

Abstract
Background and Purpose
PPAR-γ has been reported to be a protective regulator in ischaemia/reperfusion (I/R) injury. The receptor for advanced glycation end-products (RAGE) plays a major role in the innate immune response, and its expression is associated with PPAR-γ activation. Several angiotensin receptor blockers possess partial agonist activities towards PPAR-γ. Therefore, this study investigated the action of losartan, particularly with regard to PPAR-γ activation and RAGE signalling pathways during hepatic I/R.
Experimental Approach
Mice were subjected to 60 min of ischaemia followed by 6 h of reperfusion. Losartan (0.1, 1, 3 and 10 mg·kg−1) was administered 1 h prior to ischaemia and immediately before reperfusion. GW9662, a PPAR-γ antagonist, was administered 30 min prior to first pretreatment with losartan.
Key Results
Losartan enhanced the DNA-binding activity of PPAR-γ in I/R. Losartan attenuated the increased serum alanine aminotransferase activity, TNF-α and IL-6 levels, and nuclear concentrations of NF-κB in I/R. GW9662 reversed these beneficial effects. Losartan caused a decrease in apoptosis as assessed by TUNEL assay, in release of cytochrome c and in cleavage of caspase-3, and these effects were abolished by GW9662 administration. Losartan attenuated not only I/R-induced RAGE overexpression, but also its downstream early growth response protein-1-dependent macrophage inflammatory protein 2 level; phosphorylation of p38, ERK and JNK; and subsequent c-Jun phosphorylation. GW9662 reversed these effects of losartan administration.
Conclusions and Implications
Our findings suggest that losartan ameliorates I/R-induced liver damage through PPAR-γ activation and down-regulation of the RAGE signalling pathway.
Keywords: ischaemia/reperfusion, liver, losartan, RAGE, PPAR-γ
Introduction
Ischaemia/reperfusion (I/R) injury develops in the absence of exogenous antigen, and innate immunity has been recognized as playing a dominant role in its pathology (Fondevila et al., 2003). I/R causes inflammation, apoptosis and tissue damage, leading to organ dysfunction. Hepatic I/R injury is commonly encountered in a variety of clinical settings, such as trauma, shock, liver transplantation and electric liver resection.
PPAR belongs to the hormone nuclear receptor superfamily. PPAR-γ plays a critical role in lipoprotein metabolism. PPAR-γ agonists, such as pioglitazone, troglitazone and rosiglitazone, have been widely used as antidiabetic agents (Rosen et al., 1999). In addition to antidiabetic activity, PPAR-γ has been reported to be a protective regulator in tissue protection and repair, particularly in ischaemic injury (Abdelrahman et al., 2005). In the ischaemic liver, PPAR-γ rapidly decreases and remains suppressed throughout the reperfusion period (Kuboki et al., 2008). Treatment with a PPAR-γ agonist not only protected against post-ischaemic injury, but also improved survival in lethally I/R-injured mice (Akahori et al., 2007). Increasing evidence indicates that several angiotensin receptor blockers (ARBs) possess partial agonist activities towards PPAR-γ (Schupp et al., 2004; Horiuchi and Mogi, 2011; Amano et al., 2012). ARBs inhibit LPS-induced pro-inflammatory response through PPAR-γ activation in human monocytes (Pang et al., 2012). In addition, ARBs reduce oxidative stress and restore blood flow in ischaemic regions of the heart, in part via the PPAR-γ pathway (Goyal et al., 2011).
The receptor for advanced glycation end-products (RAGE), a member of the immunoglobulin superfamily, is linked to amplification of the inflammatory response and implicated in the pathogenesis of various devastating disorders, such as diabetic vascular complications, cancer growth and metastasis, and non-alcoholic fatty liver disease (Hyogo and Yamagishi, 2008; Tontonoz and Spiegelman, 2008). Some studies have found that damage-associated molecular proteins (DAMPs), released during ischaemia or tissue injury, interact with RAGE to result in propagation of stress signals including MAPK and c-Jun, thereby leading to induction of inflammatory cytokines and liver damage (Zeng et al., 2009). Indeed, soluble RAGE, consisting of the extracellular ligand-binding domain of RAGE, is highly protective against hepatocellular death and necrosis in I/R (Zeng et al., 2004). Attention has increasingly focused on the relation between RAGE and PPAR-γ activation. Marx et al. (2004) reported that a PPAR-γ agonist reduced RAGE expression levels in human endothelial cells. Further, PPAR-γ agonists inhibit RAGE expression at the site of injury, while down-regulation of RAGE by PPAR-γ activation inhibits neointimal formation in response to arterial injury (Wang et al., 2006).
Losartan (2-N-butyl-4-chloro-5-hydroxymethyl-1-[(2′-(1H-tetrazol-5-yl)biphenyl-4-yl)methyl]imidazole) is a typical angiotensin II (Ang II) type I receptor (AT1R) blocker that has been used for regulation of blood pressure and fluid homeostasis. As Ang II increases vascular permeability, stimulates inflammatory cell recruitment and activates pro-inflammatory chemokines and cytokines, several AT1R blockers, including losartan, have been shown to be protective in various experimental I/R models (Suzuki et al., 2003; Guo et al., 2004). Indeed, losartan abates hepatic I/R injury by inhibition of hepatic necrosis and apoptosis, neutrophil infiltration, and intercellular adhesion molecule (ICAM)-1 expression (Ramalho et al., 2009). Telmisartan, another ARB, inhibits advanced AGE-induced monocyte chemoattractant protein-1 expression in mesangial cells through down-regulation of RAGE via PPAR-γ activation (Matsui et al., 2007). These considerations led us to hypothesize that the hepatoprotective properties of losartan in I/R may be associated with PPAR-γ activation and RAGE down-regulation.
Therefore, the present study was undertaken to elucidate the molecular mechanisms involved in the protective effects of losartan in hepatic I/R, particularly focusing on PPAR-γ-mediated RAGE signalling.
Methods
All drug/molecular target nomenclature conforms with BJP's Guide to Receptors and Channels (Alexander et al., 2011).
Hepatic I/R procedure
Male C57BL/6 mice (8–10 weeks) were kept in a temperature- and humidity-controlled room (25 ± 1°C and 55 ± 5%, respectively) under a 12 h light–dark cycle. All animal protocols were approved by the Animal Care Committee of Sungkyunkwan University and performed in accordance with the guidelines of the National Institutes of Health. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Mice were fasted for 18 h before the experiments but were provided with tap water ad libitum. Under ketamine (55 mg·kg−1, i.p.) and xylazine (7 mg·kg−1, i.p.) anaesthesia, a midline incision was made to the abdomen, and the left branches of the portal vein and hepatic artery were clamped to induce complete ischaemia of the median and left hepatic lobes. The right lobes remained perfused to prevent intestinal congestion. After 60 min of ischaemia, the clip around the left branches of the portal vein was removed to allow reperfusion. Vehicle- and losartan-treated sham groups underwent midline laparotomy as I/R groups; however, a clip was not placed on the vasculature leading to the median and left lobes. All animals received subcutaneous administration of 0.5 mL of normal saline immediately after the operation (i.e. fluid resuscitation). After 6 h of reperfusion, the mice were killed, and blood and ischaemic liver tissue were collected. Liver tissue was analysed immediately by histological staining (aliquots from the left lobe), and the remaining major portions of the liver tissues were frozen in liquid nitrogen and kept at −75°C until biochemical analyses.
Experimental design
Losartan (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in PBS (vehicle) and administered intraperitoneally (0.1, 1, 3 and 10 mg·kg−1) 1 h prior to ischaemia and immediately before reperfusion. The dosage and time point of losartan administration were selected based on previously published reports (Kuboki et al., 2008; Zhang et al., 2012) and our preliminary study. GW9662 (2-chloro-5-nitro-N-phenylbenzamide; Sigma-Aldrich), a PPAR-γ antagonist, was dissolved in 4% dimethyl sulfoxide (Calbiochem, La Jolla, CA, USA) and administered intraperitoneally (4 mg·kg−1) 30 min prior to the first pretreatment with losartan. The animals were randomly assigned to the following nine groups: (a) vehicle-treated sham (sham; n = 5); (b) losartan-treated sham (n = 5); (c) GW9662-treated sham (GW9662; n = 5); (d) vehicle-treated I/R (I/R; n = 10); (e–h) losartan-treated I/R (0.1, 1, 3 and 10 mg·kg−1) (los + I/R; n = 10 for each group); and (i) GW9662 plus losartan-treated I/R (GW9662 + los + I/R; n = 10). As there were no differences found in any of the parameters between vehicle- and losartan-treated mice in the sham groups, the results of group (a) and (b) were pooled and are hereafter referred to as ‘sham’.
Serum aminotransferase activities
The levels of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined by standard spectrophotometric procedures using the ChemiLab ALT and AST assay kits (IVDLab Co., Ltd, Uiwang, Korea) respectively.
Histological analysis
Liver tissues were removed from a portion of the left lobe, fixed immediately in 10% neutral buffered formalin, embedded in paraffin and cut serially into 5 μm sections. Haematoxylin and eosin (H&E)-stained sections were evaluated using an optical microscope (Olympus Optical Co., Tokyo, Japan). The histological changes were evaluated at ×200 magnification by a point-counting method for severity of hepatic injury, using an ordinal scale according to the method described by Camargo et al. (1997). The stained sections were graded as follows: grade 0, minimal or no evidence of injury; grade 1, mild injury with cytoplasmic vacuolation and focal nuclear pycnosis; grade 2, moderate to severe injury with extensive nuclear pycnosis, cytoplasmic hypereosinophilia and loss of intercellular borders; grade 3, severe necrosis with disintegration of hepatic cords, haemorrhage and neutrophil infiltration. Apoptotic cells were detected by TUNEL staining with a commercially available kit (In Situ Apoptosis Detection Kit; Takara Bio Inc., Shiga, Japan). Under microscopy, the number of TUNEL-positive cells in ×200 histological fields was counted per liver section.
Isolation of total, cytosolic and nuclear proteins
PRO-PREP® (iNtRON Biotechnology, Seongnam, Korea) and NE-PER® (Pierce Biotechnology, Rockford, IL, USA) were used for extraction of total, nuclear and cytosolic fractions according to the manufacturer's instructions. Protein concentrations were determined using the BCA Protein Assay kit (Pierce Biotechnology).
Western blot analysis
Protein samples were loaded on 7.5–18% polyacrylamide gels and were then separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes using the Semi-Dry Trans-Blot Cell (Bio-Rad Laboratories, Hercules, CA, USA). After transfer, the membranes were washed with 0.1% Tween-20 in 1 × Tris-buffered saline (TBS/T) and blocked for 1 h at room temperature with 5% skim milk powder in TBS/T. Blots were then incubated overnight at 4°C with primary antibodies. After being washed three times for 5 min each in TBS/T, the membranes were incubated with appropriate secondary antibodies for 1 h at room temperature, followed by detection using an enhanced chemiluminescence detection system (iNtRON Biotechnology), according to the manufacturer's instructions. Intensity of the immune-reactive bands was determined using TotalLab TL120 software (Nonlinear Dynamics Ltd, Newcastle, UK). Primary antibodies against mouse RAGE, early growth response protein 1 (Egr-1), macrophage inflammatory protein 2 (MIP-2), c-Jun p39 phosphorylated (p-) on serine 63, NF-κB/p65, PPAR-γ (SantaCruz Biotechnology, Santa Cruz, CA, USA), cytochrome c (Abcam, Cambridge, MA, USA), caspase-3, cleaved caspase-3, p-p38, p-JNK, p-ERK, total p38, total JNK, and total ERK (Cell Signaling Technology, Beverly, MA, USA) were used, and the signals were standardized to β-actin (Sigma-Aldrich) or lamin B1 (Abcam).
Electrophoretic mobility shift assay (EMSA)
Two micrograms of nuclear protein were pre-incubated with EMSA binding buffer (Panomics, Redwood City, CA, USA) and 1 μg poly (deoxyinosinic-deoxycytidylic) acid for 5 min at room temperature. Ten nanograms of biotinylated PPAR-γ element probe (Panomics, 5′-TGAAACTAGGGTAAAGTTC-3′), with or without an excess of unlabelled competitor DNA, were added and incubated at 15°C for 30 min. Specific binding of the PPAR-γ to labelled PPAR-γ was assessed by introducing unlabelled PPAR-γ at a 66-fold molar excess. After incubation, reaction products were separated on a 6% non-denaturing polyacrylamide gel in 1 × Tris-borate EDTA buffer. The gel was run in an ice-water bath for 55 min at 120 V and transferred to a Biodyne B nylon membrane (Pierce Biotechnology) for 30 min at 300 mA. Chemiluminescent detection of biotinylated DNA was performed using the Panomics EMSA kit according to the manufacturer's directions.
Serum cytokine levels
Commercial TNF-α and IL-6 elisa kits (BD Biosciences Co., CA, USA) were used for quantification of the serum levels of TNF-α and IL-6, respectively.
siRNA gene silencing of RAGE
The siRNAs for RAGE and non-specific control were purchased from Bioneer (AccuTarget™ predesigned siRNA; Daejeon, Korea). RAGE siRNA (90 μg) was diluted in 50 μL of a 10% glucose solution, and the volume was adjusted to 100 μL using RNase/DNase-free water. In a separate tube, 14.4 μL of in vivo-jetPEI® (Polyplus Transfection, Illkirch, France) was diluted in 50 μL of 10% glucose solution, and the volume was adjusted to 100 uL. The solutions were mixed and incubated for 15 min to allow the complexes to form. RAGE or non-specific control siRNAs were injected via the tail twice in 3 days. Mice were subjected to I/R 2 days after the last siRNA injection.
Reverse transcription PCR (RT-PCR)
Tissues were homogenized in RNAiso (Takara Bio Inc.), and total RNA was prepared according to the manufacturer's protocol. Reverse transcription of total RNA was performed for synthesis of the first strand of cDNA using a Takara RNA PCR Kit (AMV) Ver. 3.0 (Takara Bio Inc.). PCR was carried out at a 20 μL reaction volume with a diluted cDNA sample. The final reaction concentrations and volume were as follows: primers, 0.5 μM; dNTP mix, 0.25 mM; 10× Ex Taq buffer, 1 μL; Ex Taq HS DNA polymerase, 0.25 U/reaction. Gene-specific primers used were RAGE (NM_007425, sense CTTGCTCTATGGGGAGCTGTA, antisense CATCGACAATTCCAGTGGCTG) and β-actin (NM_007393, sense TGGAATCCTGTGGCATCCATGAAA, antisense TAAAACGCAGCTCAGTAACAGTCCG).
Statistical analysis
All results are reported as the mean ± SEM. The overall significance of the data was examined by one- or two-way anova. The differences among the groups were considered significant at P < 0.05, with the appropriate Bonferroni correction made for multiple comparisons. All statistical analyses were performed with the Statistical Package for the Social Sciences (SPSS) 12.0 for Windows (SPSS Inc., Chicago, IL, USA).
Results
Effect of losartan on PPAR-γ activation and expression
To examine whether losartan affects the DNA binding of PPAR-γ, we used an EMSA assay. PPAR-γ was constitutively activated in normal liver, and DNA binding of PPAR-γ markedly decreased after I/R. The decrease in the DNA binding of PPAR-γ was attenuated by losartan, and GW9662, a PPAR-γ antagonist, reversed this effect (Figure 1A). However, there were no significant differences in PPAR-γ protein levels in any of the experimental groups (Figure 1B).
Figure 1.
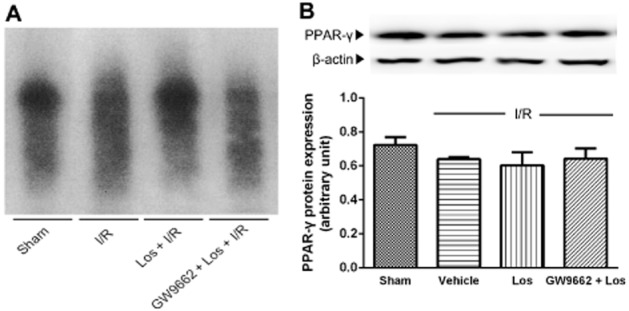
Effects of losartan (10 mg·kg−1) on PPAR-γ DNA binding activity and protein expression. (A) PPAR-γ DNA binding activity was measured by EMSA. (B) Protein expression of PPAR-γ was determined by Western blot. Mice were intraperitoneally administered GW9662 (4 mg·kg−1) 30 min prior to pretreatment with losartan. Losartan was administered intraperitoneally 1 h prior to ischaemia and immediately before reperfusion. The values are represented as mean ± SEM.
Effects of losartan and PPAR-γ antagonist on hepatic injury
We studied the impact of losartan on release of enzymes such as ALT and AST, which are serum markers of hepatocyte necrosis, to evaluate hepatic injury during I/R. The levels of serum ALT and AST activities in the sham group were 29.6 ± 1.6 U·L−1 and 139.6 ± 7.7 U·L−1 respectively. In the ischaemic group, the levels of serum ALT and AST activities showed a significant increase to 8914.6 ± 426.6 U·L−1 and 14656.2 ± 1152.7 U·L−1, respectively at 6 h after reperfusion. These increases were attenuated by losartan at doses of 1, 3 and 10 mg·kg−1 (Figure 2A,B). Moreover, the increases of serum TNF-α and IL-6 levels induced by I/R were attenuated by losartan at 1, 3 and 10 mg·kg−1 in a dose-dependent manner (Figure 2C,D). Therefore, losartan at 10 mg·kg−1 was selected as the optimal effective dose for evaluating the molecular mechanisms of losartan against I/R-induced hepatocellular damage in our study. We next examined whether this protection is related to PPAR-γ activation. After a 6 h reperfusion, the levels of serum ALT and AST activities increased to 266-fold and 108-fold of those in the sham group, respectively, and losartan attenuated these increases. GW9662 reduced the effect of losartan treatment (Figure 3A,B). The histological features shown in Figure 3E indicate normal lobular architecture and cell structure in the livers of the sham groups. However, livers exposed to I/R showed apparent broad haemorrhagic necrosis, extensive areas of portal inflammation and moderate increase in inflammatory cell infiltration at 6 h after reperfusion. This histological damage was ameliorated by losartan. GW9662 blocked this beneficial effect of losartan (Figure 3F).
Figure 2.
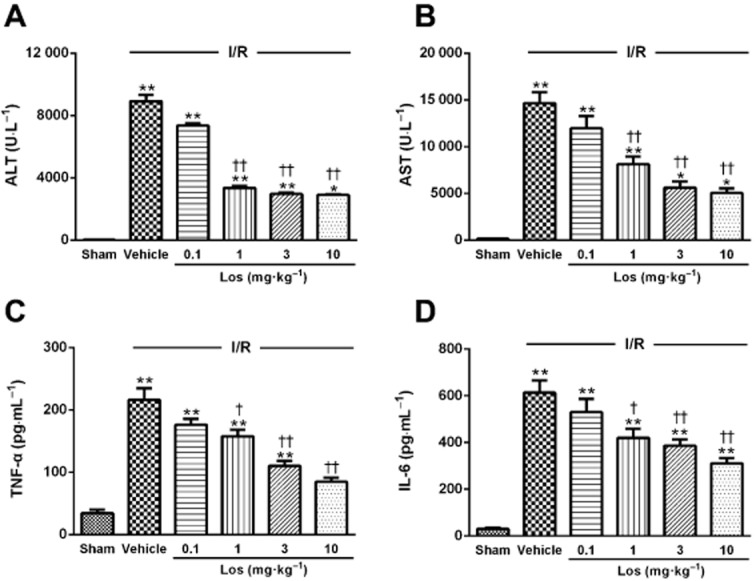
Effects of losartan on serum (A) ALT and (B) AST activities and serum (C) TNF-α and (D) IL-6 levels. The values are represented as mean ± SEM. *P < 0.05, **P < 0.01 versus sham group; †P < 0.05, ††P < 0.01 versus I/R group.
Figure 3.

Effects of losartan (10 mg·kg−1) and PPAR-γ on serum (A) ALT and (B) AST activities, serum (C) TNF-α and (D) IL-6 levels, (E) histological changes and (F) grading of histopathological damage 6 h after reperfusion. Histological features of liver sections stained with H&E at 6 h after reperfusion. Typical images were chosen from each experimental group (original magnification ×200). (i) Sham; (ii) I/R; (iii) Los (10 mg·kg−1) + I/R; and (iv) GW9662 + Los + I/R. The values are represented as mean ± SEM. **P < 0.01 versus sham group; ††P < 0.01 versus I/R group; #P < 0.05, ##P < 0.01 versus Los + I/R group.
Effects of losartan and PPAR-γ antagonist on serum TNF-α and IL-6 levels
The levels of serum TNF-α and IL-6 in the sham group were 46.7 ± 2.0 pg·mL−1 and 13.9 ± 3.5 pg·mL−1, respectively. After 6 h of reperfusion, serum levels of TNF-α and IL-6 showed significant increases, to 194.6 ± 4.9 pg·mL−1 and 509.9 ± 39.2 pg·mL−1 respectively. Losartan attenuated this increased serum TNF-α level, and GW9662 reversed the effect of losartan. The increased IL-6 level was attenuated by losartan, and GW9662 reduced the effect of losartan (Figure 3C,D). However, GW9662 alone did not affect any of the parameters used in this study.
Effects of losartan and PPAR-γ antagonist on NF-κB translocation
After 6 h of reperfusion, the level of nuclear NF-κB/p65 protein content markedly increased, to twofold compared with that of the sham group. Losartan attenuated this increased nuclear NF-κB/p65 translocation, and GW 9662 blocked the effect of losartan (Figure 4).
Figure 4.
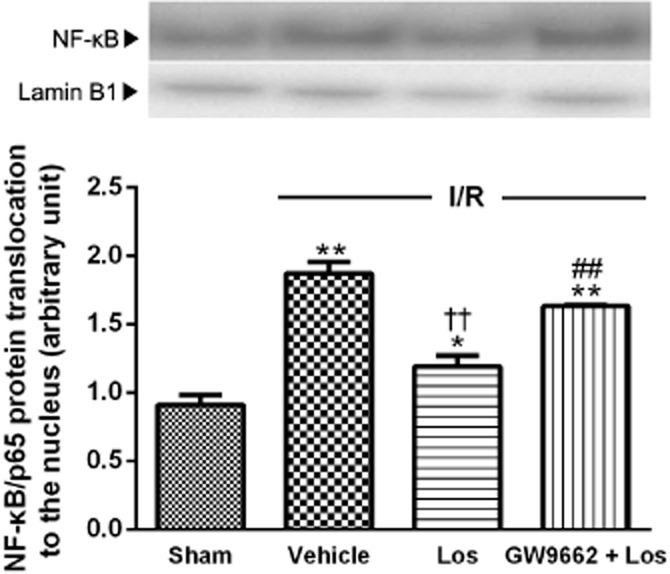
Effects of losartan (10 mg·kg−1) and PPAR-γ on nuclear NF-κB/p65 content. Western blot analysis for NF-κB was performed on nuclear extracts from the liver. The values are represented as mean ± SEM. *P < 0.05, **P < 0.01 versus sham group; ††P < 0.01 versus I/R group; ##P < 0.01 versus Los + I/R group.
Effects of losartan and PPAR-γ antagonist on apoptosis
We assessed apoptosis (by TUNEL assay), release of cytochrome c and cleavage of caspase-3 to examine the effects of losartan and PPAR-γ on apoptosis in liver remnant after I/R. In the ischaemic group, a large number of TUNEL-positive hepatocytes were observed. However, few TUNEL-positive hepatocytes were observed in livers treated with losartan. GW9662 reduced the beneficial effect of losartan (Figure 5A,B). Furthermore, I/R significantly increased the levels of cytochrome c and cleaved caspase-3 protein content, to 1.56 times and 3.85 times those of the sham group respectively (Figure 5C). The increases in release of cytochrome c and cleavage of caspase-3 were attenuated by losartan. GW9662 reversed the beneficial effect of losartan (Figure 5D).
Figure 5.

Effects of losartan (10 mg·kg−1) and PPARγ on apoptosis. (A) TUNEL-stained histology (original magnification ×200). (i) Sham; (ii) I/R; (iii) Los (10 mg·kg−1) + I/R; and (iv) GW9662 + Los (10 mg·kg−1) + I/R. (B) TUNEL-positive cell numbers were determined in randomly chosen ×200 histological fields. (C) Western blot analysis for cleaved-caspase 3 was performed in the whole liver lysate, and that for (D) cytochrome c was performed on the cytosolic extracts from liver. The values are represented as mean ± SEM. *P < 0.05, **P < 0.01 versus sham group; ††P < 0.01 versus I/R group; #P < 0.05, ##P < 0.01 versus Los + I/R group.
Effect of losartan and PPAR-γ antagonist on RAGE protein content
The level of RAGE protein content in sham animals was very low. After 6 h of reperfusion, the level of RAGE protein showed a significant increase, to 4.85 times that of sham animals. Losartan attenuated this increased RAGE protein level. However, GW 9662 blocked the effect of losartan (Figure 6).
Figure 6.
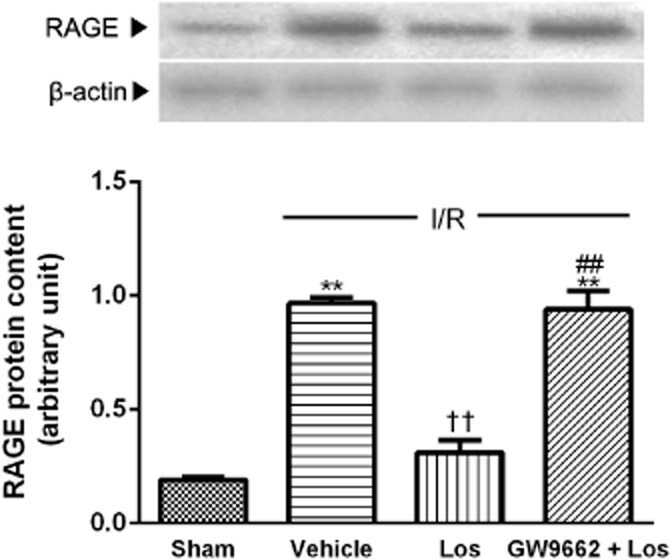
Role of PPAR-γ in effect of losartan (10 mg·kg−1) on RAGE content. RAGE protein level in the liver was measured by Western blot. The values are represented as mean ± SEM. **P < 0.01 versus sham group; ††P < 0.01 versus I/R group; ##P < 0.01 versus Los + I/R group.
Effects of losartan and PPAR-γ antagonist on MAPKs and c-Jun activation
The levels of p38, ERK and JNK phosphorylation in the sham animals were low. After 6 h of reperfusion, phosphorylation of p38, ERK and JNK showed a significant increase, to 6.08 times, 1.59 times and 3.03 times those of the sham group respectively. These increases were attenuated by losartan, and GW9662 abolished the effect of losartan (Figure 7A). Nuclear levels of phosphorylated c-Jun showed marked increase, 1.55 times that of the sham group. This increase was attenuated by losartan; the effect was reversed by GW9662 (Figure 7B).
Figure 7.

Effects of losartan (10 mg·kg−1) and PPAR-γ on MAPK and c-Jun activation. (A) P38, ERK and JNK activation in the whole liver were measured by Western blot. (B) Western blot analysis for p-c-Jun was performed on nuclear extracts from the liver. The values are represented as mean ± SEM. *P < 0.05, **P < 0.01 versus sham group; †P < 0.05, ††P < 0.01 versus I/R group; #P < 0.05, ##P < 0.01 versus Los + I/R group.
Effects of losartan and PPAR-γ antagonist on Egr-1 and MIP-2
At 6 h after reperfusion, the nuclear Egr-1 and MIP-2 protein levels increased to 2.28 times and 3.45 times those of the sham animals respectively. These increases were attenuated by losartan, and GW9662 reversed the effect of losartan (Figure 8).
Figure 8.
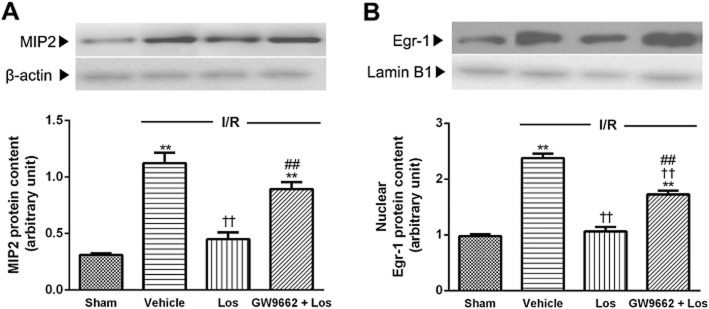
Effects of losartan (10 mg·kg−1) and PPAR-γ on MIP-2 and Egr-1. (A) Western blot analysis for MIP-2 was performed on the whole liver lysate, and that for (B) Egr-1 was performed on nuclear extracts from the liver. The values are represented as mean ± SEM. **P < 0.01 versus sham group; ††P < 0.01 versus I/R group; ##P < 0.01 versus Los + I/R group.
Effect of losartan on hepatocellular damage and MAPK and Egr-1 activation in RAGE knockdown mice
Involvement of RAGE in the protective effects of losartan on hepatic I/R was further confirmed by the transfection studies. Efficacy of transfection of siRNA is shown in Figure 9A,B. Non-specific control siRNA transfection did not affect any parameters with regard to the effect of losartan on I/R-induced hepatic injury. As shown in Figure 9C,D, RAGE siRNA blunted I/R-induced increases of serum ALT and AST activities compared with those of the group treated with non-specific control siRNA. Losartan did not affect serum aminotransferase activities in the group treated with RAGE siRNA. The increased serum TNF-α and IL-6 levels were blocked by RAGE siRNA, and losartan did not affect serum TNF-α and IL-6 levels in the group treated with RAGE siRNA (Figure 9E,F). As expected, knockdown of RAGE with siRNA modulated MAPK and Egr-1 signals. Figure 9G–K showed that transfection with RAGE siRNA reversed I/R-stimulated MAPK activation and MIP-2 and Egr-1 levels, and losartan did not affect MAPK, MIP-2 and Egr-1 levels in the group treated with RAGE siRNA.
Figure 9.

Effect of losartan (10 mg·kg−1) on hepatocellular damage, MAPK and Egr-1 activation in RAGE knockdown mice. (A) RAGE protein level and (B) mRNA level were measured by Western blot and RT-PCR respectively. The serum (C) ALT and (D) AST activities were determined by spectrophotometric procedures. The serum concentrations of (E) TNF-α and (F) IL-6 were determined using elisa. (G) P38, (H) ERK, (I) JNK activations in the whole liver were measured by Western blot. (J) Western blot analysis for Egr-1 was performed on the nuclear extracts from the liver, and that for (K) MIP2 was performed in the whole liver lysate. The values are represented as mean ± SEM. *P < 0.05, **P < 0.01 versus control siRNA-treated sham group; ††P < 0.01 versus control siRNA-treated I/R group; #P < 0.05, ##P < 0.01 versus RAGE siRNA-treated sham group.
Discussion and conclusions
In addition to being a physiological mediator restoring circulatory integrity, Ang II is involved in key events of the inflammatory process. Previous studies suggest that the renin–angiotensin system (RAS) is involved in the acute inflammatory response in liver I/R, including expression of inflammatory mediators and subsequent hepatic damage (Guo et al., 2004; Padrissa-Altes et al., 2009). Based on these studies, we examined the protective effect of losartan, an ARB, against hepatic I/R and sought molecular evidence for a role for losartan in hepatoprotection.
PPAR-γ plays important roles in the regulation of proliferation and differentiation of several cell types, including adipose cells, and has anti-inflammatory properties. In hamster liver, LPS induces a marked decrease in the expression of PPAR-γ (Beigneux et al., 2000). Previous studies have shown that endogenous PPAR-γ mediates anti-inflammatory activity in I/R injury by inhibition of the NF-κB pathway and that activation of PPAR-γ inhibits the release of TNF, IL-1 and IL-6 by macrophages (Nakajima et al., 2001). Ischaemia causes a rapid decrease in PPAR-γ DNA binding, and administration of a PPAR-γ agonist inhibits I/R injury (Kuboki et al., 2008). Losartan markedly stimulates PPAR-γ activity and attenuates LPS-induced expression of pro-inflammatory genes via PPAR-γ activation in macrophages (An et al., 2010). In the present study, I/R caused a decrease in PPAR-γ DNA binding activity; however, the reduction was not a result of decreased protein expression. These results were inconsistent with previous reports indicating increased PPAR-γ protein expression at 4 and 8 h of reperfusion after 90 min of hepatic ischaemia (Kuboki et al., 2008). These different results for PPAR-γ protein expression could be explained, at least in part, by the different ischaemia experimental models elucidated and the degree of hepatic injury. Losartan enhanced the DNA binding activity of PPAR-γ, and GW9662 blocked these effects. Although there is some disputation concerning the change in PPAR-γ protein expression in hepatic I/R injury (Kuboki et al., 2008; Shin et al., 2008), the deleterious role of decreased PPAR-γ activation is apparent (Akahori et al., 2007). The reduction of PPAR-γ DNA binding could result from either decreased endogenous PPAR-γ ligands or reduced interaction with a co-activator, such as p300 (Kuboki et al., 2008). Further studies will be required to clarify the precise mechanism by which stimulation of PPAR-γ protects the liver during I/R.
In our study, I/R markedly increased serum ALT, AST, TNF-α and IL-6 levels and hepatic nuclear translocation of NF-κB. These alterations were attenuated by losartan, and GW9662 reversed the effects of losartan. However, treatment of PPAR-γ antagonist did not completely prevent the hepatoprotective effect of losartan. Thus, we may infer that mechanisms other than PPAR-γ, such as RAS inhibition or bradykinin B2 receptor agonism, might be involved in the protection afforded by losartan (Bonde et al., 2011). The histological observations of the liver samples strongly support the release of aminotransferases and pro-inflammatory cytokines by the damaged hepatic cells as well as the protective effect of losartan. Furthermore, livers obtained from mice undergoing I/R showed multiple and extensive areas of hepatocyte necrosis randomly distributed throughout the parenchyma, while only mild centrizonal necrosis and Kupffer cell hyperplasia were observed in the losartan-treated groups. GW9662 reversed the beneficial effect of losartan. Overall, our results suggest that suppression of inflammatory responses associated with PPAR-γ activation is responsible for the hepatoprotective effect of losartan.
Apoptosis is a prominent factor contributing to cell death during hepatic I/R injury. PPAR-γ agonists induce apoptosis in a wide variety of cells including hepatocytes, as shown in several in vitro studies (Guo et al., 2006). In in vivo I/R, however, pretreatment with PPAR-γ ligands reduces apoptosis. Rosiglitazone reduces apoptotic caspases in cerebral ischaemia in a PPAR-γ-dependent manner, and trioglitazone reduces apoptosis in renal I/R injury (Doi et al., 2007; Wu et al., 2009). In our study, I/R increased release of cytochrome c and cleavage of caspase-3, and these increases were attenuated by losartan. However, the PPAR-γ antagonist blocked the effects of losartan. Our observations of the biochemical parameters for apoptotic cell death were supported by the morphological changes observed with TUNEL staining. As TUNEL staining may reflect necrotic or autolytic cells as well as apoptotic cells, the TUNEL data for the GW9662 group were weaker than those for changes in caspase-3 cleavage and cytochrome c release. This observation indicates that losartan could prevent apoptosis via PPAR-γ activation.
RAGE is a family of pattern recognition receptors that are activated by specific components of microbes, referred to as pathogen-associated molecular patterns, and certain other molecules, such as DAMPs. High mobility group box 1, released in the early phase of I/R injury, is known to stimulate RAGE signalling and amplify the inflammatory response (Schmidt et al., 2001). Pharmacological blockage of RAGE or genetic deletion or modulation of RAGE-mediated signalling reduces apoptosis in myocardial and liver I/R injuries (Bucciarelli et al., 2008; Zeng et al., 2009). Interestingly, the hepatoprotective effect of losartan was blocked by siRNA for RAGE in our study. PPAR-γ agonists down-regulate RAGE expression through NF-κB inhibition in human endothelial cells and suppress renal RAGE expression in proximal tubular cell injury (Marx et al., 2004; Tang et al., 2006). Losartan suppresses RAGE and NF-κB activation and improved endothelial function in spontaneous hypertension rats (Zhu et al., 2007). In the present study, RAGE expression showed a marked increase after reperfusion. Losartan attenuated the increased level of RAGE expression, and GW9662 reversed this effect. Our results indicate that losartan down-regulates RAGE expression through PPAR-γ activation.
RAGE activation by myriad ligands is linked to an array of signalling pathways. MAPK signal transduction pathways represent one of the most widespread mechanisms of cell regulation in response to oxidative and other environmental stress, leading to up-regulation of pro-inflammatory mediators such as cytokines and chemokines and to cell death (Bradham et al., 1997). In hepatic I/R, RAGE ligation causes cellular activation via signalling cascades, including MAPK and c-Jun, leading to induction of inflammatory cytokines and liver damage (Zeng et al., 2004). Activation of p38 and JNK MAPK is related to apoptotic cell death from reactive oxygen species (Cross et al., 2000). Although the classical ERK pathway is generally involved in cell growth, proliferation and survival, the involvement of ERK activation in the mechanism underlying ischaemia-induced cell death has been reported (Irving and Bamford, 2002). PPAR-γ agonists show anti-inflammatory effects through inhibition of p38 and p42/44 MAPK phosphorylation in cerebral I/R (Collino et al., 2006). In our study, I/R significantly increased activation of p38, ERK, and JNK MAPKs and c-Jun. These changes were attenuated by pretreatment with losartan, which was reversed by GW9662.
Egr-1, a zinc-finger transcription factor, functions as a master switch, activated by ischaemia to trigger expression of pivotal regulators of inflammation. Among the immediate response genes, Egr-1 induces various chemokines, as well as the cytokines TNF-α, IL-1β, ICAM-1 and MIP-2 (Yan et al., 2000). Regulation of Egr-1 in hypoxia is mediated by RAGE-dependent MAPK activation (Chang et al., 2008). Recently, Zeng et al. (2009) reported that Egr-1 is a downstream target of RAGE in hepatic I/R injury. In our study, the levels of Egr-1 and MIP-2 expression were increased after I/R, and losartan attenuated these increases, which was reversed by GW9662. Furthermore, I/R-induced MAPK and Egr-1 activation was blocked by siRNA for RAGE. Losartan did not affect MAPK and Egr-1 activation in animals treated with RAGE siRNA. Taken together, our results may imply that losartan inhibits the RAGE-mediated MAPK and Egr-1-pathways through PPAR-γ activation.
In conclusion, these observations suggest that losartan protects the liver against I/R injury through PPAR-γ activation, which in turn inhibits the RAGE-mediated signalling pathway.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2010-0029359).
Glossary
- ALT
alanine aminotransferase
- Ang II
angiotensin II
- ARB
angiotensin receptor blocker
- AT1R
angiotensin II type 1 receptor
- Egr
early growth response protein
- EMSA
electrophoretic mobility shift assay
- H&E
haematoxylin and eosin
- I/R
ischaemia/reperfusion
- ICAM
intercellular adhesion molecule
- MIP-2
macrophage inflammatory protein 2
- RAGE
receptor for advanced glycation end product
- RAS
renin–angiotensin system
- TBS/T
tween-20 in 1 × tris-buffered saline
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
- Abdelrahman M, Sivarajah A, Thiemermann C. Beneficial effects of PPAR-gamma ligands in ischemia-reperfusion injury, inflammation and shock. Cardiovasc Res. 2005;65:772–781. doi: 10.1016/j.cardiores.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Akahori T, Sho M, Hamada K, Suzaki Y, Kuzumoto Y, Nomi T, et al. Importance of peroxisome proliferator-activated receptor-gamma in hepatic ischemia/reperfusion injury in mice. J Hepatol. 2007;47:784–792. doi: 10.1016/j.jhep.2007.07.030. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano Y, Yamaguchi T, Ohno K, Niimi T, Orita M, Sakashita H, et al. Structural basis for telmisartan-mediated partial activation of PPAR gamma. Hypertens Res. 2012;35:715–719. doi: 10.1038/hr.2012.17. [DOI] [PubMed] [Google Scholar]
- An J, Nakajima T, Kuba K, Kimura A. Losartan inhibits LPS-induced inflammatory signaling through a PPAR gamma-dependent mechanism in human THP-1 macrophages. Hypertens Res. 2010;33:831–835. doi: 10.1038/hr.2010.79. [DOI] [PubMed] [Google Scholar]
- Beigneux AP, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. The acute phase response is associated with retinoid X receptor repression in rodent liver. J Biol Chem. 2000;275:16390–16399. doi: 10.1074/jbc.M000953200. [DOI] [PubMed] [Google Scholar]
- Bonde MM, Olsen KB, Erikstrup N, Speerschneider T, Lyngso C, Haunso S, et al. The angiotensin II type 1 receptor antagonist losartan binds and activates bradykinin B2 receptor signaling. Regul Pept. 2011;167:21–25. doi: 10.1016/j.regpep.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Bradham CA, Stachlewitz RF, Gao W, Qian T, Jayadev S, Jenkins G, et al. Reperfusion after liver transplantation in rats differentially activates the mitogen-activated protein kinases. Hepatology. 1997;25:1128–1135. doi: 10.1002/hep.510250514. [DOI] [PubMed] [Google Scholar]
- Bucciarelli LG, Ananthakrishnan R, Hwang YC, Kaneko M, Song F, Sell DR, et al. RAGE and modulation of ischemic injury in the diabetic myocardium. Diabetes. 2008;57:1941–1951. doi: 10.2337/db07-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo CA, Jr, Madden JF, Gao W, Selvan RS, Clavien PA. Interleukin-6 protects liver against warm ischemia/reperfusion injury and promotes hepatocyte proliferation in the rodent. Hepatology. 1997;26:1513–1520. doi: 10.1002/hep.510260619. [DOI] [PubMed] [Google Scholar]
- Chang JS, Wendt T, Qu W, Kong L, Zou YS, Schmidt AM, et al. Oxygen deprivation triggers upregulation of early growth response-1 by the receptor for advanced glycation end products. Circ Res. 2008;102:905–913. doi: 10.1161/CIRCRESAHA.107.165308. [DOI] [PubMed] [Google Scholar]
- Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C, et al. Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol. 2006;530:70–80. doi: 10.1016/j.ejphar.2005.11.049. [DOI] [PubMed] [Google Scholar]
- Cross TG, Scheel-Toellner D, Henriquez NV, Deacon E, Salmon M, Lord JM. Serine/threonine protein kinases and apoptosis. Exp Cell Res. 2000;256:34–41. doi: 10.1006/excr.2000.4836. [DOI] [PubMed] [Google Scholar]
- Doi S, Masaki T, Arakawa T, Takahashi S, Kawai T, Nakashima A, et al. Protective effects of peroxisome proliferator-activated receptor gamma ligand on apoptosis and hepatocyte growth factor induction in renal ischemia-reperfusion injury. Transplantation. 2007;84:207–213. doi: 10.1097/01.tp.0000269614.21367.3f. [DOI] [PubMed] [Google Scholar]
- Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic ischemia/reperfusion injury – a fresh look. Exp Mol Pathol. 2003;74:86–93. doi: 10.1016/s0014-4800(03)00008-x. [DOI] [PubMed] [Google Scholar]
- Goyal SN, Bharti S, Bhatia J, Nag TC, Ray R, Arya DS. Telmisartan, a dual ARB/partial PPAR-gamma agonist, protects myocardium from ischaemic reperfusion injury in experimental diabetes. Diabetes Obes Metab. 2011;13:533–541. doi: 10.1111/j.1463-1326.2011.01377.x. [DOI] [PubMed] [Google Scholar]
- Guo L, Richardson KS, Tucker LM, Doll MA, Hein DW, Arteel GE. Role of the renin-angiotensin system in hepatic ischemia reperfusion injury in rats. Hepatology. 2004;40:583–589. doi: 10.1002/hep.20369. [DOI] [PubMed] [Google Scholar]
- Guo L, Zhang L, Sun Y, Muskhelishvili L, Blann E, Dial S, et al. Differences in hepatotoxicity and gene expression profiles by anti-diabetic PPAR gamma agonists on rat primary hepatocytes and human HepG2 cells. Mol Divers. 2006;10:349–360. doi: 10.1007/s11030-006-9038-0. [DOI] [PubMed] [Google Scholar]
- Horiuchi M, Mogi M. Role of angiotensin II receptor subtype activation in cognitive function and ischaemic brain damage. Br J Pharmacol. 2011;163:1122–1130. doi: 10.1111/j.1476-5381.2010.01167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyogo H, Yamagishi S. Advanced glycation end products (AGEs) and their involvement in liver disease. Curr Pharm Des. 2008;14:969–972. doi: 10.2174/138161208784139701. [DOI] [PubMed] [Google Scholar]
- Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–647. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, et al. Peroxisome proliferator-activated receptor-gamma protects against hepatic ischemia/reperfusion injury in mice. Hepatology. 2008;47:215–224. doi: 10.1002/hep.21963. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx N, Walcher D, Ivanova N, Rautzenberg K, Jung A, Friedl R, et al. Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes. 2004;53:2662–2668. doi: 10.2337/diabetes.53.10.2662. [DOI] [PubMed] [Google Scholar]
- Matsui T, Yamagishi S, Ueda S, Nakamura K, Imaizumi T, Takeuchi M, et al. Telmisartan, an angiotensin II type 1 receptor blocker, inhibits advanced glycation end-product (AGE)-induced monocyte chemoattractant protein-1 expression in mesangial cells through downregulation of receptor for AGEs via peroxisome proliferator-activated receptor-gamma activation. J Int Med Res. 2007;35:482–489. doi: 10.1177/147323000703500407. [DOI] [PubMed] [Google Scholar]
- Nakajima A, Wada K, Miki H, Kubota N, Nakajima N, Terauchi Y, et al. Endogenous PPAR gamma mediates anti-inflammatory activity in murine ischemia-reperfusion injury. Gastroenterology. 2001;120:460–469. doi: 10.1053/gast.2001.21191. [DOI] [PubMed] [Google Scholar]
- Padrissa-Altes S, Franco-Gou R, Boillot O, Serafin A, Rimola A, Arroyo V, et al. Effect of angiotensin II and bradykinin inhibition in rat reduced-size liver transplantation. Liver Transpl. 2009;15:313–320. doi: 10.1002/lt.21693. [DOI] [PubMed] [Google Scholar]
- Pang T, Benicky J, Wang J, Orecna M, Sanchez-Lemus E, Saavedra JM. Telmisartan ameliorates lipopolysaccharide-induced innate immune response through peroxisome proliferator-activated receptor-gamma activation in human monocytes. J Hypertens. 2012;30:87–96. doi: 10.1097/HJH.0b013e32834dde5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho FS, Alfany-Fernandez I, Casillas-Ramirez A, Massip-Salcedo M, Serafin A, Rimola A, et al. Are angiotensin II receptor antagonists useful strategies in steatotic and nonsteatotic livers in conditions of partial hepatectomy under ischemia-reperfusion? J Pharmacol Exp Ther. 2009;329:130–140. doi: 10.1124/jpet.108.147835. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schupp M, Janke J, Clasen R, Unger T, Kintscher U. Angiotensin type 1 receptor blockers induce peroxisome proliferator-activated receptor-gamma activity. Circulation. 2004;109:2054–2057. doi: 10.1161/01.CIR.0000127955.36250.65. [DOI] [PubMed] [Google Scholar]
- Shin T, Kuboki S, Huber N, Eismann T, Galloway E, Schuster R, et al. Activation of peroxisome proliferator-activated receptor-gamma during hepatic ischemia is age-dependent. J Surg Res. 2008;147:200–205. doi: 10.1016/j.jss.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003;35:881–900. doi: 10.1016/s1357-2725(02)00271-6. [DOI] [PubMed] [Google Scholar]
- Tang SC, Leung JC, Chan LY, Tsang AW, Lai KN. Activation of tubular epithelial cells in diabetic nephropathy and the role of the peroxisome proliferator-activated receptor-gamma agonist. J Am Soc Nephrol. 2006;17:1633–1643. doi: 10.1681/ASN.2005101113. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPAR gamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhou Z, Zhang M, Fan L, Forudi F, Zhou X, et al. Peroxisome proliferator-activated receptor gamma down-regulates receptor for advanced glycation end products and inhibits smooth muscle cell proliferation in a diabetic and nondiabetic rat carotid artery injury model. J Pharmacol Exp Ther. 2006;317:37–43. doi: 10.1124/jpet.105.095125. [DOI] [PubMed] [Google Scholar]
- Wu JS, Cheung WM, Tsai YS, Chen YT, Fong WH, Tsai HD, et al. Ligand-activated peroxisome proliferator-activated receptor-gamma protects against ischemic cerebral infarction and neuronal apoptosis by 14-3-3 epsilon upregulation. Circulation. 2009;119:1124–1134. doi: 10.1161/CIRCULATIONAHA.108.812537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, et al. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat Med. 2000;6:1355–1361. doi: 10.1038/82168. [DOI] [PubMed] [Google Scholar]
- Zeng S, Feirt N, Goldstein M, Guarrera J, Ippagunta N, Ekong U, et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology. 2004;39:422–432. doi: 10.1002/hep.20045. [DOI] [PubMed] [Google Scholar]
- Zeng S, Dun H, Ippagunta N, Rosario R, Zhang QY, Lefkowitch J, et al. Receptor for advanced glycation end product (RAGE)-dependent modulation of early growth response-1 in hepatic ischemia/reperfusion injury. J Hepatol. 2009;50:929–936. doi: 10.1016/j.jhep.2008.11.022. [DOI] [PubMed] [Google Scholar]
- Zhang TL, Fu JL, Geng Z, Yang JJ, Sun XJ. The neuroprotective effect of losartan through inhibiting AT1/ASK1/MKK4/JNK3 pathway following cerebral I/R in rat hippocampal CA1 region. CNS Neurosci Ther. 2012;18:981–987. doi: 10.1111/cns.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WW, Liu XP, Wu N, Zhao TT, Zhao Y, Zhang J, et al. Beneficial effects of losartan on vascular injury induced by advanced glycosylation end products and their receptors in spontaneous hypertension rats. Mol Cell Biochem. 2007;304:35–43. doi: 10.1007/s11010-007-9483-9. [DOI] [PubMed] [Google Scholar]