/j.jneuroim.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- Kim SH, Henry EC, Kim DK, Kim YH, Shin KJ, Han MS, et al. Novel compound 2-methyl-2H-pyrazole-3-carboxylic acid (2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) prevents 2,3,7,8-TCDD-induced toxicity by antagonizing the aryl hydrocarbon receptor. Mol Pharmacol. 2006;69:1871–1878. doi: 10.1124/mol.105.021832. [DOI] [PubMed] [Google Scholar]
- Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008;105:9721–9726. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence BP, Kerkvliet NI. Immune modulation by TCDD and related polyhalogenated aromatic hydrocarbons. Immunopharmacol Immunotoxicol. 2007;3:239–258. [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou KA, Liapis V, Evdokiou A, Constantinou C, Magiatis P, Skaltsounis AL, et al. Induction of discrete apoptotic pathways by bromo-substituted indirubin derivatives in invasive breast cancer cells. Biochem Biophys Res Commun. 2008;425:76–82. doi: 10.1016/j.bbrc.2012.07.053. [DOI] [PubMed] [Google Scholar]
- Nishiumi S, Yamamoto N, Kodoi R, Fukuda I, Yoshida K, Ashida H. Antagonistic and agonistic effects of indigoids on the transformation of an aryl hydrocarbon receptor. Arch Biochem Biophys. 2012;470:187–199. doi: 10.1016/j.abb.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T-reg and Th17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:20768–20773. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy J, Illes Z, Zhang X, Encinas J, Pyrdol J, Nicholson L, et al. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2004;101:15434–15439. doi: 10.1073/pnas.0404444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renwick AB, Mistry H, Barton PT, Mallet F, Price RJ, Beamand JA, et al. Effect of some indole derivatives on xenobiotic metabolism and xenobiotic-induced toxicity in cultured rat liver slices. Food Chem Toxicol. 1999;37:609–618. doi: 10.1016/s0278-6915(99)00026-5. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- Santostefano M, Merchant M, Arellano L, Morrison V, Denison MS, Safe S. alpha-Naphthoflavone-induced CYP1A1 gene expression and cytosolic aryl hydrocarbon receptor transformation. Mol Pharmacol. 1993;43:200–206. [PubMed] [Google Scholar]
- Shertzer HG, Senft AP. The micronutrient indole-4-carbinol: implications for disease and chemoprevention. Drug Metabol Drug Interact. 2000;17:159–188. doi: 10.1515/dmdi.2000.17.1-4.159. [DOI] [PubMed] [Google Scholar]
- Singh NP, Hegde VL, Hofseth LJ, Nagarkatti M, Nagarkatti P. Resveratrol (trans-3,5,4′-trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol Pharmacol. 2007;72:1508–1521. doi: 10.1124/mol.107.038984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NP, Nagarkatti M, Nagarkatti P. Primary peripheral T cells become susceptible to 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated apoptosis in vitro upon activation and in the presence of dendritic cells. Mol Pharmacol. 2008;73:1722–1735. doi: 10.1124/mol.107.043406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NP, Singh UP, Singh B, Price RL, Nagarkatti M, Nagarkatti P. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS ONE. 2011;6:e23522. doi: 10.1371/journal.pone.0023522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stresser DM, Williams DE, Griffin DA, Bailey GS. Mechanisms of tumor modulation by indole-3-carbinol. Disposition and excretion in male Fischer 344 rats. Drug Metab Dispos. 1995;23:965–975. [PubMed] [Google Scholar]
- Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1810–1819. doi: 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Christensen J, O'Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–49. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven DTH, Goldbolm RA, van Poppel G, Verhagen H, van den Brandt PA. Epidemiological studies on brassica vegetables and cancer risk. Cancer Epidemiol Biomarkers Prev. 1996;5:733–748. [PubMed] [Google Scholar]
- Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Quintana FJ, da Cunha AP, Dake BT, Koeglsperger T, Starossom SC, et al. In vivo induction of Tr1 cells via mucosal dendritic cells and AhR signaling. PLoS ONE. 2011;6:e23618. doi: 10.1371/journal.pone.0023618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Hao Y, Liu B, Qian Y. Indirubin and meisoindigo in the treatment of chronic myelogeneous leukemia in China. Leuk Lymphoma. 2002;43:1763–1768. doi: 10.1080/1042819021000006295. [DOI] [PubMed] [Google Scholar]
- Zhao B, Degroot DE, Hayashi A, He G, Denison MS. CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol Sci. 2010;117:393–403. doi: 10.1093/toxsci/kfq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Nagarkatti P, Zhong Y, Nagarkatti M. Immune modulation by chondroitin sulfate and its degraded disaccharide product in the development of an experimental model of multiple sclerosis. J Neuroimmunol. 2010;223:55–64. doi: 10.1016/j.jneuroim.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.