

Abstract
Background and Purpose
Normal pregnancy is associated with decreased vascular resistance and increased release of vasodilators. Endothelin-1 (ET-1) causes vasoconstriction via endothelin receptor type A (ETAR), but could activate ETBR in the endothelium and release vasodilator substances. However, the roles of ETBR in the regulation of vascular function during pregnancy and the vascular mediators involved are unclear.
Experimental Approach
Pressurized mesenteric microvessels from pregnant and virgin Sprague–Dawley rats were loaded with fura-2/AM for simultaneous measurement of diameter and [Ca2+]i.
Key Results
High KCl (51 mM) and phenylephrine (PHE) caused increases in vasoconstriction and [Ca2+]i that were similar in pregnant and virgin rats. ET-1 caused vasoconstriction that was less in pregnant than virgin rats, with small increases in [Ca2+]i. Pretreatment with the ETBR antagonist BQ-788 caused greater enhancement of ET-1-induced vasoconstriction in pregnant rats. ACh caused endothelium-dependent relaxation and decreased [Ca2+]i, and was more potent in pregnant than in virgin rats. ET-1 + ETAR antagonist BQ-123, and the ETBR agonists sarafotoxin 6c (S6c) and IRL-1620 caused greater vasodilation in pregnant than in virgin rats with no changes in [Ca2+]i, suggesting up-regulated ETBR-mediated relaxation pathways. ACh-, S6c- and IRL-1620-induced relaxation was reduced by the NO synthase inhibitor Nω-nitro-l-arginine methyl ester, and abolished by tetraethylammonium or endothelium removal. Western blots revealed greater amount of ETBR in intact microvessels of pregnant than virgin rats, but reduced levels in endothelium-denuded microvessels, supporting a role of endothelial ETBR.
Conclusions and Implications
The enhanced ETBR-mediated microvascular relaxation may contribute to the decreased vasoconstriction and vascular resistance during pregnancy.
Keywords: endothelin, endothelium, pregnancy, endothelin type B receptor, vascular relaxation, endothelium-derived hyperpolarizing factor, nitric oxide, intracellular free Ca2+ concentration
Introduction
The maternal cardiovascular system undergoes marked adaptive changes during pregnancy in order to meet the metabolic demands of the growing fetus. During pregnancy, there is an increase in maternal blood volume and cardiac output, decreased systemic vascular resistance, and increased blood flow to the uteroplacental circulation (Poston et al., 1995; Thornburg et al., 2000). To accommodate for the increased blood volume during pregnancy, vascular tone is shifted towards vasodilation, and BP may remain unchanged or is slightly decreased (Thornburg et al., 2000). Studies have shown that the vasoconstriction responses to vasopressors are attenuated in some vascular beds during pregnancy (Crandall et al., 1990; Davidge and McLaughlin, 1992; Mazzuca et al., 2012). Furthermore, the responses to vasodilator stimuli are enhanced during pregnancy via both endothelium-dependent and endothelium-independent mechanisms (Davidge and McLaughlin, 1992; Gerber et al., 1998; Cooke and Davidge, 2003; Mazzuca et al., 2012). The endothelium is known to release endothelium-derived vasodilators such as NO, prostacyclin (PGI2) and endothelium-derived hyperpolarizing factor (EDHF) (Vanhoutte et al., 2009; Flammer and Luscher, 2010). These endothelium-derived vasodilators act on vascular smooth muscle (VSM) causing a decrease in VSM intracellular free Ca2+ concentration ([Ca2+]i) and vascular relaxation.
In addition to endothelium-derived vasodilators, the endothelium releases contracting factors such as thromboxane A2, angiotensin II (ANG II) and endothelin-1 (ET-1). ET-1 is a major vasoconstrictor and regulator of BP (Yanagisawa et al., 1988; Schiffrin, 2005). The vascular effects of ET-1 are mediated by at least two receptor subtypes: endothelin receptor type A (ETAR) and endothelin receptor type B (ETBR) (Arai et al., 1990; Sakurai et al., 1990). ETARs are mainly expressed in VSM to induce vasoconstriction (Davenport and Maguire, 2011). On the other hand, ETBRs are largely expressed in endothelial cells and mediate the release of endothelium-derived vasodilator substances such as NO, PGI2, and EDHF, and could also play a role in ET-1 clearance (Nakashima and Vanhoutte, 1993; Tirapelli et al., 2005). Although a role of ET-1 and ETAR in modulation of cardiovascular and renal function during normal and hypertensive pregnancy has been suggested (Conrad et al., 1999; Thaete and Neerhof, 2000; Alexander et al., 2001; Gandley et al., 2001; Roberts et al., 2006; George and Granger, 2011; Tam Tam et al., 2011; George et al., 2012), the role of ETBR in mediating the enhanced vasodilator response during pregnancy, the vascular mediators involved, and the underlying changes in vascular [Ca2+]i are unclear.
The present study was designed to test the hypothesis that ETBR-mediated signalling plays a role in the regulation of microvascular function during pregnancy. We used mesenteric microvessels from late pregnant and virgin rats to determine whether (i) normal pregnancy is associated with attenuated vasoconstriction, enhanced microvascular relaxation and parallel changes in underlying microvascular [Ca2+]i; (ii) the pregnancy-associated changes in microvascular function involve enhanced endothelial ETBR activity; (iii) the pregnancy-associated changes in ETBR activity involve changes in endothelium-derived vasodilators; and (iv) the pregnancy-associated changes in ETBR activity reflect changes in microvascular ETBR levels.
Methods
Animals and tissue preparation
Time-pregnant (day 11 of gestation) and age-matched virgin female Sprague–Dawley rats (12 weeks of age) were purchased from Charles River Laboratories (Wilmington, MA, USA). The rats were housed in the animal facility for at least 1 week and had access to ad libitum standard rat chow and tap water in 12 h light/dark cycle. Virgin rats were studied during oestrus in order to control for reproductive and endocrine confounders. Oestrus cycle was determined by taking a vaginal smear with a pasteur pipette (Mazzuca et al., 2010). An oestrus smear primarily contained a nucleated cornified squamous cell and was confirmed prior to all experiments (Yener et al., 2007). Late pregnant rats (day 19 of gestation) and age-matched virgin rats were killed by inhalation of CO2. The abdominal cavity was opened and the small intestine, adjacent mesentery and mesenteric arterial arcade were rapidly excised and placed in ice-cold oxygenated Krebs physiological solution. With the aid of a dissection microscope, small second- and third-order mesenteric arteries were carefully isolated and cleaned of surrounding fat and adipose tissue and used for microvascular functional studies. The remainder of the vascular tissue was stored at −80°C for Western blot experiments. All experimental procedures followed the guidelines of the Institutional Animal Care and Use Committee at Harvard Medical School.
Microvessel cannulation and pressurization
Segments of small mesenteric microvessels [outside diameter (OD) ∼300 μm], ∼4–5 mm in length, were transferred to a temperature-controlled perfusion chamber and mounted between two glass micropipettes (cannulas), and secured with 10–0 ophthalmic nylon monofilament (Living Systems Instrumentation, Burlington, VT, USA), as previously described (Chen and Khalil, 2008). One end of the microvessel was mounted on a glass micropipette and the lumen was gently rinsed with Krebs solution to ensure removal of any remaining blood, then the other end of the microvessel was mounted on a second cannula and tied in place. The microvessel segment in the perfusion chamber was placed on an inverted microscope (TE300; Nikon, Melville, NY, USA). A stopcock located distal to the vessel was closed, the proximal end was connected to a pressure transducer pressure-servo control system (Living Systems Instrumentation), and the vessel was gradually pressurized to 60 mmHg and then maintained at constant pressure with the pressure-servo control unit. The microvessel was bathed in 5 mL of Krebs bubbled with 95% O2 and 5% CO2 at 37°C and was continuously superfused with fresh Krebs at a rate of 1 mL·min−1 using a peristaltic mini-pump (Master-Flex; Cole-Parmer, Vernon Hills, IL, USA). Drugs were added abluminally to the bath solution. The vessel was allowed to equilibrate for 60 min before testing its functional viability using high potassium chloride (KCl) depolarizing solution (51 mM), phenylephrine (PHE, 10−5 M) and ACh (10−5 M). Microvessels were unacceptable if they showed leaks or if they failed to produce maintained constriction to KCl and PHE or dilation to ACh.
The mesenteric microvessels were continuously monitored using a video camera connected to a monitor, and the microvessel diameter was measured using automatic edge-detection system (Crescent Electronics, Sandy, UT, USA) and digitized at 1 Hz using a personal computer as previously described (Chen and Khalil, 2008). Snap pictures of the microvessel were taken at rest and following steady-state constriction to different vasoconstrictor stimuli using a digital camera (Cool-Snap; Photometrics, Tucson, AZ, USA).
Fura-2 loading and [Ca2+]i measurement
For measurement of [Ca2+]i, microvessels were incubated in Krebs solution containing the cell permeable Ca2+ indicator fura-2/AM (5 μM) and the mild detergent cremophor EL (0.25%) for 1 h, as previously described (Chen and Khalil, 2008). The microvessel was washed three times in Krebs solution to remove extracellular fura-2/AM, and incubated in normal Krebs for an additional 30 min to allow for deesterification of the trapped intracellular fura-2/AM into the Ca2+-sensitive fura-2. The fura-2-loaded microvessel was excited alternately at 340 and 380 nm, the emitted light was collected at 510 nm every 1 s, and the fluorescence signal was measured using Felix fluorescence data acquisition and analysis software (Photon Technology International, Birmingham, NJ, USA). The 340/380 ratio was calculated and represented the changes in [Ca2+]i. The signal-to-noise ratio was improved by averaging 10 consecutive 340/380 fluorescence ratio readings.
Simultaneous measurement of microvessel diameter and [Ca2+]i
Our initial experiments demonstrated that KCl- and PHE-induced changes in [Ca2+]i were rather small. Furthermore, the ET-1 response was relatively slow in onset, particularly at low concentrations, and a cumulative-constriction response curve would require prolonged exposure to excitation light, which would cause significant photobleaching of fura-2 and affect the accuracy of [Ca2+]i measurements. Therefore, to accurately compare the [Ca2+]i-dependent constriction induced by various agonists, we used maximal concentrations and an 8 min exposure time. The maximal concentrations of KCl, PHE and ET-1 used were based on previous reports from our laboratory (Chen and Khalil, 2008).
In all experiments, the microvessel from pregnant or virgin rats was first stimulated with 51 mM KCl and the simultaneous changes in constriction and 340/380 ratio (indicative of [Ca2+]i) were recorded. The microvessel was washed with Krebs solution and allowed to recover for 20 min. The microvessel was then stimulated with PHE (10−5 M) or ET-1 (10−7 M) and the simultaneous changes in constriction and 340/380 ratio were recorded. In a separate set of microvessels, the role of ETBR in modulating ET-1 responses was tested by pretreating the microvessel with the selective ETBR antagonist BQ-788 (10−6 M) for 30 min, then ET-1 (10−7 M) was added and the microvessel constriction and 340/380 ratio were recorded.
To test for endothelial function, microvessels were submaximally preconstricted (∼70% of maximum) with PHE (6 × 10−6 M), then ACh (10−5 M) was added and the simultaneous changes in microvessel diameter and 340/380 ratio were recorded over an 8 min exposure time. PHE (6 × 10−6 M) produced similar preconstriction in microvessels from pregnant and virgin rats. To further test endothelium-dependent vasodilation, experiments were conducted using increasing concentrations of ACh (10−9–10−5 M), added cumulatively 2 min for each concentration.
To test ETBR-mediated relaxation, microvessels were pretreated with the ETAR antagonist BQ-123 (10−6 M) for 30 min to block ETAR-mediated vasoconstriction, preconstricted with PHE (6 × 10−6 M), then stimulated with increasing concentrations of ET-1 (10−11–10−7 M), and the microvascular relaxation and 340/380 ratio were recorded. To further test ETBR-mediated relaxation, mesenteric microvessels were preconstricted with PHE (6 × 10−6 M) then stimulated with increasing concentrations of the ETBR agonist sarafotoxin 6c (S6c, 10−11–10−7 M) or IRL-1620 (10−11–10−7 M), and the percentage relaxation and underlying [Ca2+]i were measured. Each agonist dose was applied cumulatively, allowing each relaxation to plateau between successive doses.
To elucidate the vasodilator mediator released during stimulation by ACh, S6c and IRL-1620, concentration–relaxation curves were repeated in the presence of Nω-nitro-l-arginine methyl ester (L-NAME, 3 × 10−4 M) and indomethacin (INDO, 10−6 M) to block NOS and COX respectively. Previous studies have shown minimal contribution of PGI2 to vasodilation in rat mesenteric arteries (Wigg et al., 2001). Therefore, in all experiments, both NOS and COX activities were blocked simultaneously. To test for a hyperpolarization pathway, experiments were repeated in the presence of tetraethylammonium chloride (TEA, 30 mM), a non-selective K+ channel blocker.
To confirm the role of endothelium, the endothelium was removed by gently injecting air bubbles through the microvessels while still mounted in the arterial chamber (total volume of injected air bubbles ∼0.3 mL). Endothelium removal was determined by the absence of vasodilator responses (<10%) to ACh (10−5 M), and the integrity of VSM function was confirmed by the maintenance of the constrictor response to PHE (10−5 M).
To directly test the ability of VSM to relax, microvessels were preconstricted with PHE (6 × 10−6 M), then stimulated with the exogenous NO donor sodium nitroprusside (SNP, 10−5 M), and the simultaneous changes in microvessel diameter and 340/380 ratio were recorded over an 8 min exposure time. To further test VSM function, experiments were repeated using increasing SNP concentrations (10−9–10−5 M) applied cumulatively, allowing each relaxation to plateau between successive doses.
Western blot analysis
Protein was extracted by homogenizing mesenteric arteries from three samples, each sample containing arteries pooled from 4 rats per group. In some samples, the endothelium was removed by passing bubbles through the lumen of the artery while mounted in the arterial chamber then pinning down the excised mesenteric artery in a Petri dish and inserting a thin wire (OD 150 μm) into the vessel lumen and carefully rubbing the microvessel interior three times forwards and backwards. Arteries were then homogenized in a homogenization buffer containing 20 mM 3-[N-morpholino] propane sulfonic acid, 4% SDS, 10% glycerol, 2.3 mg dithiothreitol, 1.2 mM EDTA, 0.02% BSA, 5.5 M leupeptin, 5.5 M pepstatin, 2.15 M aprotinin and 20 M 4-(2-aminoethyl)-benzenesulfonyl fluoride, using a 2 mL tight-fitting homogenizer (Kontes Glass Co., Vineland, NJ, USA). The homogenate was centrifuged at 10 000× g for 5 min. The supernatant was collected, and protein concentration was determined using a protein assay kit (Bio-Rad, Hercules, CA, USA). Protein extracts (20 μg) were combined with an equal volume of 2X Laemmli loading buffer, boiled for 5 min, and size fractionated by electrophoresis on 8% SDS-polyacrylamide gels. Proteins were transferred from the gel to a nitrocellulose membrane by electroblotting. Membranes were incubated in 5% non-fat dry milk in TBS-Tween for 1 h and then overnight at 4°C with polyclonal rabbit anti-ETAR antibody or anti-ETBR antibody (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were washed five times for 15 min each in TBS-Tween then incubated with HRP-conjugated secondary antibody (1:1000) for 1.5 h, and the immunoreactive bands were detected using enhanced chemiluminescence Western blotting detection reagent (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). The blots were subsequently reprobed for β-actin (1:2000). Data were analysed by optical densitometry and ImageJ software (National Institutes of Health, Bethesda, MD, USA). The densitometry values represented the pixel intensity normalized to β-actin to correct for loading as previously described (Yin et al., 2012).
Solutions and drugs
Krebs solution contained 120 mM NaCl, 5.9 mM KCl, 25 mM NaHCO3, 1.2 mM NaH2PO4, 11.5 mM dextrose, 2.5 mM CaCl2, 1.2 mM MgCl2, at pH 7.4, and bubbled with 95% O2 and 5% CO2. The 51 mM KCl was prepared as Krebs solution with equimolar substitution of NaCl with KCl. Stock solutions of PHE, ACh, SNP, ET-1 and L-NAME (10−1 M; Sigma-Aldrich, St. Louis, MO, USA), S6c and IRL-1620 (10−3 M; Tocris Bioscience, Hallsville, MO, USA) were prepared in distilled water. TEA (30 mM, Sigma-Aldrich) was prepared as Krebs solution with equimolar substitution of NaCl with TEA. INDO (10−2 M; Sigma-Aldrich), the ETAR antagonist BQ-123, the ETBR antagonist BQ-788 (10−3 M; Tocris Bioscience) and the Ca2+ indicator fura-2/AM (10−3 M; Invitrogen, Carlsbad, CA, USA) were prepared in dimethylsulfoxide (DMSO). The final concentration of DMSO in the experimental solution was <0.1%. All other chemicals were of reagent grade or better. All drugs and molecular targets nomenclature used in this manuscript conform to BJP's Guide on Receptors and Channels (Alexander et al., 2011).
Statistical analysis
Experiments were conducted on mesenteric microvessels isolated from 4 to 12 different rats per group (virgin and pregnant), and cumulative data were presented as means ± SEM, with the ‘n’ value representing the number of rats per group. Time-dependent constriction was measured as [(resting diameter − constriction diameter)/resting diameter] × 100. Time-dependent relaxation was measured as [(relaxation diameter − PHE preconstriction diameter)/(resting diameter − PHE preconstriction diameter)] × 100. Concentration-dependent relaxations were expressed as percentage of maximum relaxation to the specific vasodilator, concentration–relaxation curves were constructed, sigmoidal curves were fitted to the data using the least squares method, and the pD2 values (−log EC50) were measured using Prism (v.5.01; GraphPad Software, San Diego, CA, USA). For vascular function experiments, data were first analysed using repeated measures anova (response to drug in a specific group vs. time or concentration). When a statistical difference was observed, data were further analysed using Bonferroni's post hoc correction for multiple comparisons. Student's unpaired t-test was used for comparison of two means. ETAR and ETBR protein levels were analysed and compared between groups using Student's unpaired t-test. Differences were statistically significant when P < 0.05.
Results
Effect of KCl and PHE
In mesenteric microvessels of both virgin (Figure 1A) and pregnant rats (Figure 1B), membrane depolarization by KCl (51 mM) caused a rapid initial decrease in diameter (time-to-maximum vasoconstriction: virgin = 18.7 ± 2.4 s, pregnant = 22.3 ± 1.8 s) that reached steady state within 33.6 ± 5.4 s in virgin rats and 40.4 ± 4.2 s in pregnant rats (Table 1), and was maintained for the 8 min duration. Furthermore, in microvessels of both virgin and pregnant rats, KCl caused a slight change in the 340 nm fura-2 fluorescence signal, a significant decrease in the 380 nm fluorescence signal, and an increase in the 340/380 fluorescence ratio, indicating a simultaneous increase in [Ca2+]i. The initial KCl-induced increase in [Ca2+]i was rapid (time to peak: virgin = 11.2 ± 0.7 s, pregnant = 11.3 ± 1.0 s), reached steady state within 36.7 ± 2.9 s in virgin rats and 34.4 ± 5.4 s in pregnant rats (Table 1), and was maintained for the 8 min duration. Cumulative data indicated that the initial and maintained KCl-induced vasoconstriction and [Ca2+]i were not significantly different in mesenteric microvessels of virgin and pregnant rats. To further test the Ca2+ dependency of the KCl response, the Δ vasoconstriction per Δ [Ca2+]i (340/380 ratio) was calculated. The KCl-induced Δ vasoconstriction per Δ 340/380 ratio was not significantly different in microvessels of virgin (Figure 1A, bottom panel) and pregnant rats (Figure 1B, bottom panel).
Figure 1.

Effect of KCl (51 mM) (A, B) and PHE (10−5 M) (C, D) on vasoconstriction and [Ca2+]i in mesenteric microvessels of virgin and pregnant (Preg) rats. Images of fura-2 loaded microvessels from virgin and pregnant rats were taken at rest and after stimulation with KCl or PHE. Simultaneous measurements of microvessel diameter, fura-2340 and 380 nm fluorescence signal, and 340/380 ratio were recorded. Cumulative KCl or PHE-induced vasoconstriction, 340/380 ratio, and vasoconstriction over Δ 340/340 ratio in mesenteric microvessels of virgin and pregnant rats were presented as means ± SEM, n = 8–12 different rats per group. # Measurements during stimulation with PHE (C and D) are significantly different (P < 0.05) from corresponding KCl measurements (A and B).
Table 1.
Time-to-peak and time-to-steady-state vasoconstriction and [Ca2+]i in response to KCl (51 mM), PHE (10−5 M) and ET-1 (10−7 M) in mesenteric microvessels of virgin and pregnant rats
| Virgin | Pregnant | |||||
|---|---|---|---|---|---|---|
| KCl | PHE | ET-1 | KCl | PHE | ET-1 | |
| Time-to-maximum constriction (s) | 18.7 ± 2.4 | 16.6 ± 2.5 | 69.4 ± 10.9# | 22.3 ± 1.8 | 19.8 ± 2.8 | 39.7 ± 6.7*# |
| Time-to-peak [Ca2+]i (s) | 11.2 ± 0.7 | 10.9 ± 1.4 | 24.3 ± 3.6# | 11.3 ± 1.0 | 12.9 ± 1.7 | 18.9 ± 2.7 |
| Time-to-steady-state constriction (s) | 33.6 ± 5.4 | 31.9 ± 4.6 | 88.7 ± 14.8# | 40.4 ± 4.2 | 37.3 ± 2.7 | 76.3 ± 10.7# |
| Time-to-steady-state [Ca2+]i (s) | 36.7 ± 2.9 | 42.6 ± 8.6 | 95.3 ± 9.4# | 34.4 ± 5.4 | 36.7 ± 3.1 | 102.0 ± 14.3# |
s, sec.
n = 8–12 different rats per group.
P < 0.05 pregnant versus virgin.
P < 0.05, measurements during stimulation with ET-1 are significantly different from corresponding measurements during stimulation with KCl or PHE.
In mesenteric microvessels of both virgin (Figure 1C) and pregnant rats (Figure 1D), stimulation with the α-adrenergic receptor agonist PHE (10−5 M) caused a rapid initial decrease in diameter (time-to-maximum vasoconstriction: virgin = 16.6 ± 2.5 s, pregnant = 19.8 ± 2.8 s) that reached steady state within 31.9 ± 4.6 s in virgin rats and 37.3 ± 2.7 s in pregnant rats (Table 1), and was maintained for the 8 min duration. The PHE-induced vasoconstriction was preceded by a rapid initial spike in [Ca2+]i (time to peak: virgin = 10.9 ± 1.4 s, pregnant = 12.9 ± 1.7 s), followed by a smaller increase in [Ca2+]i that reached steady state within 42.6 ± 8.6 s in virgin rats and 36.7 ± 3.1 s in pregnant rats (Table 1), and was maintained for the 8 min duration. Cumulative data demonstrated no significant differences in PHE-induced initial or maintained vasoconstriction or [Ca2+]i in mesenteric microvessels of virgin (Figure 1C) and pregnant rats (Figure 1D). Furthermore, the PHE-induced Δ vasoconstriction per Δ 340/380 ratio was not significantly different between virgin (Figure 1C, bottom panel) and pregnant rats (Figure 1D, bottom panel). However, the PHE-induced Δ vasoconstriction per Δ 340/380 ratio (Figure 1C, bottom panel) was significantly greater (P < 0.05) than that induced by KCl (Figure 1A, bottom panel) in microvessels of virgin rats. Similarly, the PHE-induced Δ vasoconstriction per Δ 340/380 ratio (Figure 1D, bottom panel) was significantly greater (P < 0.05) than that induced by KCl (Figure 1B, bottom panel) in microvessels of pregnant rats.
Effect of ET-1
In mesenteric microvessels of both virgin (Figure 2A) and pregnant rats (Figure 2B), stimulation with ET-1 (10−7 M) caused an initial decrease in diameter that was relatively slower (P < 0.05) than that induced by KCl or PHE (time-to-maximum vasoconstriction: virgin = 69.4 ± 10.9 s, pregnant = 39.7 ± 6.7 s; time to steady state: virgin = 88.7 ± 14.8 s, pregnant = 76.3 ± 10.7 s) (Table 1), and was maintained for the 8 min duration. The ET-1-induced time-to-maximum constriction was faster (P < 0.05) in microvessels of pregnant than virgin rats (Table 1). The ET-1-induced vasoconstriction was associated with an initial peak in [Ca2+]i that was slower (P < 0.05) than that induced by KCl or PHE (time to peak: virgin = 24.3 ± 3.6 s, pregnant = 18.9 ± 2.7 s), followed by a smaller increase in [Ca2+]i that was slower in reaching steady state within 95.3 ± 9.4 s in virgin rats (Figure 2C) and 102.0 ± 14.3 s in pregnant rats (Figure 2D) (Table 1), and was maintained for the 8 min duration. Cumulative data showed that ET-1-induced vasoconstriction was significantly reduced (P < 0.05) in microvessels of pregnant (Figure 2F) compared with virgin rats (Figure 2E). On the other hand, the ET-1-induced initial and maintained [Ca2+]i was not significantly different in microvessels of pregnant (Figure 2F) compared with virgin rats (Figure 2E). The ET-1-induced Δ vasoconstriction per Δ 340/380 ratio (Figure 2G, bottom panel) was significantly greater (P < 0.05) than that induced by KCl in microvessels of virgin rats (Figure 1A, bottom panel). Similarly, the ET-1-induced Δ vasoconstriction per Δ 340/380 ratio (Figure 2H, bottom panel) was significantly greater (P < 0.05) than that induced by KCl in microvessels of pregnant rats (Figure 1B, bottom panel). Also, the ET-1-induced Δ vasoconstriction per Δ 340/380 ratio was significantly reduced (P < 0.05) in microvessels of pregnant (Figure 2H) compared with virgin rats (Figure 2G).
Figure 2.
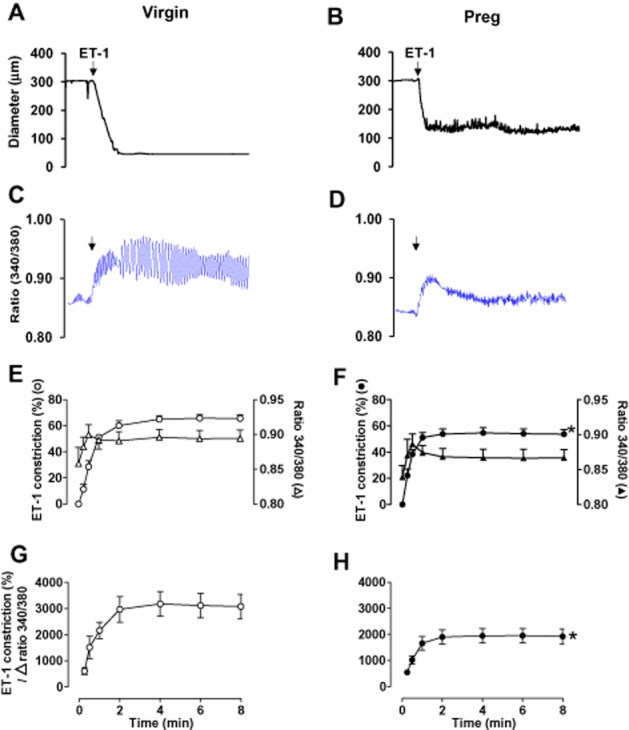
Effect of ET-1 (10−7 M) on vasoconstriction and [Ca2+]i in mesenteric microvessels of virgin and pregnant rats. Simultaneous measurements of ET-1-induced changes in diameter (A, B) and fura-2 340/380 fluorescence ratio (C, D) were recorded in isolated microvessels from virgin (A, C) and pregnant rats (B, D). Cumulative ET-1-induced vasoconstriction, [Ca2+]i, and vasoconstriction per Δ 340/340 ratio in mesenteric microvessels of virgin (E, G) and pregnant rats (F, H) were presented as means ± SEM, n = 8–12 different rats per group. * Measurements in pregnant rats (F and H) are significantly different (P < 0.05) from corresponding measurements in virgin rats (E and G).
Effect of ETBR blockade
When compared with control ET-1 experiments in the absence of blockers, pretreatment with the selective ETBR antagonist BQ-788 (10−6 M) significantly enhanced (P < 0.05) ET-1-induced vasoconstriction in microvessels of both virgin (Figure 3A) and pregnant rats (Figure 3B). In the presence of BQ-788, the maximal ET-1-induced vasoconstriction was not significantly different in pregnant (Figure 3B) versus virgin rats (Figure 3A). However, when compared with control experiments in the absence of blockers, the ET-1-induced vasoconstriction in the presence of BQ-788 was ∼1.7-fold greater in pregnant (Figure 3B) compared with ∼1.2-fold greater in virgin rats (Figure 3A). The enhanced ET-1-induced vasoconstriction in the presence of BQ-788 was not associated with significant changes in [Ca2+]i in microvessels of either virgin (Figure 3C) or pregnant rats (Figure 3D).
Figure 3.
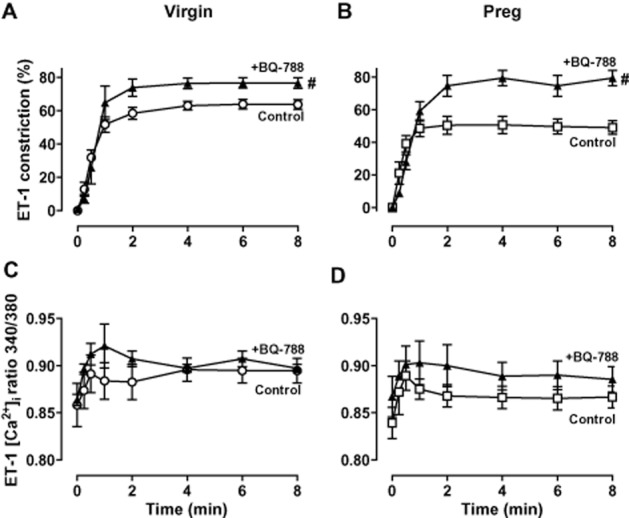
Effect of ETBR blockade on ET-1-induced constriction and [Ca2+]i. Mesenteric microvessels of virgin and pregnant rats were either non-treated or pretreated with BQ-788 (10−6 M), then stimulated with ET-1 (10−7 M), and vasoconstriction and [Ca2+]i measurements were recorded in virgin (A, C) and pregnant rats (B, D). Line graphs represent means ± SEM, n = 6 different rats per group. # Measurements in the presence of BQ-788 are significantly different (P < 0.05) from corresponding control measurements in the absence of BQ-788.
Effect of ACh and SNP
In microvessels of both virgin (Figure 4A) and pregnant rats (Figure 4B) preconstricted with PHE (6 × 10−6 M), ACh (10−5 M) caused rapid relaxation (time-to-maximum vasodilation: virgin = 59.1 ± 5.0 s, pregnant = 54.6 ± 4.8 s) that reached steady state within 75.8.1 ± 4.8 s in virgin rats and 68.2 ± 5.2 s in pregnant rats, and was maintained for the 8 min duration. ACh-induced vasodilation was associated with a rapid initial decrease in [Ca2+]i within 24.7 ± 1.9 s in virgin rats and 26.5 ± 3.9 s in pregnant rats that reached steady state in 59.7 ± 5.0 s in virgin rats and 51.0 ± 12.2 s in pregnant rats, and was maintained for the 8 min duration. Cumulative data demonstrated no significant differences in ACh-induced initial or maintained vasodilation or [Ca2+]i in mesenteric microvessels of virgin (Figure 4A) and pregnant rats (Figure 4B). ACh also evoked concentration-dependent relaxation in mesenteric microvessels of both virgin and pregnant rats (Figure 4E). Although the maximal ACh-induced relaxation was not different, ACh was significantly more potent (P < 0.05) in inducing vasodilation in microvessels of pregnant (pD2 = 6.69 ± 0.14) compared with virgin rats (pD2 = 6.14 ± 0.16) (Figure 4E).
Figure 4.

Effect of ACh (10−5 M) on endothelium-dependent vasodilation, and SNP (10−5 M) on endothelium-independent VSM relaxation and [Ca2+]i in mesenteric microvessels of virgin (A, C) and pregnant rats (B, D). Simultaneous measurements of microvessel diameter and fura-2 340/380 fluorescence ratio were recorded, and cumulative ACh- and SNP-induced time-dependent changes in microvascular relaxation and [Ca2+]i were presented. Concentration–relaxation curves to ACh (10−9–10−5 M) (E) and SNP (10−9–10−5 M) (F) were also constructed in mesenteric microvessels of virgin and pregnant rats, and the pD2 value was measured. Line graphs represent means ± SEM of vasodilation, n = 8–12 different rats per group. *P < 0.05, pregnant versus virgin.
In microvessels of both virgin (Figure 4C) and pregnant rats (Figure 4D), the exogenous NO donor SNP (10−5 M) caused rapid relaxation (time-to-maximum vasodilation: virgin = 66.8 ± 6.9 s, pregnant = 60.4 ± 8.6 s) that reached steady state within 77.5 ± 8.1 s in virgin rats and 73.0 ± 7.1 s in pregnant rats, and was maintained for the 8 min duration. SNP-induced vasodilation was associated with a rapid initial decrease in [Ca2+]i within 30.0 ± 3.2 s in virgin rats and 25.3 ± 4.8 s in pregnant rats, reached steady state within 77.5 ± 8.1 s in virgin rats and 73.0 ± 7.1 s in pregnant rats, and was maintained for the 8 min duration. Cumulative data demonstrated no significant differences in SNP-induced initial or maintained vasodilation or [Ca2+]i in mesenteric microvessels of virgin (Figure 4C) and pregnant rats (Figure 4D). SNP also evoked concentration-dependent relaxation, and the maximal SNP-induced relaxation was not different in microvessels of virgin and pregnant rats (Figure 4F). Furthermore, SNP was equally potent in inducing relaxation in microvessels of virgin (pD2 = 6.51 ± 0.14) and pregnant rats (pD2 = 6.49 ± 0.23) (Figure 4F).
Effect of ETBR activation
In microvessels pretreated with the ETAR antagonist BQ-123 (10−6 M) and preconstricted with PHE (6 × 10−6 M), ET-1 induced concentration-dependent relaxation that was significantly greater (P < 0.05) in pregnant than in virgin rats (Figure 5A). The BQ-123 + ET-1-induced relaxation was not associated with significant changes in [Ca2+]i in microvessels of virgin or pregnant rats (Figure 5B). Furthermore, the selective ETBR agonists S6c (Figure 5C) and IRL-1620 (Figure 5E) caused concentration-dependent relaxation that was significantly greater (P < 0.05) in microvessels of pregnant compared with virgin rats. The differences in S6c and IRL-1620-induced relaxation in microvessels of pregnant versus virgin rats were not associated with significant differences in the underlying [Ca2+]i between virgin (Figure 5D) and pregnant rats (Figure 5F).
Figure 5.
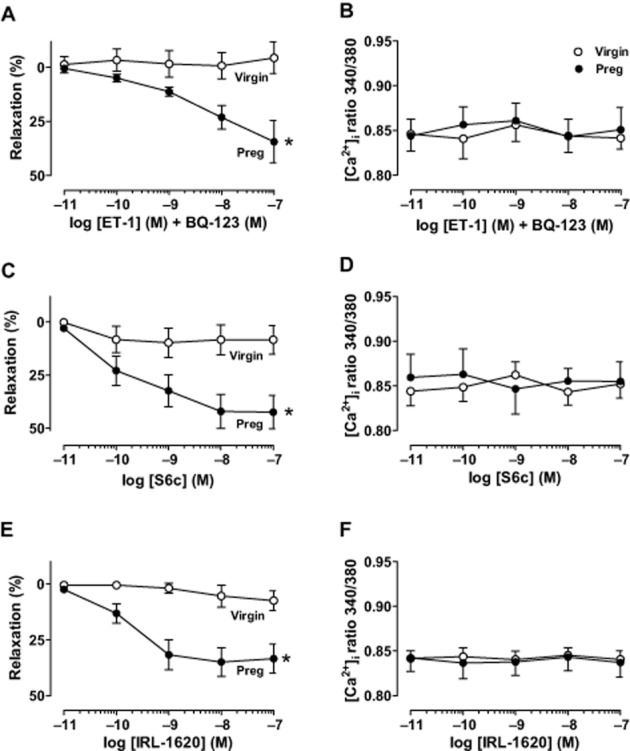
Effect of ETBR-mediated activation. Microvessels of virgin and pregnant rats were pretreated with the ETAR antagonist BQ-123 (10−6 M) to block ETAR-mediated vasoconstriction. Microvessels were then preconstricted with PHE (6 × 10−6 M), then stimulated with increasing concentrations of ET-1 (10−11–10−7 M), and the % relaxation of PHE contraction (A) and underlying [Ca2+]i (B) was recorded. Concentration–relaxation curves (C, E) and underlying [Ca2+]i (D, F) in response to the ETBR agonist S6c (10−11–10−7 M) (C, D) and IRL-1620 (10−11–10−7 M) (E, F) were compared in microvessels of virgin and pregnant rats. Line graphs represent means ± SEM of vasodilation and [Ca2+]i, n = 8–12 different rats per group. *P < 0.05, pregnant versus virgin.
Role of endothelium in microvascular relaxation
To test the role of the endothelium, microvessel relaxation was compared in endothelium-denuded and intact microvessels. ACh-induced relaxation was abolished in endothelium-denuded compared with intact microvessels of virgin (Figure 6A) and pregnant rats (Figure 6B). Furthermore, microvascular relaxation in response to BQ123 + ET-1 (data not shown), S6c (Figure 6C) and IRL-1620 (Figure 6D) was abolished in endothelium-denuded compared with intact microvessels of pregnant rats.
Figure 6.
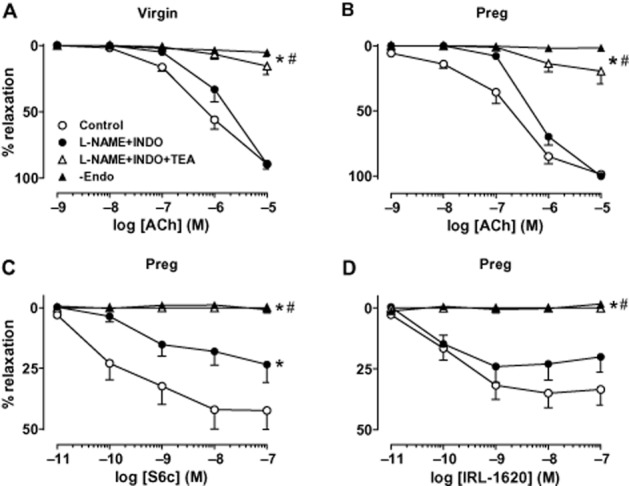
Effect of blocking NO, PGI2 and EDHF in mesenteric microvessels of virgin and pregnant rats. Mesenteric microvessels from virgin (A) and pregnant rats (B, C, D) were preconstricted with PHE (6 × 10−6 M) and concentration–relaxation curves in response to ACh (10−9–10−5 M) (A, B), S6c (10−11–10−7 M) (C), and IRL-1620 (10−11–10−7 M) (D), and in the absence and presence of L-NAME (3 × 10−4 M) and INDO (10−6 M) and TEA (30 mM) or in endothelium-denuded microvessels (−Endo) were recorded. Line graphs represent means ± SEM of vasodilation, n = 8–12 different rats per group. −Endo experiments were n = 4 rats per group. *P < 0.05 versus control measurements in the absence of blockers. #P < 0.05 versus measurements in the presence of L-NAME + INDO.
Effect of blocking NO, PGI2 and EDHF
To investigate the vascular mediator released during microvessel relaxation, we tested the effects of blockade of NOS and COX with L-NAME (3 × 10−4 M) and INDO (10−6 M) respectively. Pretreatment with L-NAME + INDO did not cause significant changes in maximal ACh-induced relaxation, but caused a significant shift to the right (P < 0.05) in the concentration–relaxation curves in microvessels of virgin (Figure 6A) and pregnant rats (Figure 6B). The L-NAME + INDO-resistant component of ACh relaxation was attributed to EDHF, and ACh was more potent (P < 0.05) in stimulating this EDHF-mediated component in microvessels of pregnant (pD2 = 6.24 ± 0.05) (Figure 6B) compared with virgin rats (pD2 = 5.58 ± 0.56) (Figure 6A). The remaining L-NAME + INDO-resistant and EDHF-mediated component of ACh relaxation was almost abolished by the non-selective K+ channel blocker TEA (30 mM) in microvessels of both virgin (Figure 6A) and pregnant rats (Figure 6B).
To test the vascular mediators involved in ETBR-mediated relaxation, microvessels were treated with blockers of NO, PGI2 and EDHF pathway. In mesenteric arteries of virgin rats, L-NAME and INDO insignificantly reduced the small S6c and IRL-1620-induced maximal relaxation (S6c without blockers = 8.39 ± 6.79%, with L-NAME + INDO = 4.01 ± 3.70%, P = 0.58; IRL-1620 without blockers = 7.50 ± 4.49%, with L-NAME + INDO = 1.28 ± 0.97, P = 0.21), and any remaining S6c or IRL-1620-induced relaxation in microvessels treated with L-NAME + INDO was almost negligible (<4%), suggesting a little role of EDHF. In contrast, in microvessels of pregnant rats, pretreatment with L-NAME + INDO caused significant reduction in S6c-induced maximal relaxation (23.42 ± 7.39%) when compared with control relaxation in the absence of blockers (42.45 ± 7.85%, P < 0.05, Figure 6C). Pretreatment with L-NAME + INDO caused insignificant reduction in IRL-1620-induced maximal relaxation in microvessels of pregnant rats (20.08 ± 6.21%) when compared with that in the absence of blockers (33.46 ± 6.46, P = 0.15, Figure 6D). Importantly, the remaining S6c or IRL-1620-induced relaxation in mesenteric arteries of pregnant rats treated with L-NAME + INDO was still significant (>20%), suggesting a greater role of EDHF-dependent component of ETBR-mediated relaxation in microvessels of pregnant compared with virgin rats. Additional pretreatment with the non-selective K+ channel blocker TEA abolished the remaining relaxation to S6c (Figure 6C) and IRL-1620 (Figure 6D) in microvessels of pregnant rats.
ETAR and ETBR protein level
Western blot analysis revealed ETAR (Figure 7A) and ETBR proteins (Figure 7B) in mesenteric microvessels of both virgin and pregnant rats. ETAR protein level was not significantly different in endothelium-intact or endothelium-denuded microvessels of pregnant compared with virgin rats, or between endothelium-denuded and intact microvessels of either virgin or pregnant rats (Figure 7A). In contrast, ETBR protein level was significantly greater (P < 0.05) in intact microvessels of pregnant compared with virgin rats (Figure 7B). Furthermore, ETBR protein level was significantly reduced (P < 0.05) in endothelium-denuded microvessels of pregnant compared with virgin rats, and in endothelium-denuded compared with intact microvessels of pregnant but not virgin rats (Figure 7B).
Figure 7.
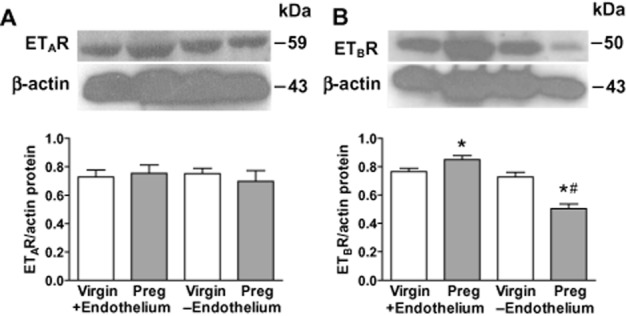
Western blot analysis of ETAR and ETBR. Mesenteric microvessels with (+) and without (−) endothelium of both virgin and pregnant rats were homogenized and prepared for Western blot analysis of protein levels of ETAR (A) and ETBR (B). Bar graphs represent means ± SEM. n = 3 samples from pooled mesenteric arteries from 4 different rats per group. *P < 0.05 pregnant versus virgin. #P < 0.05 −Endothelium versus +Endothelium.
Discussion and conclusions
The main findings of the present study are: (i) ET-1-induced vasoconstriction is reduced in mesenteric microvessels of pregnant compared with virgin rats, with small increases in [Ca2+]i; (ii) the pregnancy-associated changes in microvascular function partly involve enhanced endothelial ETBR-mediated relaxation; and (iii) the pregnancy-associated increase in ETBR-mediated relaxation involves changes in endothelium-dependent NO- and EDHF-mediated pathways, and reflects increases in microvascular endothelial ETBR protein levels.
Normal pregnancy is associated with increased maternal blood volume and cardiac output and decreased systemic vascular resistance (Poston et al., 1995; Thornburg et al., 2000), but BP may remain unchanged or slightly decreased (Khalil et al., 1998; Thornburg et al., 2000). BP regulation is a multifactorial process involving neuronal, renal and vascular control mechanisms (Hall et al., 1986; Lohmeier, 2001; Cain and Khalil, 2002), and the lack of detectable changes in BP during pregnancy does not rule out possible changes in blood vessels. Pregnancy is associated with increased flow-mediated vasodilation, arterial compliance, and Gq-endothelial cell and Gs-VSM receptor-coupled vasodilation pathways, as well as decreased Gq-VSM-coupled receptor-mediated vasoconstriction (van Drongelen et al., 2012). Furthermore, in vivo studies have shown that the pressor response to PHE and ANG II is reduced in the systemic and uterine circulations of pregnant sheep and rats (Naden and Rosenfeld, 1981; Paller, 1984; Magness and Rosenfeld, 1986; Rosenfeld and Naden, 1989). The blunted vasoconstrictor responses in vivo may be compensated by cardiac baroreceptor reflexes (D'Angelo and Osol, 1993), thereby normalizing any changes in BP during pregnancy. In light of this, we directly assessed vascular function in isolated mesenteric vessels because they contribute to vascular resistance in conscious rats (Christensen and Mulvany, 1993), and previous studies have reported pregnancy-associated alterations in these vessels (Anderson et al., 2006a; Chen and Khalil, 2008). Furthermore, studies have measured [Ca2+]i in blood vessels loaded with fura-2, and suggested that the measured [Ca2+]i reflected mainly changes in VSM (Himpens et al., 1990; Khalil and van Breemen, 1990; Bolz et al., 1999; Chen and Khalil, 2008). In the present study, the increases in vasoconstriction and [Ca2+]i during microvessel activation by KCl, PHE and ET-1, and the parallel vasodilation and decreased [Ca2+]i during stimulation by ACh and SNP, support that the detected changes in microvessel [Ca2+]i originated mainly from VSM.
We found that KCl, which stimulates Ca2+ entry through voltage-gated Ca2+ channels (Khalil and van Breemen, 1988), caused microvascular constriction and increased [Ca2+]i that were not different in pregnant and virgin rats. Our data agree with other reports (D'Angelo and Osol, 1993; Mazzuca et al., 2012), and support that in the mesenteric vascular bed Ca2+ influx through voltage-gated Ca2+ channels is not altered during late pregnancy. Furthermore, consistent with other studies (Parent et al., 1990; D'Angelo and Osol, 1993; Pascoal et al., 1995), maximal doses of the α-adrenergic receptor agonist PHE (10−5 M) caused vasoconstriction that was not different in pregnant and virgin rats. In order to accurately measure [Ca2+]i, we used the maximal PHE concentration, and found that PHE-induced [Ca2+]i was not different in pregnant versus virgin rats. Although we did not test a dose-dependent response to PHE, other studies have shown that the sensitivity to PHE either increased (D'Angelo and Osol, 1993), decreased (Parent et al., 1990; Davidge and McLaughlin, 1992) or unchanged (Pascoal et al., 1995; Mazzuca et al., 2012) in mesenteric arteries of late pregnant compared with virgin rats. The cause of these discrepancies is unclear but may be related to differences in methodology, rat strain, gestational time or the vessel order along the vascular tree (van Drongelen et al., 2012).
The KCl- and PHE-induced constriction in microvessels of pregnant and virgin rats was associated with increases in [Ca2+]i, suggesting activation of Ca2+-dependent mechanisms of VSM contraction. While in both virgin and pregnant rats the maximal PHE-induced constriction was similar to that of KCl, [Ca2+]i was smaller during activation by PHE compared with KCl. Assuming that KCl mainly stimulates Ca2+ entry, Ca2+-calmodulin, and myosin light chain phosphorylation, the similar magnitude of PHE contraction despite the significantly less [Ca2+]i and greater Δ constriction to Δ [Ca2+]i ratio suggests activation of additional mechanisms that increase the myofilament force sensitivity to [Ca2+]i such as rho kinase, MAPK (Somlyo and Somlyo, 2003; Hilgers and Webb, 2005; Puetz et al., 2009) and PKC (Khalil et al., 1992; Khalil, 2010).
ET-1 is a major regulator of vascular function, acting via both ETAR and ETBR (Yanagisawa et al., 1988; Arai et al., 1990; Kohan et al., 2011). ET-1 activates ETAR in VSM cells causing vasoconstriction and increases in [Ca2+]i, PKC, MAPK, and other pathways of VSM contraction and cell growth (Schiffrin and Touyz, 1998; Cain et al., 2002; Dallas and Khalil, 2003; Touyz et al., 2004). ETBR in endothelial cells may promote vascular relaxation (Giardina et al., 2001; Tirapelli et al., 2005; Tykocki et al., 2009) and has been identified in the systemic, renal, pulmonary, coronary and cerebral circulation (Mazzuca and Khalil, 2012). We found that ET-1 induced vasoconstriction in mesenteric microvessels of virgin and pregnant rats. Similar to PHE, ET-1-induced constriction was similar to KCl, but associated with smaller increases in [Ca2+]i, and greater Δ vasoconstriction per Δ [Ca2+]i, supporting that ET-1 causes VSM contraction not only by increasing [Ca2+]i but also by activating Ca2+ sensitization pathways such as PKC and rho kinase (Dallas and Khalil, 2003; Tykocki and Watts, 2010; Lima et al., 2011). ET-1-induced constriction was reduced in microvessels of pregnant versus virgin rats and associated with small increases in [Ca2+]i that were not different in virgin and pregnant rats. Furthermore, the ET-1-induced Δ vasoconstriction per Δ [Ca2+]i was reduced in microvessels of pregnant compared with virgin rats. These findings suggest that the reduced ET-1-induced vasoconstriction in pregnant rats may not be exclusively due to changes in VSM [Ca2+]i but could involve reduced [Ca2+]i sensitization pathways such as PKC or rho kinase, and further analysis of these pathways should be examined in future studies.
Because endothelial ETBR could have opposing effects to VSM ETAR, we investigated whether the reduced ET-1 vasoconstriction in pregnant rats was due to up-regulation of endothelial ETBR. We first tested the effects of blocking ETBR using BQ-788 on ET-1-induced vasoconstriction. We reasoned that if BQ-788 blocks endothelial ETBR-mediated vasodilation, then it will allow ET-1 to mainly activate ETAR and cause greater vasoconstriction. BQ-788 enhanced ET-1-induced vasoconstriction. While maximal ET-1-induced constriction was not different in virgin and pregnant rats, the fold change in ET-1 constriction with ETBR blockade was greater in pregnant than in virgin rats, suggesting enhanced ETBR-mediated vasodilation during pregnancy. Interestingly, [Ca2+]i was not altered by BQ-788, suggesting that the increase in ET-1 vasoconstriction may be largely due to activation of Ca2+-sensitization pathways.
We then examined the effects of activation of ETBR on microvascular function. Microvascular ETBR activity was enhanced during pregnancy because pretreatment with the ETAR antagonist BQ-123 then adding ET-1, or treatment with the ETBR agonists S6c and IRL-1620 caused greater relaxation in microvessels of pregnant than virgin rats. The enhanced ETBR activity could be partly due to increased endothelial ETBR expression because (i) removal of the endothelium abolished BQ-123 + ET-1-, S6c- and IRL-1620-induced relaxation in pregnant rats; (ii) Western blots showed greater ETBR levels in endothelium-intact microvessels of pregnant than virgin rats; and (iii) removal of the endothelium did not show a difference in ETAR levels, but reduced ETBR expression in pregnant versus virgin rats, supporting that endothelial ETBR plays a role in promoting vasodilation during pregnancy. The enhanced ETBR dilation is unlikely due to increased VSM sensitivity to vasodilators because SNP-induced relaxation was not different in pregnant and virgin rats, which is in accordance with other studies (Gerber et al., 1998; Cooke and Davidge, 2003; Mazzuca et al., 2012).
In search for the factors involved in the enhanced ETBR-mediated relaxation in pregnant rats, we examined the various endothelium-dependent relaxation factors released by known vasodilators such as ACh. Although the maximal ACh-induced relaxation was not different, ACh was more potent in inducing vasodilation in microvessels of pregnant than virgin rats, which is consistent with other studies (Davidge and McLaughlin, 1992; Kim et al., 1994; Gerber et al., 1998; Mazzuca et al., 2012). ACh-induced relaxation was abolished in endothelium-denuded microvessels of virgin and pregnant rats, supporting endothelium-dependent release of vasodilator substances. In many blood vessels, NO is a major endothelium-derived vasodilator (Fleming and Busse, 2003; Félétou and Vanhoutte, 2006; Flammer and Luscher, 2010), while PGI2 may be important in specialized blood vessels such as the pulmonary artery (Majed and Khalil, 2012). The NOS inhibitor L-NAME did not inhibit maximal ACh relaxation in microvessels of virgin or pregnant rats, but reduced the sensitivity to ACh, suggesting that the increased sensitivity to ACh-induced relaxation in microvessels of pregnant rats may be partly due to changes in NO release. This is supported by reports that endothelial NOS (eNOS) is up-regulated in the systemic circulation during pregnancy (Magness et al., 2001). The pregnancy-associated increase in NO synthesis could be partly due to up-regulation of oestrogen, which is known to increase eNOS expression/activity (Magness et al., 2001; Rupnow et al., 2001). Activation of endothelial cells is known to increase [Ca2+]i, which in turn activates eNOS and increases NO production. In addition to increases in endothelial [Ca2+]i, phosphorylation by MAPK and Akt could lead to further activation of eNOS. NO is known to diffuse into VSM and increase cGMP production, which in turn decreases VSM [Ca2+]i by inhibiting Ca2+ influx and promoting Ca2+ extrusion (Félétou and Vanhoutte, 2006). In the present study, ACh-stimulated increase in endothelial [Ca2+]i could not be detected either because any increase in endothelial Ca2+ is masked by the concomitant decrease in VSM [Ca2+]i with the net observation of vasodilation and simultaneous decrease in [Ca2+]i, or because eNOS activation is less dependent on Ca2+ and more dependent on other pathways such as MAPK and Akt, and these possibilities need to be further examined. The increased sensitivity to ACh in mesenteric arteries of pregnant rats may also be due to increased cGMP production in the systemic circulation during pregnancy (Conrad et al., 1993), although this is less likely because the microvascular relaxation to SNP, an exogenous NO donor and a stimulator of cGMP production, was not different in virgin and pregnant rats.
In addition to NO and possibly PGI2, endothelium-dependent relaxation may involve EDHF, an important vasodilator in small resistance arteries that contributes to the regulation of vascular resistance and blood flow (Shimokawa et al., 1996; Busse et al., 2002; Feletou and Vanhoutte, 2009). EDHF produces relaxation by membrane hyperpolarization, inactivation of voltage-gated Ca2+ channels (Nelson et al., 1990) and decreasing the activity of PLC (Itoh et al., 1992). In the presence of L-NAME and INDO to block NOS and COX, respectively, ACh still caused microvascular relaxation and the sensitivity to ACh was enhanced in pregnant compared with virgin rats, supporting that EDHF-mediated pathways are up-regulated in the mesenteric circulation during pregnancy (Gerber et al., 1998; Dalle Lucca et al., 2000; Mazzuca et al., 2012). The observations that blockade with L-NAME + INDO did not inhibit maximal ACh-induced relaxation but reduced the sensitivity to ACh, while ACh relaxation was abolished by blocking EDHF with TEA and the sensitivity to EDHF-mediated relaxation was greater in pregnant than virgin rats, suggest that ACh-induced mesenteric microvascular relaxation was only partly due to changes in NO synthesis/release and, to a large extent, due to release of EDHF.
Similar to ACh, the microvessel relaxation in response to ETBR activation by BQ-123 + ET-1 or the ETBR agonists S6c and IRL-1620 was abolished by endothelium removal, supporting an endothelium-dependent ETBR-mediated vasodilation. In comparison with ACh, the enhanced endothelium-dependent ETBR-mediated relaxation in microvessels of pregnant rats was partially inhibited by L-NAME supporting a role of NO. S6c and IRL-1620 did not cause detectable change in [Ca2+]i likely because the increase in ETBR-mediated [Ca2+]i in endothelial cells is cancelled by the concomitant decrease in [Ca2+]i in VSM, or perhaps due to eNOS activation by other pathways such as MAPK and Akt. In microvessels of pregnant rats, a significant S6c- and IRL-1620-induced relaxation remained in the presence of L-NAME and INDO, supporting a role of EDHF. The remaining EDHF-mediated S6c- and IRL-1620-induced relaxation was abolished by TEA, a non-selective K+ channel blocker. Although the present study did not identify which ETBR-mediated EDHF(s) were up-regulated in the mesenteric arteries during pregnancy, studies have shown that the up-regulation of EDHF is due to enhanced myoendothelial gap junction communication in subcutaneous and myometrial arteries of pregnant women (Kenny et al., 2002; Luksha et al., 2004; Lang et al., 2007) and aortic rings from pregnant rats (Dantas et al., 1999). Other studies have shown that 4-aminopyridine, but not apamin, charybdotoxin or glibenclamide reduces IRL-1620-induced relaxation, indicating that voltage-dependent K+ channel (Kv) but not small-conductance Ca2+-activated K+ channel (KCa2.3), intermediate-conductance Ca2+-activated K+ channel (KCa3.1) or ATP-sensitive K+ channel (KATP), respectively, contributes to the ETBR/EDHF-mediated response (Tirapelli et al., 2005). These channels could be involved in the prominent EDHF-dependent ETBR-mediated relaxation in mesenteric microvessels of pregnant rats. Other proposed EDHF mechanisms including cytochrome p450 pathway of arachidonic acid such as epoxyeicosatrienoic acids, inward rectifier K+ channel (KIR), Na+/K+-ATPase, hydrogen peroxide (H2O2), and C-type natriuretic peptide may be involved, and warrant further investigation.
An important question is what causes the enhanced endothelial ETBR activity during pregnancy. The pregnancy-associated increase in microvascular relaxation may take place at the ETBR level, and several factors such as oestrogen and relaxin may affect the expression and activity of ETBR during pregnancy (Nuedling et al., 2003; Conrad, 2011). Furthermore, post-translation modification and O-GlcNAcylation may modulate the vascular effects of ET-1 (Lima et al., 2011), and may affect ETBR activity during pregnancy.
We should caution that ETBRs have been identified not only in the endothelium, but have also been proposed to be expressed in VSM, and to promote vasoconstriction by mechanisms similar to those activated by ETAR (Touyz et al., 1995; Tostes et al., 2008), and therefore the net physiological impact of ETBR stimulation may depend on the balance between the opposing effects of ETBR in endothelial cells and VSM. Importantly, in freshly isolated male Wistar-Kyoto rat mesenteric arteries mounted on wire or perfusion myographs, stimulation with the ETBR agonists S6c and IRL-1620 produced negligible vasoconstrictor effects (Adner et al., 1998a,b), suggesting little role of VSM ETBR in mesenteric artery vasoconstriction or that any VSM ETBR-mediated contraction mechanisms are counterbalanced by endothelial ETBR-mediated vasodilation. Different effects of ETBR activation have also been observed in various vascular beds and in health and disease (Schneider et al., 2007; Davenport and Maguire, 2011; Mazzuca and Khalil, 2012). However, the observation that ETBR agonists caused vasodilation in mesenteric microvessels of pregnant rats supports greater amount of endothelial than VSM ETBR. This is further supported by the Western blot data that ETBR levels were greater in endothelium-intact than endothelium-denuded microvessels of pregnant rats. Conversely, in virgin rats, ETBR levels were not different in endothelium-denuded and intact mesenteric arteries, suggesting that most of the detected ETBR represents VSM ETBR as opposed to endothelial ETBR. This may also explain the minimal ETBR-mediated vasodilator response to BQ-123 + ET-1, S6c and IRL-1620 in mesenteric microvessels of virgin rats.
The present findings could have important physiological and pathological implications. Up-regulation of endothelial ETBR could lead to enhanced microvascular relaxation, attenuated vasoconstriction, and may play a protective role in modulating BP during normal pregnancy. Conversely, decreased expression/activity of ETBR may play a role in the endothelial dysfunction and vasoconstriction in hypertension in pregnancy and pre-eclampsia. Mesenteric microvascular constriction is enhanced in rat models of hypertension in pregnancy (Anderson et al., 2006b). Because endothelial dysfunction is a major factor in the pathogenesis of pre-eclampsia (Maynard et al., 2003; Reslan and Khalil, 2010), it could cause down-regulation of endothelial ETBR leading to decreased vasodilation and increased BP. Interestingly, some studies have examined the effects of ETBR modulation in vivo and demonstrated that chronic subcutaneous infusion of the ETBR antagonist A-192621 caused transient increases in BP in pregnant rats (Thaete and Neerhof, 2000; Madsen et al., 2001), which could be well explained by blockade of endothelial ETBR-mediated vasodilation. However, in vivo changes in BP should be interpreted with caution as BP regulation involves multiple renal, hormonal, neuronal and vascular control mechanisms. For instance, ETBR in the renal medulla may play a role in salt and water excretion, and blockade of renal ETBR in vivo could cause salt and water retention and hypertension (Kohan et al., 2011). Furthermore, ETBR may mediate ET-1 clearance, and blockade of these receptors in vivo would allow more ET-1 to activate ETAR to promote vasoconstriction and increase BP. One approach to circumvent the long-term renal effects on BP is to study the acute effects of intravenous infusion of an ETBR receptor antagonist over the course of minutes in pregnant rats. Over such a short time course, it is unlikely that the renal haemodynamics would play a significant role in modulating the BP response. However, during the acute infusion of an ETBR modulator in vivo, any changes in vascular reactivity may be compensated by baroreceptor reflexes, which could mask any vascular effects on BP (D'Angelo and Osol, 1993). The present study was pivotal in that it addressed the ‘direct effects’ of ETBR agonists and antagonists in the mesenteric vascular bed, and highlighted the important role of endothelial ETBR activation in enhancing the vasodilatory response commonly observed during pregnancy.
ET-1 has been suggested to play a role in pre-eclampsia (Maynard et al., 2003). ET-1 levels are increased in pre-eclampsia, particularly when accompanied with intrauterine growth restriction (McQeen et al., 1993; Furuhashi et al., 1995), and modulation of the ET-1 system may be an important target in the management of pre-eclampsia. Studies have shown that infusion of ETAR antagonist reduces BP in the L-NAME-infused reduced uterine perfusion pressure and sFlt-1-infused rat models of hypertension in pregnancy (Olson et al., 1999; Alexander et al., 2001; Murphy et al., 2010; 2012; Tam Tam et al., 2011). While the decrease in BP could be due to blockade of ETAR in VSM and reduction of vasoconstriction, it could also be due to the possibility that most ET-1 would be directed towards endothelial ETBR and promote vasodilation. Endothelial ETBR agonists or conditional genetic activation of endothelial ETBR may represent novel approaches in the management of hypertension in pregnancy and pre-eclampsia.
In summary, the present study provided evidence that ETBR expression and activity are enhanced during pregnancy, and may explain the blunted vasoconstrictor response to ET-1 during pregnancy. The role of endothelial ETBR needs to be further evaluated in the pathogenesis and management of hypertension in pregnancy and pre-eclampsia.
Acknowledgments
This work was supported by grants from the National Heart, Lung, and Blood Institute (HL-65998, HL-98724) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (HD-60702). Dr. Y. D. was a visiting scholar from Union Hospital, Huazhong University of Science & Technology, Wuhan, Hubei Province, China, and a recipient of a scholarship from the China Scholarship Council. We would like to thank Mr. Sridhar Rangan for his assistance in analysing the Western blot data.
Glossary
- BQ-123
cyclo[D-Trp-D-Asp-Pro-D-Val-Leu]
- BQ-788
N-[(cis-2,6-dimethyl-1-piperidinyl)carbonyl]-4-methyl-L-leucyl-1-(methoxycarbonyl)-D-tryptophyl-D-norleucine sodium salt
- [Ca2+]i
intracellular free Ca2+ concentration
- EDHF
endothelium-derived hyperpolarizing factor
- eNOS
endothelial NOS
- ET-1
endothelin-1
- ETAR
endothelin receptor type A
- ETBR
endothelin receptor type B
- INDO
indomethacin
- IRL-1620
Suc-Asp-Glu-Glu-Ala-Val-Tyr-Phe-Ala-His-Leu-Asp-Ile-Ile-Trp
- L-NAME
Nω-nitro-l-arginine methyl ester
- pD2 (−log EC50)
concentration of drug required to evoke a half-maximal response
- PGI2
prostacyclin
- PHE
phenylephrine
- S6c
sarafotoxin 6c
- SNP
sodium nitroprusside
- TEA
tetraethylammonium chloride
- VSM
vascular smooth muscle
Conflict of interest
The authors declare no conflict of interest.
References
- Adner M, Geary GG, Edvinsson L. Appearance of contractile endothelin-B receptors in rat mesenteric arterial segments following organ culture. Acta Physiol Scand. 1998a;163:121–129. doi: 10.1046/j.1365-201X.1998.00369.x. [DOI] [PubMed] [Google Scholar]
- Adner M, Uddman E, Cardell LO, Edvinsson L. Regional variation in appearance of vascular contractile endothelin-B receptors following organ culture. Cardiovasc Res. 1998b;37:254–262. doi: 10.1016/s0008-6363(97)00206-x. [DOI] [PubMed] [Google Scholar]
- Alexander BT, Rinewalt AN, Cockrell KL, Massey MB, Bennett WA, Granger JP. Endothelin type a receptor blockade attenuates the hypertension in response to chronic reductions in uterine perfusion pressure. Hypertension. 2001;37:485–489. doi: 10.1161/01.hyp.37.2.485. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Lopez F, Zhang H-Y, Shirasawa Y, Pavlish K, Benoit JN. Mestenteric vascular responsiveness in a rat model of pregnancy-induced hypertension. Exp Biol Med. 2006a;231:1398–1402. doi: 10.1177/153537020623100813. [DOI] [PubMed] [Google Scholar]
- Anderson CM, Lopez F, Zimmer A, Benoit JN. Placental insufficiency leads to developmental hypertension and mesenteric artery dysfunction in two generations of Sprague-Dawley rat offspring. Biol Reprod. 2006b;74:538–544. doi: 10.1095/biolreprod.105.045807. [DOI] [PubMed] [Google Scholar]
- Arai H, Hori S, Aramori I, Ohkubo H, Nakanishi S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature. 1990;348:730–732. doi: 10.1038/348730a0. [DOI] [PubMed] [Google Scholar]
- Bolz SS, de Wit C, Pohl U. Endothelium-derived hyperpolarizing factor but not NO reduces smooth muscle Ca2+ during acetylcholine-induced dilation of microvessels. Br J Pharmacol. 1999;128:124–134. doi: 10.1038/sj.bjp.0702775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- Cain AE, Khalil RA. Pathophysiology of essential hypertension: role of the pump, the vessel, and the kidney. Semin Nephrol. 2002;22:3–16. [PubMed] [Google Scholar]
- Cain AE, Tanner DM, Khalil RA. Endothelin-1–induced enhancement of coronary smooth muscle contraction via MAPK-dependent and MAPK-independent [Ca(2+)](i) sensitization pathways. Hypertension. 2002;39:543–549. doi: 10.1161/hy0202.103129. [DOI] [PubMed] [Google Scholar]
- Chen W, Khalil RA. Differential [Ca2+]i signaling of vasoconstriction in mesenteric microvessels of normal and reduced uterine perfusion pregnant rats. Am J Physiol. 2008;295:R1962–R1972. doi: 10.1152/ajpregu.90523.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen KL, Mulvany MJ. Mesenteric arcade arteries contribute substantially to vascular resistance in conscious rats. J Vasc Res. 1993;30:73–79. doi: 10.1159/000158978. [DOI] [PubMed] [Google Scholar]
- Conrad KP. Maternal vasodilation in pregnancy: the emerging role of relaxin. Am J Physiol. 2011;301:R267–R275. doi: 10.1152/ajpregu.00156.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP, Joffe GM, Kruszyna H, Kruszyna R, Rochelle LG, Smith RP, et al. Identification of increased nitric oxide biosynthesis during pregnancy in rats. FASEB J. 1993;7:566–571. [PubMed] [Google Scholar]
- Conrad KP, Gandley RE, Ogawa T, Nakanishi S, Danielson LA. Endothelin mediates renal vasodilation and hyperfiltration during pregnancy in chronically instrumented conscious rats. Am J Physiol. 1999;276:F767–F776. doi: 10.1152/ajprenal.1999.276.5.F767. [DOI] [PubMed] [Google Scholar]
- Cooke CL, Davidge ST. Pregnancy-induced alterations of vascular function in mouse mesenteric and uterine arteries. Biol Reprod. 2003;68:1072–1077. doi: 10.1095/biolreprod.102.009886. [DOI] [PubMed] [Google Scholar]
- Crandall ME, Keve TM, McLaughlin MK. Characterization of norepinephrine sensitivity in the maternal splanchnic circulation during pregnancy. Am J Obstet Gynecol. 1990;162:1296–1301. doi: 10.1016/0002-9378(90)90040-e. [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Osol G. Regional variation in resistance artery diameter responses to alpha-adrenergic stimulation during pregnancy. Am J Physiol. 1993;264:H78–H85. doi: 10.1152/ajpheart.1993.264.1.H78. [DOI] [PubMed] [Google Scholar]
- Dallas A, Khalil RA. Ca2+ antagonist-insensitive coronary smooth muscle contraction involves activation of epsilon-protein kinase C-dependent pathway. Am J Physiol. 2003;285:C1454–C1463. doi: 10.1152/ajpcell.00066.2003. [DOI] [PubMed] [Google Scholar]
- Dalle Lucca JJ, Adeagbo AS, Alsip NL. Influence of oestrous cycle and pregnancy on the reactivity of the rat mesenteric vascular bed. Hum Reprod. 2000;15:961–968. doi: 10.1093/humrep/15.4.961. [DOI] [PubMed] [Google Scholar]
- Dantas MF, Urban M, Spray D, Catelli De Carvalho MH, Passaglia RD. Increased acetylcholine-induced vasodilation in pregnant rats: a role for gap junctional communication. Hypertension. 1999;34:937–942. doi: 10.1161/01.hyp.34.4.937. [DOI] [PubMed] [Google Scholar]
- Davenport AP, Maguire JJ. Pharmacology of renal endothelin receptors. Contrib Nephrol. 2011;172:1–17. doi: 10.1159/000328678. [DOI] [PubMed] [Google Scholar]
- Davidge ST, McLaughlin MK. Endogenous modulation of the blunted adrenergic response in resistance-sized mesenteric arteries from the pregnant rat. Am J Obstet Gynecol. 1992;167:1691–1698. doi: 10.1016/0002-9378(92)91763-z. [DOI] [PubMed] [Google Scholar]
- van Drongelen J, Hooijmans CR, Lotgering FK, Smits P, Spaanderman ME. Adaptive changes of mesenteric arteries in pregnancy: a meta-analysis. Am J Physiol. 2012;303:H639–H657. doi: 10.1152/ajpheart.00617.2011. [DOI] [PubMed] [Google Scholar]
- Félétou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture) Am J Physiol. 2006;291:H985–H1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM. EDHF: an update. Clin Sci. 2009;117:139–155. doi: 10.1042/CS20090096. [DOI] [PubMed] [Google Scholar]
- Flammer AJ, Luscher TF. Human endothelial dysfunction: EDRFs. Pflugers Arch. 2010;459:1005–1013. doi: 10.1007/s00424-010-0822-4. [DOI] [PubMed] [Google Scholar]
- Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol. 2003;284:R1–R12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- Furuhashi N, Kimura H, Nagae H, Yajima A. Maternal plasma endothelin levels and fetal status in normal and preeclamptic pregnancies. Gynecol Obstet Invest. 1995;39:88–92. doi: 10.1159/000292386. [DOI] [PubMed] [Google Scholar]
- Gandley RE, Conrad KP, McLaughlin MK. Endothelin and nitric oxide mediate reduced myogenic reactivity of small renal arteries from pregnant rats. Am J Physiol. 2001;280:R1–R7. doi: 10.1152/ajpregu.2001.280.1.R1. [DOI] [PubMed] [Google Scholar]
- George EM, Granger JP. Endothelin: key mediator of hypertension in preeclampsia. Am J Hypertens. 2011;24:964–969. doi: 10.1038/ajh.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George EM, Palei AC, Granger JP. Endothelin as a final common pathway in the pathophysiology of preeclampsia: therapeutic implications. Curr Opin Nephrol Hypertens. 2012;21:157–162. doi: 10.1097/MNH.0b013e328350094b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber RT, Anwar MA, Poston L. Enhanced acetylcholine induced relaxation in small mesenteric arteries from pregnant rats: an important role for endothelium-derived hyperpolarizing factor (EDHF) Br J Pharmacol. 1998;125:455–460. doi: 10.1038/sj.bjp.0702099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardina JB, Green GM, Rinewalt AN, Granger JP, Khalil RA. Role of endothelin B receptors in enhancing endothelium-dependent nitric oxide-mediated vascular relaxation during high salt diet. Hypertension. 2001;37:516–523. doi: 10.1161/01.hyp.37.2.516. [DOI] [PubMed] [Google Scholar]
- Hall JE, Guyton AC, Coleman TG, Mizelle HL, Woods LL. Regulation of arterial pressure: role of pressure natriuresis and diuresis. Fed Proc. 1986;45:2897–2903. [PubMed] [Google Scholar]
- Hilgers RHP, Webb RC. Molecular aspects of arterial smooth muscle contraction: focus on Rho. Exp Biol Med. 2005;230:829–835. doi: 10.1177/153537020523001107. [DOI] [PubMed] [Google Scholar]
- Himpens B, Kitazawa T, Somlyo AP. Agonist-dependent modulation of Ca2+-sensitivity in rabbit pulmonary artery smooth muscle. Pflugers Arch. 1990;417:21–28. doi: 10.1007/BF00370764. [DOI] [PubMed] [Google Scholar]
- Itoh T, Seki N, Suzuki S, Ito S, Kajikuri J, Kuriyama H. Membrane hyperpolarization inhibits agonist-induced synthesis of inositol 1,4,5-trisphosphate in rabbit mesenteric artery. J Physiol. 1992;451:307–328. doi: 10.1113/jphysiol.1992.sp019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny LC, Baker PN, Kendall DA, Randall MD, Dunn WR. The role of gap junctions in mediating endothelium-dependent responses to bradykinin in myometrial small arteries isolated from pregnant women. Br J Pharmacol. 2002;136:1085–1088. doi: 10.1038/sj.bjp.0704817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil RA. Regulation of vascular smooth muscle function. In: Granger N, Granger JP, editors. Colloquium Series on Integrated Systems Physiology: From Molecule to Function. San Rafael: Morgan & Claypool Life Sciences; 2010. pp. 1–62. [PubMed] [Google Scholar]
- Khalil RA, van Breemen C. Sustained contraction of vascular smooth muscle: calcium influx or C-kinase activation? J Pharmacol Exp Ther. 1988;244:537–542. [PubMed] [Google Scholar]
- Khalil RA, van Breemen C. Intracellular free calcium concentration/force relationship in rabbit inferior vena cava activated by norepinephrine and high K+ Pflugers Arch. 1990;416:727–734. doi: 10.1007/BF00370622. [DOI] [PubMed] [Google Scholar]
- Khalil RA, Lajoie C, Resnick MS, Morgan KG. Ca(2+)-independent isoforms of protein kinase C differentially translocate in smooth muscle. Am J Physiol. 1992;263:C714–C719. doi: 10.1152/ajpcell.1992.263.3.C714. [DOI] [PubMed] [Google Scholar]
- Khalil RA, Crews JK, Novak J, Kassab S, Granger JP. Enhanced vascular reactivity during inhibition of nitric oxide synthesis in pregnant rats. Hypertension. 1998;31:1065–1069. doi: 10.1161/01.hyp.31.5.1065. [DOI] [PubMed] [Google Scholar]
- Kim TH, Weiner CP, Thompson LP. Effect of pregnancy on contraction and endothelium-mediated relaxation of renal and mesenteric arteries. Am J Physiol. 1994;267:H41–H47. doi: 10.1152/ajpheart.1994.267.1.H41. [DOI] [PubMed] [Google Scholar]
- Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011;91:1–77. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang NN, Luksha L, Newby DE, Kublickiene K. Connexin 43 mediates endothelium-derived hyperpolarizing factor-induced vasodilatation in subcutaneous resistance arteries from healthy pregnant women. Am J Physiol. 2007;292:H1026–H1032. doi: 10.1152/ajpheart.00797.2006. [DOI] [PubMed] [Google Scholar]
- Lima VV, Giachini FR, Carneiro FS, Carvalho MH, Fortes ZB, Webb RC, et al. O-GlcNAcylation contributes to the vascular effects of ET-1 via activation of the RhoA/Rho-kinase pathway. Cardiovasc Res. 2011;89:614–622. doi: 10.1093/cvr/cvq338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmeier TE. The sympathetic nervous system and long-term blood pressure regulation. Am J Hypertens. 2001;14:147S–154S. doi: 10.1016/s0895-7061(01)02082-9. [DOI] [PubMed] [Google Scholar]
- Luksha L, Nisell H, Kublickiene K. The mechanism of EDHF-mediated responses in subcutaneous small arteries from healthy pregnant women. Am J Physiol. 2004;286:R1102–R1109. doi: 10.1152/ajpregu.00550.2003. [DOI] [PubMed] [Google Scholar]
- McQeen J, Kingdom JC, Connell JM, Whittle MJ. Fetal endothelin levels and placental vascular endothelin receptors in intrauterine growth retardation. Obstet Gynecol. 1993;82:992–998. [PubMed] [Google Scholar]
- Madsen KM, Neerhof MG, Wessale JL, Thaete LG. Influence of ET(B) receptor antagonism on pregnancy outcome in rats. J Soc Gynecol Investig. 2001;8:239–244. doi: 10.1016/s1071-5576(01)00120-4. [DOI] [PubMed] [Google Scholar]
- Magness RR, Rosenfeld CR. Systemic and uterine responses to alpha-adrenergic stimulation in pregnant and nonpregnant ewes. Am J Obstet Gynecol. 1986;155:897–904. doi: 10.1016/s0002-9378(86)80047-3. [DOI] [PubMed] [Google Scholar]
- Magness RR, Sullivan JA, Li Y, Phernetton TM, Bird IM. Endothelial vasodilator production by uterine and systemic arteries. VI. Ovarian and pregnancy effects on eNOS and NO(x) Am J Physiol. 2001;280:H1692–H1698. doi: 10.1152/ajpheart.2001.280.4.H1692. [DOI] [PubMed] [Google Scholar]
- Majed BH, Khalil RA. Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol Rev. 2012;64:540–582. doi: 10.1124/pr.111.004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzuca MQ, Khalil RA. Vascular endothelin receptor type B: structure, function and dysregulation in vascular disease. Biochem Pharmacol. 2012;84:147–162. doi: 10.1016/j.bcp.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzuca MQ, Wlodek ME, Dragomir NM, Parkington HC, Tare M. Uteroplacental insufficiency programs regional vascular dysfunction and alters arterial stiffness in female offspring. J Physiol. 2010;588:1997–2010. doi: 10.1113/jphysiol.2010.187849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzuca MQ, Tare M, Parkington HC, Dragomir NM, Parry LJ, Wlodek ME. Uteroplacental insufficiency programmes vascular dysfunction in non-pregnant rats: compensatory adaptations in pregnancy. J Physiol. 2012;590:3375–3388. doi: 10.1113/jphysiol.2012.230011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SR, LaMarca BB, Cockrell K, Granger JP. Role of endothelin in mediating soluble fms-like tyrosine kinase 1-induced hypertension in pregnant rats. Hypertension. 2010;55:394–398. doi: 10.1161/HYPERTENSIONAHA.109.141473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SR, LaMarca B, Cockrell K, Arany M, Granger JP. L-arginine supplementation abolishes the blood pressure and endothelin response to chronic increases in plasma sFlt-1 in pregnant rats. Am J Physiol. 2012;302:R259–R263. doi: 10.1152/ajpregu.00319.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naden RP, Rosenfeld CR. Effect of angiotensin II on uterine and systemic vasculature in pregnant sheep. J Clin Invest. 1981;68:468–474. doi: 10.1172/JCI110277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima M, Vanhoutte PM. Endothelin-1 and -3 cause endothelium-dependent hyperpolarization in the rat mesenteric artery. Am J Physiol. 1993;265:H2137–H2141. doi: 10.1152/ajpheart.1993.265.6.H2137. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol. 1990;259:C3–C18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- Nuedling S, van Eickels M, Allera A, Doevendans P, Meyer R, Vetter H, et al. 17 Beta-estradiol regulates the expression of endothelin receptor type B in the heart. Br J Pharmacol. 2003;140:195–201. doi: 10.1038/sj.bjp.0705409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson GL, Saade GR, Buhimschi I, Chwalisz K, Garfield RE. The effect of an endothelin antagonist on blood pressure in a rat model of preeclampsia. Am J Obstet Gynecol. 1999;181:638–641. doi: 10.1016/s0002-9378(99)70506-5. [DOI] [PubMed] [Google Scholar]
- Paller MS. Mechanism of decreased pressor responsiveness to ANG II, NE, and vasopressin in pregnant rats. Am J Physiol. 1984;247:H100–H108. doi: 10.1152/ajpheart.1984.247.1.H100. [DOI] [PubMed] [Google Scholar]
- Parent A, Schiffrin EL, St Louis J. Role of the endothelium in adrenergic responses of mesenteric artery rings of pregnant rats. Am J Obstet Gynecol. 1990;163:229–234. doi: 10.1016/s0002-9378(11)90703-0. [DOI] [PubMed] [Google Scholar]
- Pascoal IF, Lindheimer MD, Nalbantian-Brandt C, Umans JG. Contraction and endothelium-dependent relaxation in mesenteric microvessels from pregnant rats. Am J Physiol. 1995;269:H1899–H1904. doi: 10.1152/ajpheart.1995.269.6.H1899. [DOI] [PubMed] [Google Scholar]
- Poston L, McCarthy AL, Ritter JM. Control of vascular resistance in the maternal and feto-placental arterial beds. Pharmacol Ther. 1995;65:215–239. doi: 10.1016/0163-7258(94)00064-a. [DOI] [PubMed] [Google Scholar]
- Puetz S, Lubomirov LT, Pfitzer G. Regulation of smooth muscle contraction by small GTPases. Physiology (Bethesda) 2009;24:342–356. doi: 10.1152/physiol.00023.2009. [DOI] [PubMed] [Google Scholar]
- Reslan OM, Khalil RA. Molecular and vascular targets in the pathogenesis and management of the hypertension associated with preeclampsia. Cardiovasc Hematol Agents Med Chem. 2010;8:204–226. doi: 10.2174/187152510792481234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts L, LaMarca BB, Fournier L, Bain J, Cockrell K, Granger JP. Enhanced endothelin synthesis by endothelial cells exposed to sera from pregnant rats with decreased uterine perfusion. Hypertension. 2006;47:615–618. doi: 10.1161/01.HYP.0000197950.42301.dd. [DOI] [PubMed] [Google Scholar]
- Rosenfeld CR, Naden RP. Uterine and nonuterine vascular responses to angiotensin II in ovine pregnancy. Am J Physiol. 1989;257:H17–H24. doi: 10.1152/ajpheart.1989.257.1.H17. [DOI] [PubMed] [Google Scholar]
- Rupnow HL, Phernetton TM, Shaw CE, Modrick ML, Bird IM, Magness RR. Endothelial vasodilator production by uterine and systemic arteries. VII. Estrogen and progesterone effects on eNOS. Am J Physiol. 2001;280:H1699–H1705. doi: 10.1152/ajpheart.2001.280.4.H1699. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Yanagisawa M, Takuwa Y, Miyazaki H, Kimura S, Goto K, et al. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature. 1990;348:732–735. doi: 10.1038/348732a0. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL. Vascular endothelin in hypertension. Vascul Pharmacol. 2005;43:19–29. doi: 10.1016/j.vph.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL, Touyz RM. Vascular biology of endothelin. J Cardiovasc Pharmacol. 1998;32(Suppl. 3):S2–S13. [PubMed] [Google Scholar]
- Schneider MP, Boesen EI, Pollock DM. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annu Rev Pharmacol Toxicol. 2007;47:731–759. doi: 10.1146/annurev.pharmtox.47.120505.105134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa H, Yasutake H, Fujii K, Owada MK, Nakaike R, Fukumoto Y, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. 1996;28:703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Tam Tam KB, George E, Cockrell K, Arany M, Speed J, Martin JN, Jr, et al. Endothelin type A receptor antagonist attenuates placental ischemia-induced hypertension and uterine vascular resistance. Am J Obstet Gynecol. 2011;204:330–334. doi: 10.1016/j.ajog.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaete LG, Neerhof MG. Endothelin and blood pressure regulation in the female rat: studies in normal pregnancy and with nitric oxide synthase inhibition-induced hypertension. Hypertens Pregnancy. 2000;19:233–247. doi: 10.1081/prg-100100139. [DOI] [PubMed] [Google Scholar]
- Thornburg KL, Jacobson SL, Giraud GD, Morton MJ. Hemodynamic changes in pregnancy. Semin Perinatol. 2000;24:11–14. doi: 10.1016/s0146-0005(00)80047-6. [DOI] [PubMed] [Google Scholar]
- Tirapelli CR, Casolari DA, Yogi A, Montezano AC, Tostes RC, Legros E, et al. Functional characterization and expression of endothelin receptors in rat carotid artery: involvement of nitric oxide, a vasodilator prostanoid and the opening of K+ channels in ETB-induced relaxation. Br J Pharmacol. 2005;146:903–912. doi: 10.1038/sj.bjp.0706388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tostes RC, Fortes ZB, Callera GE, Montezano AC, Touyz RM, Webb RC, et al. Endothelin, sex and hypertension. Clin Sci (Lond) 2008;114:85–97. doi: 10.1042/CS20070169. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Deng LY, Schiffrin EL. Endothelin subtype B receptor-mediated calcium and contractile responses in small arteries of hypertensive rats. Hypertension. 1995;26:1041–1045. doi: 10.1161/01.hyp.26.6.1041. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens. 2004;22:1141–1149. doi: 10.1097/00004872-200406000-00015. [DOI] [PubMed] [Google Scholar]
- Tykocki NR, Watts SW. The interdependence of endothelin-1 and calcium: a review. Clin Sci (Lond) 2010;119:361–372. doi: 10.1042/CS20100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tykocki NR, Gariepy CE, Watts SW. Endothelin ET(B) receptors in arteries and veins: multiple actions in the vein. J Pharmacol Exp Ther. 2009;329:875–881. doi: 10.1124/jpet.108.145953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf) 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- Wigg SJ, Tare M, Tonta MA, O'Brien RC, Meredith IT, Parkington HC. Comparison of effects of diabetes mellitus on an EDHF-dependent and an EDHF-independent artery. Am J Physiol. 2001;281:H232–H240. doi: 10.1152/ajpheart.2001.281.1.H232. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- Yener T, Turkkani TA, Aslan H, Aytan H, Cantug CA. Determination of oestrous cycle of the rats by direct examination: how reliable? Anat Histol Embryol. 2007;36:75–77. doi: 10.1111/j.1439-0264.2006.00743.x. [DOI] [PubMed] [Google Scholar]
- Yin Z, Sada AA, Reslan OM, Narula N, Khalil RA. Increased MMPs expression and decreased contraction in the rat myometrium during pregnancy and in response to prolonged stretch and sex hormones. Am J Physiol. 2012;303:E55–E70. doi: 10.1152/ajpendo.00553.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]