

Abstract
Background
Whole-cell redox biocatalysis has been intensively explored for the production of valuable compounds because excellent selectivity is routinely achieved. Although the cellular cofactor level, redox state and the corresponding enzymatic activity are expected to have major effects on the performance of the biocatalysts, our ability remains limited to predict the outcome upon variation of those factors as well as the relationship among them.
Results
In order to investigate the effects of cofactor availability on whole-cell redox biocatalysis, we devised recombinant Escherichia coli strains for the production of dihydroxyacetone (DHA) catalyzed by the NAD+-dependent glycerol dehydrogenase (GldA). In this model system, a water-forming NAD+ oxidase (NOX) and a NAD+ transporter (NTT4) were also co-expressed for cofactor regeneration and extracellular NAD+ uptake, respectively. We found that cellular cofactor level, NAD+/NADH ratio and NOX activity were not only strain-dependent, but also growth condition-dependent, leading to significant differences in specific DHA titer among different whole-cell biocatalysts. The host E. coli DH5α had the highest DHA specific titer of 0.81 g/gDCW with the highest NAD+/NADH ratio of 6.7 and NOX activity of 3900 U. The biocatalyst had a higher activity when induced with IPTG at 37°C for 8 h compared with those at 30°C for 8 h and 18 h. When cells were transformed with the ntt4 gene, feeding NAD+ during the cell culture stage increased cellular NAD(H) level by 1.44 fold and DHA specific titer by 1.58 fold to 2.13 g/gDCW. Supplementing NAD+ during the biotransformation stage was also beneficial to cellular NAD(H) level and DHA production, and the highest DHA productivity reached 0.76 g/gDCW/h. Cellular NAD(H) level, NAD+/NADH ratio, and NOX and GldA activity dropped over time during the biotransformation process.
Conclusions
High NAD+/NADH ratio driving by NOX was very important for DHA production. Once cofactor was efficiently cycled, high cellular NAD(H) level was also beneficial for whole-cell redox biocatalysis. Our results indicated that NAD+ transporter could be applied to manipulate redox cofactor level for biocatalysis. Moreover, we suggested that genetically designed redox transformation should be carefully profiled for further optimizing whole-cell biocatalysis.
Keywords: Cofactor engineering, NAD(H) level, NAD+ transporter, Escherichia coli, Dihydroxyacetone, Whole-cell biocatalysis
Background
Cofactor-dependent redox biocatalysis has been shown as a powerful strategy for the production of valuable chemicals that are otherwise difficult to be synthesized [1,2]. Whole cells are preferred for industrial application because cofactors can be regenerated more efficiently [1]. To drive the redox chemistry to a specified direction, it is essential to manipulate intracellular redox state as well as cofactor levels [3]. Thus, various strategies have been applied to control the cofactor regeneration system or balance the enzyme activities of redox reactions. For example, H2O-forming NADH oxidase (NOX) has been applied for cofactor regeneration by engineered whole-cell biocatalyst for chiral compound production [4]. The intracellular cofactor concentration was also important to attain high efficiency especially in the case that the redox enzymes had high apparent Km values to the cofactor [5]. Previous reports showed that exogenous supplied cofactors could improve the reaction rates under whole-cell catalysis conditions, although cells were permeated and external cofactor concentrations were applied at concentrations of over 0.5 mM [6-9]. We recently found that the nucleotide transporter NTT4 encoded by the ntt4 gene from the chlamydial endosymbiont Protochlamydia amoebophila UWE25 [10] could enable Escherichia coli cells to uptake NAD(H) from the culture broth [11]. The NAD+ auxotrophic E. coli YJE003 cells expressing NTT4 cultivated in the media containing 40 μM NAD+ could realize the intracellular NAD(H) pool of 5.1 mM, which was 5.8-fold more than that of the wild-type cells [11]. We reasoned that such a unique NAD(H) supplementation system could be further explored to drive cellular redox chemistry.
Glycerol has become an inexpensive and readily available commodity as a byproduct of biodiesel industry, which makes it attractive to convert glycerol into higher-value products, such as 1,3-propanediol [12], glyceric acid [13], and dihydroxyacetone (DHA) [14]. Besides the clinical and biological applications, DHA is widely used as building blocks in chemical industry and as an artificial suntan in cosmetic industry [15]. Gluconobacter oxydans has been shown as an excellent DHA producer by the oxidation of glycerol using the membrane-bound pyrroloquinoline quinone-dependent glycerol dehydrogenase (GDH) [16]. Native GDH contains two subunits SldA and SldB and their membrane-associated feature may cause problems for heterologous expression. In E. coli, the gldA gene (ID: 6058353) encodes an endogenous NAD+-dependent GDH, which catalyzes reversible reactions for the interconversion of glycerol and DHA [17]. However, GldA has higher affinity and specificity (Km (DHA) = 0.3 mM vs. Km(Glycerol) = 56 mM) towards DHA under normal physiological conditions, such that the conversion from DHA to glycerol (kcat/Km = 5.7 × 104 M-1 s-1) by GldA is far more efficient than the reverse reaction of glycerol to DHA (kcat/Km = 4.0 × 102 M-1 s-1) [17]. Here, we explored the possibility to engineer the NAD+ availability to reverse the GldA reaction direction for DHA production. For this purpose, we constructed recombinant E. coli strains that co-expressed two genes gldA and nox, and three genes gldA, nox and ntt4, for the production of DHA (Figure 1). Using this system as a model, we show that cellular NAD+ availability could be manipulated by different strategies and that the overall DHA specific titer was influenced by enzyme activity, cellular NAD+/NADH ratio, as well as cellular cofactor level. Our results provided useful information for the design and evaluation of redox biocatalyst to produce value-added chemicals.
Figure 1.

Construction of recombinant E. coli strains for DHA production. (A) Schematic representation of the engineered E. coli for DHA production by oxidizing glycerol with NAD+ regeneration and uptaking system; (B) Genetic arrangement of plasmids used for DHA production. Trc P, Trc promoter; gnt105 P, gluconate transporter promoter 105 mutation; RBS, ribosome binding site; rrnB T, rrnB terminator; B0015, synthetic artificial terminator B0015 from IGEM.
Results
Construction of plasmids for DHA production
As no discernible DHA was observed when GldA alone was overexpressed in E. coli whole cells (date not shown), we introduced NOX as well as the NAD+ transporter NTT4 to increase NAD+ availability. Thus, two plasmids were constructed for DHA production from glycerol (Figure 1). For the plasmid pTrc99A-gldA-nox, the gldA gene from E. coli and the nox gene from Enterococcus faecalis were cloned into the vector pTrc99A with ribosome binding sites (RBS), under the control of the Trc promoter and the lacI repressor (Figure 1B). An rrnbT transcription terminator was located downstream of nox. For the plasmid pTrc99A-gldA-nox + ntt4, the NTT4 expression cassette in which the ntt4 gene from P. amoebophila UWE25 was regulated by the promoter gntT105 P was constructed, and inserted into the pTrc99A-gldA-nox backbone downstream of the rrnBT terminator using the RF cloning strategy [18]. Both plasmids ensured the expression of GldA for the oxidation of glycerol to DHA and NOX for NAD+ regeneration. In the case of pTrc99A-gldA-nox + ntt4, NTT4 was expressed to enable NAD+ uptake.
Strain dependence of DHA production
In E. coli, intracellular cofactor concentrations varied widely between different strains even under identical culture conditions in previous reports (Additional file 1: Table S1), which might affect the performance of whole-cell biocatalysts. Such phenomena may be used as a basis for host strain selection. A recent study showed that the efficiencies of different E. coli strains varied largely in the synthesis of (S)-1-(2-chlorophenyl)ethanol [19]. In addition, our previously constructed recombinant Bl21(DE3) whole-cells produced only 0.07 g/gDCW DHA without external NAD+ supplementation [9]. Thus, we transformed pTrc99A-gldA-nox into 6 E. coli strains and investigated their biocatalysis profiles (Figure 2). DHA specific titers varied greatly among different hosts (Figure 2A). Recombinant E. coli DH5α had the highest DHA specific titer of 0.81 g/gDCW. Recombinant DH10B and DH1 had similar specific titer of 0.08 g/gDCW. However, when MG1655, BW2513 or BL21(DE3) was transformed with pTrc99A-gldA-nox, no DHA was detectable. Enzyme activity assay showed that NOX activity was well correlated with DHA titer (Figure 2B). While DH5α had a NOX activity of 3900 U/gDCW, DH10B and DH1 had much lower NOX activity. NOX activities for the other three strains were below 150 U/gDCW. A high NOX activity was beneficial for NAD+ regeneration, which was exemplified by NAD+/NADH ratios of different strains (Figure 2C). Indeed, DH5α, DH10B and DH1 had a relatively higher NAD+/NADH ratios, and the ratio reached 6.7 for DH5α. Intracellular NAD(H) levels were shown in Figure 2D. It was apparent that DH5α and DH10B had similar NAD(H) levels, and the other four strains had slightly lower yet similar values. Thus, high intracellular NAD(H) levels seemed necessary [20], but not sufficient for high DHA productivity. Instead, high initial NAD+/NADH ratio was well correlated with DHA titer (Figure 2A and C) and NOX activity was important to maintain high NAD+/NADH ratio. It remains puzzling that enzymatic activities, NAD+/NADH ratios and DHA productivities varied in different E. coli strains regardless of the presence of the same plasmid. However, similar phenomenon was reported recently that tyrosine production varied largely in 14 different E. coli strains containing the same pathway [21].
Figure 2.
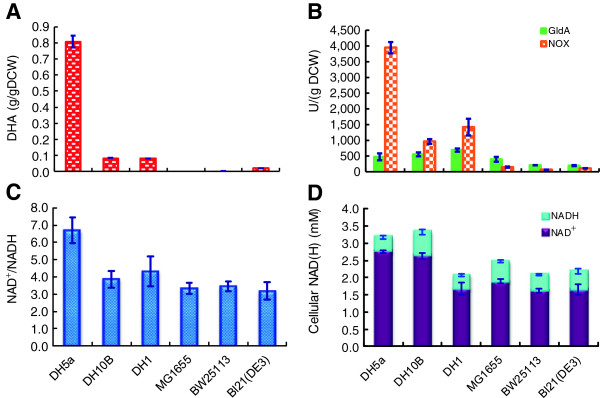
DHA production by different E. coli strains harboring the plasmid pTrc99A-gldA-nox. Cells were induced by IPTG for 20 h at 30°C, and collected for biotransformation experiments in 5 mL of reaction buffer with 20 g/L glycerol in 50-mL test tubes at 37°C, 200 rpm for 10 h. (A) DHA specific titer; (B) Initial enzymatic activity of GldA and NOX; (C) Initial NAD+/NADH ratio; (D) Initial intracellular NAD(H) level. The data represent the averages ± standard deviations (SDs) from three independent clones.
Effects of cell growth on DHA production
To test whether expression levels of GldA and NOX have an effect on DHA production, pTrc99A-gldA-nox transformed E. coli DH5α cells were cultivated under different conditions after being induced by IPTG. At 30°C, the DHA production was increased over time to 18 h. When the cell cultivated at 37°C, the DHA production was increased when the induction time extended from 4 h (0.62 g/g DCW) to 8 h, and dropped to below detection after 12 h (Figure 3A). So we chose the representative turning time point (8 h, 18 h) for enzyme activity and NAD(H) assay and investigating their correlation with DHA production. When cells were cultivated at 30°C for 8 h or 18 h after being induced by IPTG, intracellular NOX activities were similar. However, GldA activity of 290 U/gDCW for the latter sample was 2.1-fold higher than that of the former (Figure 3B). As NAD+/NADH ratios for both samples were also similar (Figure 3C), a higher DHA specific titer of 0.97 g/gDCW for the latter was likely resulted from a higher GldA activity. When cells were cultivated at 37°C for 8 h, the highest DHA specific titer of 1.78 g/gDCW was obtained together with the highest GldA activity of 440 U/gDCW. The NAD+/NADH ratio for this sample was also slightly higher than that of the cells cultivated at 30°C. However, for the cells being cultivated at 37°C for 18 h, GldA activity, NOX activity and NAD+/NADH ratio were 290 U/gDCW, 39 U/gDCW, and 3.1, respectively, which were all substantially lower than those of other samples. As a result, DHA specific titer was only 0.01 g/gDCW. The reason for a reduced NOX activity might be due to the formation of protein inclusion bodies at a higher culture temperature, because NOX overexpression was toxic to the cell [22]. These results suggested that GldA determined the catalyst efficiency in the presence of adequate NOX for NAD+ regeneration. Although cells were harboring an identical plasmid, these results clearly demonstrated that protein expression levels could be significantly different under varied culture conditions, leading to drastic performance differences for whole-cell biocatalysis. Similar phenomena of culture conditions affecting biocatalyst efficiency have been observed in a recent study on L-glyceraldehyde production where glucose dehydrogenase was used for NAD+ regeneration [7].
Figure 3.

DHA production by E. coli DH5α cells harboring the plasmid pTrc99A-gldA-nox. Cells were cultivated at 30°C or 37°C for 8 h or 18 h after being induced by IPTG, and collected for biotransformation experiments in 5 mL of reaction buffer with 20 g/L glycerol at 37°C, 200 rpm for 10 h. (A) DHA specific titer; (B) Initial enzymatic activity of GldA and NOX; (C) Initial NAD+/NADH ratio. All data represent the averages ± SDs for three independent samples.
Promoter selection for NAD+ transporter expression
As has been demonstrated that intracellular NAD+ level and NAD+/NADH ratio had major effects on DHA production, we decided to further increase intracellular NAD+ level by NAD+ feeding. We [11] and others [10] showed that nucleotide transporter NTT4 could import NAD+ into E. coli cells. In this study, ntt4 gene was cloned into vectors pBCTC and pBCTD, downstream constitutive promoters gapA P1 [23] and gntT105 P [24], to give plasmids pBCTC-ntt4 and pBCTD-ntt4, respectively. Together with our previous construct pET15K-ntt4 in which T7 promoter was used [11], three promoters were tested. These three constructs and their corresponding backbone plasmids were transformed into E. coli DH5α cells. As shown in Table 1, when transformants were cultivated in the presence of 0.2 mM NAD+, pET15K-ntt4 led to 7.9% intracellular NAD+ level increment upon IPTG induction. Similarly, pBCTC-ntt4 led to 8.3% increment. However, pBCTD-ntt4 transformed cells accumulated NAD+ to 4.47 mM, which was 2.41-fold higher than that of pBCTD transformed cells. These results suggested that the promoter gntT105 P was by far the most effective one in terms of NTT4 expression for NAD+ uptake. It should be pointed out that NTT4 is a membrane protein such that a high NTT4 expression seems toxic to the hosts which inhibited the cell growth (Additional file 1: Figure S1).
Table 1.
Intracellular NAD(H) levels of E. coli DH5α cells a harboring NTT4 expression and corresponding empty plasmids
| Plasmid | Promoter |
Cellular NAD(H) level (mM)
b
|
Increment | |
|---|---|---|---|---|
| Empty vector | NTT4 expression | |||
| pET15k |
T7 |
1.72 ± 0.03 |
1.85 ± 0.01 |
7.9% |
| pBCTC |
gap P1 |
1.96 ± 0.00 |
2.13 ± 0.01 |
8.3% |
| pBCTD | gntT105 P | 1.86 ± 0.05 | 4.47 ± 0.14 | 141.0% |
aCells were cultivated at 37°C for 12 h in LB media containing 0.2 mM NAD+, collected by centrifugation and washed twice with PBS buffer.
bAll data represent the averages ± SDs for three independent samples.
Enhancing NAD+ supply for DHA production by using a NAD+ transporter NTT4
E. coli DH5α cells were transformed with plasmids pTrc99A-gldA-nox and pTrc99A-gldA-nox + ntt4 to give strains YJE005 and YJE006, respectively. Both strains were cultivated at 37°C for 8 h in the presence of 0.2 mM NAD+, and whole-cell biocatalysts were prepared. DHA specific titer for YJE006 was 2.13 g/gDCW, which was 1.58-fold higher than that of YJE005 (Figure 4A), suggesting that NAD+ feeding during the cell culture stage was beneficial to DHA production. Initial GldA activity of YJE006 (250 U/gDCW) was slightly lower than that of YJE005 (310 U/gDCW), while initial NOX activity of YJE006 (3500 U/gDCW) was slightly higher than that of YJE005 (3100 U/gDCW). After being used for 10 h, both strains had significantly reduced NOX activities. While GldA activity of YJE006 dropped to 59 U/gDCW, GldA activity of YJE005, in sharp contrast, increased to 565 U/gDCW (Figure 4B). Initial cellular NAD(H) level and NAD+/NADH ratio were 1.44 mM and 10.0 in YJE006, which were 1.44-fold and 1.34-fold higher than those of the YJE005, respectively (Figure 4C). Cellular NAD(H) level of YJE005 dropped by only 8% to 0.92 mM, but the NAD+/NADH ratio drastically dropped to 2.11 after 10 h. For YJE006, cellular NAD(H) level and the NAD+/NADH ratio were dropped by 78% and 87%, respectively, compared with those of the initial values. The fact that YJE006 cells had lower initial GldA activity but higher initial NOX activity, cellular NAD(H) level and NAD+/NADH ratio suggested that sufficient NAD+ supply and higher cellular redox state were more important than GldA activity to drive the system for DHA production. However, YJE006 cells appeared to lose enzyme activities and cellular cofactor more rapidly than YJE005 cells.
Figure 4.

DHA production by E. coli YJE005 and YJE006. Cells were cultivated at 37°C for 8 h after being induced by IPTG, and collected for biotransformation experiments with 20 g/L glycerol and 0.2 mM NAD+ in 5 mL of reaction buffer in 50-mL test tubes at 37°C, 200 rpm for 10 h. (A) DHA specific titer; (B) Initial and end-point enzymatic activity of GldA and NOX; (C) Initial and end-point cellular NAD(H) level. All data represent the averages ± SDs for three independent clones.
DHA production under shake-flask conditions
In order to further investigate the NAD+ effect on the durability of the biocatalyst, the performance of YJE006 cells for DHA production was examined under shake-flask conditions after 2 h. When the reaction was initiated with 2.0 gDCW/L cells and 2.5 g/L glycerol, DHA concentration reached 2.08 g/L after 2 h (Figure 5A, Line a) and the biocatalyst activity reached 96 U/gDCW. When 5.0 g/L glycerol was used, the whole-cell catalyst produced 3.02 g/L DHA after 2 h with a higher biocatalyst activity of 140 U/gDCW (Figure 5A, Line b). In both cases, DHA concentration dropped slowly over time, suggesting that DHA may be further consumed (Figure 1A). When the reaction was initiated with 2.0 gDCW/L and 5.0 g/L glycerol in the presence of 0.2 mM external NAD+, DHA production was burst to 3.05 g/L within 2 h, but it was slowly increasing for up to 10 h with a final titer of 3.69 g/L (Figure 5A, Line c). However, NAD+ supplementation failed to further improve the initial biocatalyst activity (141 U/gDCW). Again, activities of GldA and NOX dropped dramatically after 10 h (Figure 5B). However, cells had higher activities of GldA and NOX as well as NAD(H) level (b-10 h vs. c-10 h) after 10 h in the presence of external NAD+ supplementation. These results indicated NAD+ feeding at the biotransformation stage was also slightly beneficial to DHA production in NTT4 expressed recombinant E. coli cells.
Figure 5.

DHA production by E. coli YJE006 whole cells in 20 mL of reaction buffer in 500-mL shake-flasks at 37°C, 200 rpm. (A) The time course of DHA formation; (B) Initial and end-point enzymatic activity of GldA and NOX; (C) Initial and end-point cellular NAD(H) level. The data represent the averages standard deviations for three independent samples. All data represent the averages ± SDs for three independent samples.
Discussion
Cellular redox state, roughly indicated by the NAD+/NADH ratio, is important for whole-cell redox biocatalysis. In E. coli, NAD+/NADH ratios ranging from 3 to 10 were previously documented [25]. It was expected that cellular NAD+/NADH ratio as well as NAD(H) level should have major effects on whole-cell biocatalysis. And co-expressing the nox gene encoding a H2O-forming NOX with GldA is expected to be helpful for reversing GldA activity toward DHA biosynthesis by efficient regenerating NAD+. Indeed, NOX has been widely used to enhance NAD+-dependent biosynthesis in both resting cells [4] and growing cells [26]. In this study, we found NOX activity was not only strain-dependent (Figure 2B), but also growth condition-dependent (Figure 3B), though cells were transformed with the same plasmid pTrc99A-gldA-nox. This may be attributed to the adaption ability difference against NOX expression among these strains because high NOX activity retarded cell growth and affected the metabolism [22]. Indeed, we also found that cell growth was negatively correlated with NOX activity (date not shown). Thus, while NOX expression provided driving force to maintain a higher NAD+ level and cellular NAD+/NADH ratio, it should be carefully tuned to avoid toxic effects on cellular physiology. It was found that DHA specific titer was positively correlated with the cellular NAD+/NADH ratio but not the cellular NAD(H) level (Figure 2). Similarly, introducing robust cofactor regeneration system increased the whole-cell biocatalysts efficiency [19,27]. However, enhancing the cellular NADP(H) pool alone without efficient cofactor regeneration failed to improve the redox biocatalyst efficiency [28]. Together, NAD+/NADH ratio driven by a cofactor regeneration system is more important than cellular cofactor level for an efficient oxidative bioreaction.
Once cofactor was efficiently regenerated, cellular cofactor level could also affect the biocatalyst efficiency [20]. External cofactor supplementation has been applied to increase intracellular NAD+ levels and NAD+/NADH ratios [6-8]. In those cases, cells were treated with permeating agents and external cofactor concentrations were high. We recently showed that NTT4 expression in E. coli led to uptaking NAD(H) from the culture broth [11]. Thus, ntt4 was used to improve E. coli whole-cell biocatalytic efficiency by enhancing the cellular NAD(H) level. Actually, ntt4 expression in YJE006 increased cellular NAD(H) level by 1.44 fold, indicating that NTT4 was functional in terms of uptaking external NAD+. As NTT4 could transport NADH efficiently [10] and increase the cellular reduction state with external NADH supplementation [11], it could also be used for increase the cellular NADH level for reduction biocatalyst such as asymmetric reduction of o-chloroacetophenone [8].
Although NTT4 expression strain YJE006 had a higher DHA production, it appeared to lose enzyme activities and cellular cofactor much more rapidly than YJE005. One possible reason was that NTT4 expression led to increased membrane permeability and other toxic effects. Therefore, it seemed challenging to maintain good NAD(H) durability for NTT4 expressions cells that were capable of uptaking external NAD+. As it requires alive cells to mediate continuous NAD+ transportation, NTT4 may have better potential profiting for NAD+-dependent multi-step biosynthesis in living cells such as 1-butanol production from glucose [29]. Although the NAD+ feeding strategy is cost prohibitive for bulk chemicals production, it may be useful for the production of high-value chemicals and pharmaceuticals. Alternatively, reconstruction of efficient heterologous NAD+ biosynthesis pathway [30] may increase cellular NAD(H) level for enhanced NAD(H)-dependent biosynthesis.
The highest specific DHA productivity reached 0.76 g/gDCW/h under shake-flask conditions for the first 2 h (Figure 5A), which was lower compared with other studies of more than 2 g/gDCW/h using G. oxydans as the hosts in small scale bioreactor [31], but much higher than that recombinant Saccharomyces cerevisiae of 0.03 g/gDCW/h from sugar [32]. The high DHA productivity of G. oxydans is attributed to the membrane-bound GDH, which can directly oxidize glycerol to DHA without material transfer across cell membrane [16]. However, our attempt, in reversing an endogenous cytosolic GldA catalysis for DHA production in recombinant E. coli by engineering NAD+ availability, provided some insights on optimizing whole-cell redox biocatalyst for other valuable chemicals and pharmaceuticals production.
As DHA concentration dropped over time after 2 h, it was possible that other metabolic steps consumed DHA. In this regard, disruption of the DHA and glycerol catabolic pathway related genes such as dhaK and glpK be useful (Figure 1A). Interestingly, external NAD+ supplementation during the whole-cell catalysis ensured a higher NOX activity and continuous accumulation of DHA for up to 10 h. These results indicated that NOX activity was beneficial to maintain oxidative ability of the NAD+-dependent whole-cell biocatalysis. It is worth mentioning that the initial specific biocatalyst activity (141 U/gDCW) was half to the initial GldA activity of 249 U/gDCW. This might be attributed to the substrate diffusion, product consumption, etc.
In summary, using oxidation of glycerol to DHA by recombinant E. coli whole-cells as a model system, we enhanced oxidation state and cellular cofactor level for increasing the catalytic efficiency by expressing NADH oxidase and NAD(H) transporter, respectively. As the overall biocatalytic performance is dependent upon the cellular cofactor level, redox state and the corresponding enzymatic activity, genetically designed redox transformation should be systematically profiled to identify optimal whole-cell biocatalysis.
Material and methods
Bacterial strains and plasmids
Cloning and plasmid propagation were performed with E. coli DH5α. The strains and plasmids are listed in Table 2. E. coli was routinely cultivated with agitation at 37°C or 30°C , 200 rpm in LB broth (10 g tryptone, 5 g yeast extract, 10 g NaCl per liter water) containing appropriate antibiotics (Kanamycin sulfate, 50 μg/mL; ampicillin, 100 μg/mL) if necessary.
Table 2.
Strains and plasmids used in this study
| Strains or plasmids | Genotype or characteristic | Resources or references |
|---|---|---|
|
E. coli
Strains |
|
|
| DH5α |
F-, φ80d/lacZ∆M15, ∆(lacZYA-argF)U169, deoR, recA1, endA1, hsdR17(rk-, mk+), phoA, supE44, λ-, thi-1, gyrA96, relA1 |
TaKaRa |
| DH10B |
F-,mcrA, ∆(mrr-hsdRMS-mcrBC), φ80lacZ∆M15, ∆lacX74, recA1, endA1, araD139, ∆ (ara, leu)7697, galU, galK, λ-, rpsL, nupG/pMON14272/pMON7124 |
Invitrogen |
| DH1 |
F-, glnV44(AS), λ
-
, rfbC1, gyrA96(NalR), recA1, endA1, thi-1, hsdR17 |
CGSC |
| (No. 6040) | ||
| MG1655 |
F-, λ
-
, rph-1 |
CGSC |
| (No. 6300) | ||
| BW25113 |
rrnB3, ∆lacZ4787, hsdR514, ∆(araBAD)567, ∆(rhaBAD)568 rph-1 |
CGSC |
| (No. 7636) | ||
| Bl21(DE3) |
F–, dcm, ompT, hsdS(rB–, mB–), gal, λ(DE3) |
Novagen |
| YJE005 |
DH5α/pTrc99A-gldA-nox |
This study |
| YJE006 |
DH5α/pTrc99A-gldA-nox + ntt4 |
This study |
|
Plasmids |
|
|
| pMD18-T |
lacZ, pBR322 ori, bla, cloning vector |
TaKaRa |
| pTrc99A |
lacI, pBR322 ori, bla, expression vector |
Amersham Pharmacia |
| pET15K-ntt4 |
ntt4 inserted within NdeI and BamH I sites, kan |
[11] |
| pBCTC-ntt4 |
ntt4 inserted within Sac I and BamH I sites, kan |
This study |
| pBCTD-ntt4 |
ntt4 inserted within Sac I and BamH I sites, kan |
This study |
| pTrc99A-gldA-nox |
gldA and nox transcription under Trc promoter |
This study |
| pTrc99A-gldA-nox + ntt4 | gldA and nox transcription under Trc promoter, ntt4 under gntT105p promoter. | This study |
Reagents
All primers used in this study (Table 3) were custom synthesized from Invitrogen (Shanghai, China) and DNA sequencing was performed in TaKaRa (Dalian, China). Dpn I, PrimeSTAR HS DNA polymerase and all other reagents for genetic manipulation were purchased from TaKaRa (Dalian, China). DNA gel purification kit and plasmid extraction kit were purchased from Beyotime (Haimen, China). All chemicals were purchased from Sigma (Shanghai, China).
Table 3.
Primers used in this study a
| Primer | Sequence(5′-3′) | Function |
|---|---|---|
| gldA-F0 |
TGCTGTATATAGCGCCGCACAAG |
gldA_cloning |
| gldA-R0 |
AGGTTGGTATTGGCCTGGATTTG |
|
| gldA-F1 |
CAATTTCACACAGGAAACAGACCATGGACCGCATTATTCAATCAC |
gldA_amplification |
| gldA-R1 |
GTGTATATCTCCTTCTCTAGTAGCGATCTATTATTCCCACTCTTGCAGG |
|
| Nox-F1 |
CTACTAGAGAAGGAGATATACACATGAAAGTCGTAGTCGTAGG |
nox_amplification |
| Nox-R1 |
CAAAACAGCCAAGCTTGCATGCCTGCAGTTACATATTTTCTAAAGCGGCTTG |
|
| gapAP1-F1 |
TCGGATATCGAGGCGAGTCAGTCGCGTAATGC |
Promoter gapAP1 amplification |
| gapAP1-R1 |
ACCGAATTCGATCTCATATGTTCCACCAGCTATTTGTTAG |
|
| gntT105P-F1 |
CCGTTGATATCTGAAAGGTGTGCGCGATCTCAC |
PromotergntT105P amplification |
| gntT105P-R1 |
GGAATTCTATCTCCTTATTCATTTGCGCTGGGTAACGTCAATTT |
|
| ntt4-F1 |
TTCGAGCTCATGAGTAAAACAAACCAGG |
|
| ntt4-R1 |
AGAGGATCCTTAGTGATGATGATGATGATGTTTTTTTATAAAAG |
ntt4 amplification |
| gntT105P-F2 |
CAAACTCTTTTTGTTTATTTTTCTAAATACATGAAAGGTGTGCGCGATCTC |
gntT105P + ntt4 amplification |
| ntt4-R2 |
CGTTTTATTTGATGCCTGGATCCGCGTCGACTCTAGAGGATCC |
|
| T-F1 |
GGATCCTCTAGAGTCGACGCGGATCCAGGCATCAAATAAAACG |
Terminator BBa_B0015 amplification |
| T-R1 | GTATTTAGAAAAATAAACATATAAACGCAGAAAGGCCCAC |
aThe restriction sites were bolded and the ribosome binding sites (RBS) were underlined.
DNA manipulation
The gldA (NCBI GeneID: 6058353) gene was amplified from E. coli DH5α genomic DNA using primer pair gldA-F0/gldA-R0 and cloned into pMD18T to give pMD18T-gldA. The nox (NCBI GeneID: 1200486) gene was amplified from pMD18T-nox, which was constructed by cloning the nox gene from Enterococcus faecalis (CGMCC 1.130) into the pMD18T. The gldA-nox co-expression cassette was constructed with the modified one-step overlap extension (SOE) PCR strategy described previously [33]. Briefly, gldA and nox were amplified with primer pairs gldA-F1/gldA-R1 and nox-F1/nox-R1, and purified gldA and nox fragments (molar ratio 1:1, about 200 ng each) were mixed. To the mixture were added 3 μL of dNTP (2.5 mM each), 5 μL 5 × PrimerStar buffer, 1.25 U PrimeSTAR HS DNA polymerase, and H2O to a total volume of 25 μL. PCR amplification was performed according to the thermocycle conditions of 95°C for 5 min, 15 cycles of 98°C for 10 s, 68°C for 3 min, and 68°C for 10 min. Next, 2 μL of unpurified PCR products was used as the template using the primer pair gldA-F1/nox-R1 for normal PCR amplification in a total volume of 100 μL. The purified gldA-nox cassette was cloned into the pTrc99A using the restriction-free (RF) cloning strategy [11] to give plasmid pTrc99A-gldA-nox.
The ntt4 (NCBI GeneID: 2780098) gene containing the 3'-end 6 × His-tag encoding sequence was cloned from the vector pET15k-ntt4[11]. The constitutive glyceraldehyde-3-phosphate dehydrogenase promoter P1 gapA P1 [23] and the internal operator of gluconate transporter promoter 105 mutant gntT105 P [24] was cloned from E. coli DH5α genomic DNA using primer pairs gapAP1-F/gapAP1-R and gntT105P-F1/gntT105P-R1, respectively. Then, EcoR V-EcoR I digested promoters were cloned into the EcoR V-EcoR I site of pTrc99A to substitute the Trc promoter, resulting in the constitutive expression vectors pBCTA and pBCTB. The function of these two vectors was checked by constitutive expression of red fluorescent protein (date not shown). Lastly, ntt4 was cloned into the Sac I-BamH I site locating downstream of the constitutive promoter of pBCTA and pBCTB after cloning with the primer pair ntt4-F1/ntt4-R1, and the bla was replaced by the kan using a RF cloning strategy, resulting in plasmids pBCTC-ntt4 and pBCTD-ntt4, respectively.
The ntt4 constitutive expression cassette was also constructed and cloned into pTrc99A-gldA-nox. The gntT105P-ntt4 was amplified from the pBCTD-ntt4 using the primer pair gntT105P-F2/ntt4-R2 and terminator B0015 cloned using the primer pair T-F1/T-R1 from the international genetically engineered machine competition (IGEM, http://partsregistry.org/Part:BBa_B0015). These two DNA fragments were fused with a modified SOE PCR approach (33), and cloned into pTrc99A-gldA-nox locating downstream of the rrnBT terminator, resulting in plasmid pTrc99A-gldA-nox + ntt4.
Whole-cell biocatalyst preparation
Recombinant E. coli cells harboring appropriate plasmid were cultivated in LB medium supplemented with appropriate antibiotics at 37°C, 200 rpm, to the early exponential phase (OD600 = 0.2–0.4). Cultures were induced by adding IPTG to a final concentration of 0.1 mM (and 0.2 mM NAD+ if needed), and cultivated for additional 8 h or 18 h at appropriate temperature (37°C or 30°C), 200 rpm. Cells were harvested by centrifugation (2,000 g, 5 min) and washed twice with 0.1 M potassium phosphate buffer (pH 9.0).
DHA production
As GldA had a higher activity toward glycerol dehydrogenation [34] and our previous study [9] showed DHA production reached the highest at pH 9.0. The IPTG induced E. coli cells were resuspended in 5 mL of 0.1 M potassium phosphate buffer (pH 9.0) for 10 h in 5 mL of potassium phosphate buffer containing 20 g/L glycerol in 50-mL test tubes; or in 20 mL of the buffer containing 2–5 g/L glycerol in 500-mL shake flasks. NAD+ was added into the reaction to a final concentration of 0.2 mM when necessary. All reactions were performed at 37°C, 200 rpm. All the data represent the averages standard deviations from at least three independent samples.
DHA quantification
DHA was assayed according to a known method with minor modifications [35]. Briefly, biotransformation mixtures were centrifuged at 10,000 g for 2 min. Exactly 20 μL of supernatants were mixed with 180 μL diphenylamine reagent containing 1% (w/v) diphenylamine and 10% (v/v) sulfuric acid in acetic acid, and heated at 105°C for 20 min. Then, the absorbance at 620 nm were recorded after cooling to room temperature, and DHA concentrations were quantified according to a standard curve obtained under identical conditions.
Enzyme activity assay
E. coli dry cell weight (DCW) was weight by converting OD600 value with a coefficient of 0.275 gDCW/(L × OD600), which was determined by freezer drying the E. coli cells according to our recently report [36]. As the DHA production was performed at 0.1 M potassium phosphate buffer (pH 9.0) due to GldA activity has higher activity as mentioned above, all enzymatic assays were performed at consistent pH of 9.0. About 2 × 109E. coli cells were harvested, washed twice with 0.1 M potassium phosphate buffer (pH 9.0), and stored as cell pellets at -80°C. For enzyme assays, cell pellets were resuspended in 0.2 mL of lysis buffer (10 mM Tris-Cl, 1.0 mM MgCl2, 1 mg/mL lysozyme and 0.1 mg/mL DNase, pH 8.0) and incubated at 37°C for 30 min. GldA activity was estimated by recording the absorbance increase at 340 nm and assays were performed at 25°C, in 100 μL of 0.1 M potassium carbonate buffer (pH 9.0) containing 5 mM NAD+, 100 mM glycerol and 5 μL of crude cell lysates, which was with minor modifications from a previous report [37]. NOX activity was measured by recording the absorbance decrease at 340 nm and assays were performed at 25°C, in 100 μL of 0.1 M potassium phosphate buffer (pH 9.0) containing 0.4 mM NADH and 1 μL (if the cell NOX activity was more than 1000 U/gDCW) or 5 μL of (if the cell NOX activity was less than 1000 U/gDCW) crude cell lysates.
Cofactor measurement
Cell pellets (containing about 2 × 109 cells) were washed twice with 0.1 M potassium phosphate buffer, and then treated at 55°C for 10 min in 150 μl of 0.2 M NaOH (for NADH extraction) or 150 μl of 0.2 M HCl (for NAD+ extraction). The extracts were neutralized by adding 150 μl of 0.1 M HCl (for NADH extraction) or 150 μl of 0.1 M NaOH (for NAD+ extraction). The cellular debris was removed by centrifuging at 12,000 g for 5 min. Supernatants were transferred to new tubes and stored at -80°C until assay. NAD(H) was quantified using a sensitive enzymatic cycling assay as reported previously [11].
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YJZ and ZKZ conceived and designed the experiments. YJZ and WY performed the experiments. YJZ, LW, ZZ, SZ and ZKZ analyzed the data. YJZ and ZKZ wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Table S1. Cellular NAD(H) level of different E. coli strains from literatures. Figure S1. NTT4 expression under gntT105P strongly retarded E. coli DH5α growth.
Contributor Information
Yongjin J Zhou, Email: yongjin@chalmers.se.
Wei Yang, Email: hi.yangwei@163.com.
Lei Wang, Email: skywise@dicp.ac.cn.
Zhiwei Zhu, Email: zwzhu@dicp.ac.cn.
Sufang Zhang, Email: zsfjxy@dicp.ac.cn.
Zongbao K Zhao, Email: zhaozb@dicp.ac.cn.
Acknowledgements
We are indebted to Prof. Qin Ye (East China University of Science and Technology, China) for kindly providing pTrc99A. This work was supported by the National Basic Research and Development Program of China (No. 2012CB721103) and the State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, CAS (R201306).
References
- Schmid A, Dordick JS, Hauer B, Kiener A, Wubbolts M, Witholt B. Industrial biocatalysis today and tomorrow. Nature. 2001;409:258–268. doi: 10.1038/35051736. [DOI] [PubMed] [Google Scholar]
- Jiang T, Gao C, Dou P, Ma C, Kong J, Xu P. Rationally re-designed mutation of NAD-independent L-lactate dehydrogenase: high optical resolution of racemic mandelic acid by the engineered Escherichia coli. Microb Cell Fact. 2012;11:151. doi: 10.1186/1475-2859-11-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank LM, Ebert BE, Buehler K, Bühler B. Redox biocatalysis and metabolism: molecular mechanisms and metabolic network analysis. Antioxid Redox Signal. 2010;13:349–394. doi: 10.1089/ars.2009.2931. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Lv C, Gao C, Qin J, Ma C, Liu Z, Liu P, Li L, Xu P. A novel whole-cell biocatalyst with NAD + regeneration for production of chiral chemicals. PLoS One. 2010;5:e8860. doi: 10.1371/journal.pone.0008860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bühler B, Park JB, Blank LM, Schmid A. NADH availability limits asymmetric biocatalytic epoxidation in a growing recombinant Escherichia coli strain. Appl Environ Microbiol. 2008;74:1436–1446. doi: 10.1128/AEM.02234-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Witholt B, Li Z. Coupling of permeabilized microorganisms for efficient enantioselective reduction of ketone with cofactor recycling. Chem Commun (Camb) 2006;42:398–400. doi: 10.1039/b515721h. [DOI] [PubMed] [Google Scholar]
- Richter N, Neumann M, Liese A, Wohlgemuth R, Weckbecker A, Eggert T, Hummel W. Characterization of a whole-cell catalyst co-expressing glycerol dehydrogenase and glucose dehydrogenase and its application in the synthesis of L-glyceraldehyde. Biotechnol Bioeng. 2010;106:541–552. doi: 10.1002/bit.22714. [DOI] [PubMed] [Google Scholar]
- Kratzer R, Pukl M, Egger S, Vogl M, Brecker L, Nidetzky B. Enzyme identification and development of a whole-cell biotransformation for asymmetric reduction of o-chloroacetophenone. Biotechnol Bioeng. 2011;108:797–803. doi: 10.1002/bit.23002. [DOI] [PubMed] [Google Scholar]
- Yang W, Zhou YJ, Zhao ZK. Production of dihydroxyacetone from glycerol by engineered Escherichia coli cells co-expressing gldA and nox genes. Afr J Biotechnol. 2013;12:4387–4392. [Google Scholar]
- Haferkamp I, Schmitz-Esser S, Linka N, Urbany C, Collingro A, Wagner M, Horn M, Neuhaus HE. A candidate NAD+ transporter in an intracellular bacterial symbiont related to Chlamydiae. Nature. 2004;432:622–625. doi: 10.1038/nature03131. [DOI] [PubMed] [Google Scholar]
- Zhou YJ, Wang L, Yang F, Lin XP, Zhang SF, Zhao ZK. Determining the extremes of the cellular NAD(H) level by using an Escherichia coli NAD+-auxotrophic mutant. Appl Environ Microbiol. 2011;77:6133–6140. doi: 10.1128/AEM.00630-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Tan Y, Zhu H, Zhao K, Shen W. Microbial conversion of glycerol to 1,3-propanediol by an engineered strain of Escherichia coli. Appl Environ Microbiol. 2009;75:1628–1634. doi: 10.1128/AEM.02376-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habe H, Shimada Y, Yakushi T, Hattori H, Ano Y, Fukuoka T, Kitamoto D, Itagaki M, Watanabe K, Yanagishita H. et al. Microbial production of glyceric acid, an organic acid that can be mass produced from glycerol. Appl Environ Microbiol. 2009;75:7760–7766. doi: 10.1128/AEM.01535-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MH, Wu J, Liu X, Lin JP, Wei DZ, Chen H. Enhanced production of dihydroxyacetone from glycerol by overexpression of glycerol dehydrogenase in an alcohol dehydrogenase-deficient mutant of Gluconobacter oxydans. Bioresour Technol. 2010;101:8294–8299. doi: 10.1016/j.biortech.2010.05.065. [DOI] [PubMed] [Google Scholar]
- Mishra R, Jain SR, Kumar A. Microbial production of dihydroxyacetone. Biotechnol Adv. 2008;26:293–303. doi: 10.1016/j.biotechadv.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Gatgens C, Degner U, Bringer-Meyer S, Herrmann U. Biotransformation of glycerol to dihydroxyacetone by recombinant Gluconobacter oxydans DSM 2343. Appl Microbiol Biotechnol. 2007;76:553–559. doi: 10.1007/s00253-007-1003-z. [DOI] [PubMed] [Google Scholar]
- Subedi KP, Kim I, Kim J, Min B, Park C. Role of GldA in dihydroxyacetone and methylglyoxal metabolism of Escherichia coli K12. FEMS Microbiol Lett. 2008;279:180–187. doi: 10.1111/j.1574-6968.2007.01032.x. [DOI] [PubMed] [Google Scholar]
- Zhou YJ, Yang F, Zhang S, Tan H, Zhao ZK. Efficient gene disruption in Saccharomyces cerevisiae using marker cassettes with long homologous arms prepared by the restriction-free cloning strategy. World J Microbiol Biotechnol. 2011;27:2999–3003. doi: 10.1007/s11274-011-0756-9. [DOI] [Google Scholar]
- Mädje K, Schmölzer K, Nidetzky B, Kratzer R. Host cell and expression engineering for development of an E. coli ketoreductase catalyst: enhancement of formate dehydrogenase activity for regeneration of NADH. Microb Cell Fact. 2012;11:7. doi: 10.1186/1475-2859-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Jiang J, Chen Y. Correlation between intracellular cofactor concentrations and biocatalytic efficiency: coexpression of diketoreductase and glucose dehydrogenase for the preparation of chiral diol for statin drugs. ACS Catal. 2011;1:1661–1664. doi: 10.1021/cs200408y. [DOI] [Google Scholar]
- Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol. 2013;31:170–174. doi: 10.1038/nbt.2461. [DOI] [PubMed] [Google Scholar]
- Holm AK, Blank LM, Oldiges M, Schmid A, Solem C, Jensen PR, Vemuri GN. Metabolic and transcriptional response to cofactor perturbations in Escherichia coli. J Biol Chem. 2010;285:17498–17506. doi: 10.1074/jbc.M109.095570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier B, Branlant C. The Escherichia coli gapA gene is transcribed by the vegetative RNA polymerase holoenzyme E sigma 70 and by the heat shock RNA polymerase E sigma 32. J Bacteriol. 1994;176:830–839. doi: 10.1128/jb.176.3.830-839.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peekhaus N, Conway T. Positive and negative transcriptional regulation of the Escherichia coli gluconate regulon gene gntT by GntR and the cyclic AMP (cAMP)-cAMP receptor protein complex. J Bacteriol. 1998;180:1777–1785. doi: 10.1128/jb.180.7.1777-1785.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardo MR, Dailly Y, Clark DP. Role of NAD in regulating the adhE gene of Escherichia coli. J Bacteriol. 1996;178:6013–6018. doi: 10.1128/jb.178.20.6013-6018.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri GN, Eiteman MA, Altman E. Increased recombinant protein production in Escherichia coli strains with overexpressed water-forming NADH oxidase and a deleted ArcA regulatory protein. Biotechnol Bioeng. 2006;94:538–542. doi: 10.1002/bit.20853. [DOI] [PubMed] [Google Scholar]
- Zhang R, Xu Y, Xiao R, Zhang B, Wang L. Efficient one-step production of (S)-1-phenyl-1,2-ethanediol from (R)-enantiomer plus NAD+-NADPH in-situ regeneration using engineered Escherichia coli. Microb Cell Fact. 2012;11:167. doi: 10.1186/1475-2859-11-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser F, Schroer K, Lutz S, Bringer-Meyer S, Sahm H. Enhancement of the NAD(P)(H) pool in Escherichia coli for biotransformation. Eng Life Sci. 2007;7:343–353. doi: 10.1002/elsc.200720203. [DOI] [Google Scholar]
- Shen CR, Lan EI, Dekishima Y, Baez A, Cho KM, Liao JC. Driving forces enable high-titer anaerobic 1-butanol synthesis in Escherichia coli. Appl Environ Microbiol. 2011;77:2905–2915. doi: 10.1128/AEM.03034-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorci L, Martynowski D, Rodionov DA, Eyobo Y, Zogaj X, Klose KE, Nikolaev EV, Magni G, Zhang H, Osterman AL. Nicotinamide mononucleotide synthetase is the key enzyme for an alternative route of NAD biosynthesis in Francisella tularensis. Proc Natl Acad Sci USA. 2009;106:3083–3088. doi: 10.1073/pnas.0811718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZC, Zheng YG, Shen YC. Use of glycerol for producing 1,3-dihydroxyacetone by Gluconobacter oxydans in an airlift bioreactor. Bioresour Technol. 2011;102:7177–7182. doi: 10.1016/j.biortech.2011.04.078. [DOI] [PubMed] [Google Scholar]
- Nguyen HT, Nevoigt E. Engineering of Saccharomyces cerevisiae for the production of dihydroxyacetone (DHA) from sugars: a proof of concept. Metab Eng. 2009;11:335–346. doi: 10.1016/j.ymben.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Zhou YJ, Gao W, Rong Q, Jin G, Chu H, Liu W, Yang W, Zhu Z, Li G, Zhu G. et al. Modular pathway engineering of diterpenoid synthases and the mevalonic acid pathway for miltiradiene production. J Am Chem Soc. 2012;134:3234–3241. doi: 10.1021/ja2114486. [DOI] [PubMed] [Google Scholar]
- Tang CT, Ruch FE, Lin ECC. Purification and properties of a nicotinamide adenine dinucleotide-linked dehydrogenase that serves an Escherichia coli mutant for glycerol catabolism. J Bacteriol. 1979;140:182–187. doi: 10.1128/jb.140.1.182-187.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZQ, Hu ZC, Zheng YG, Shen YC. Optimization of cultivation conditions for the production of 1,3-dihydroxyacetone by Pichia membranifaciens using response surface methodology. Biochem Eng J. 2008;38:285–291. doi: 10.1016/j.bej.2007.07.015. [DOI] [Google Scholar]
- Wang L, Zhou YJ, Ji D, Zhao ZK. An accurate method for estimation of the intracellular aqueous volume of Escherichia coli cells. J Microbiol Methods. 2013;93:73–76. doi: 10.1016/j.mimet.2013.02.006. [DOI] [PubMed] [Google Scholar]
- Gonzalez R, Murarka A, Dharmadi Y, Yazdani SS. A new model for the anaerobic fermentation of glycerol in enteric bacteria: trunk and auxiliary pathways in Escherichia coli. Metab Eng. 2008;10:234–245. doi: 10.1016/j.ymben.2008.05.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Cellular NAD(H) level of different E. coli strains from literatures. Figure S1. NTT4 expression under gntT105P strongly retarded E. coli DH5α growth.