

Abstract
Pancreatic cancer, one of the deadliest human malignancies, is almost invariably associated with the presence of an oncogenic form of Kras. Mice expressing oncogenic Kras in the pancreas recapitulate the step-wise progression of the human disease. The inflammatory cytokine interleukin 6 (IL6) is often expressed by multiple cell types within the tumor microenvironment. Here, we show that IL6 is required for the maintenance and progression of pancreatic cancer precursor lesions. In fact, the lack of IL6 completely ablates cancer progression even in presence of oncogenic Kras. Mechanistically, we show that IL6 synergizes with oncogenic Kras to activate the reactive oxygen species (ROS) detoxification program downstream of the MAPK/ERK signaling cascade. In addition, IL6 regulates the inflammatory microenvironment of pancreatic cancer throughout its progression, providing several signals that are essential for carcinogenesis. Thus, IL6 emerges as a key player at all stages of pancreatic carcinogenesis, and a potential therapeutic target.
Keywords: Kras, MAPK, pancreatic cancer, Interleukin 6, ROS
Introduction
Pancreatic cancer is one of the deadliest human malignancies, with a 5-year survival rate of less than 6% (1–3). The dismal survival rate has remained essentially unchanged over the course of the past 40 years, highlighting the need for a deeper understanding of the biology of this disease, which might lead to new targeting strategies. Recent sequencing studies (4) have confirmed the decades old observation that KRAS is the most commonly mutated gene in pancreatic cancer (5, 6). KRAS mutations occur early during disease progression, in pancreatic cancer precursor lesions known as Pancreatic Intraepithelial Neoplasias (PanINs) (7). However, KRAS mutations are found in healthy pancreata at a much higher rate than the incidence of pancreatic cancer (8, 9), suggesting that additional genetic, epigenetic or environmental factors are required for tumorigenesis.
Chronic pancreatitis confers a significantly increased life-time risk to develop pancreatic cancer, and is thus one of the highest known risk factors (10). The relationship between acute pancreatitis and carcinogenesis is less well established; however, acute pancreatitis can progress to chronic pancreatitis in individuals carrying additional risk factors (such as smoking or alcohol abuse) (11). In mice, both chronic and acute pancreatitis synergize with the presence of oncogenic Kras to drive formation of PanINs (12, 13). The cytokine interleukin 6 (IL6) is up-regulated during pancreatitis in mice and humans (14). IL6 plays an essential pro-carcinogenic function in colon and liver cancer (15, 16). In contrast, at least in mice, its role is secondary to the closely related cytokine IL11 in gastric cancer (17). Thus, the relevance of IL6 in carcinogenesis is tissue-specific. Previous studies have identified IL6 and its downstream effector Stat3 as being important for pancreatic cancer initiation in mouse models of this disease (18–20). However, whether IL6 plays a role in inflammation-driven pancreatic carcinogenesis, as well as its role at later stages of carcinogenesis, was not known. These questions have therapeutic relevance, as pancreatitis patients are a population where preventive strategies could be successfully employed to avoid progression to cancer. Preventive strategies that block PanIN progression to cancer could conceivably also be useful in familial pancreatic cancer, as well as to prevent recurrence in patients that have undergone resection of the primary tumor.
In this study, we set out to determine whether sustained IL6 expression was required to initiate pancreatitis-associated pancreatic cancer. We utilized a genetically engineered mouse model of pancreatic cancer, the iKras* mouse, based on pancreas-specific, inducible and reversible expression of oncogenic KrasG12D (Kras*) recently described by our group (21). This model develops pancreatic cancer in a step-wise manner within an intact microenvironment. Our data show that IL6 was dispensable for the initiation of pancreatic cancer precursor lesions in the presence of inflammation. However, we uncovered a previously unrecognized role for IL6 in the maintenance of these precursor lesions and progression to cancer. Thus, our data set the rationale for exploring IL6 as a therapeutic target in pancreatic cancer.
Materials and Methods
Mouse Strains
We generated iKras*;IL6−/− mice by crossing previously described triple transgenic mice iKras* (p48-Cre;R26-rtTa-IRES-EGFP;TetO-KrasG12D) (21) with IL6-deficient mice (B6;129S6-Il6tm1Kopf, Jackson Laboratory). Combinations of single or double mutant littermates were used as controls. Animals were housed in specific pathogen-free facilities of the University of Michigan Comprehensive Cancer Center. Studies were conducted in compliance with University of Michigan University Committee on Use and Care of Animals (UCUCA) guidelines. Pdx1-Cre;KrasLSL-G12D/+;p53fl/+;Rosa26LSL-YFP/+ (KPCY) mice (22) were housed in a specific pathogen-free facility at the University of Pennsylvania and in compliance with Penn IACUC guidelines.
Doxycycline Treatment
iKras* or iKras*;IL6−/− mice were treated with Doxycycline to induce KrasG12D expression. Doxycycline was administered in the low-dose doxycycline chow (50mg/kg) or drinking water, at a concentration of 0.2g/L in a solution of 5% sucrose, and replaced every 3–4 days.
Primary Tumor Cells
Primary tumor specimen implantation and preparation of single-cell suspensions of tumor cells were performed as previously described (23). All samples derived from human subjects were approved by University of Michigan and the Federal Institutional Review Board (IRB). Establishment of primary mouse pancreatic cancer cell line from iKras* p53* mouse tumor was previously described (24).
Histopathological and Histological Analysis
The histopathological analysis was performed as previously described (21). The data was expressed as percentage of total counted clusters. Error bars represent standard error.
Flow Cytometry
Single-cell suspensions of fresh spleen and pancreas were prepared as follows: spleens were crushed and passed through a 40 μm cell strainer, washed once with RPMI/10% FCS, and treated with RBC lysis buffer (eBioscience) to eliminate RBC. Pancreata were minced using sterile scalpels, then incubated in 1 mg/mL collagenase (Sigma-Aldrich) in HBSS for 15 mins at 37°C before passing through a 40 μm cell strainer. Single-cell suspensions were stained in HBSS/2% FCS with the following antibodies: CD3 (17A2), CD4 (RM4-5), CD8α (53-6.7), CD25 (PC61), CD11b (M1/70), F4/80 (BM8), CD11c (HL3), Gr-1 (RB6-8C5) and Foxp3 (FJK-16s; all from BD Pharmingen) and CD45 (MCD4530, Invitrogen). Flow cytometry was done using a CyAn™ ADP Analyzer (Beckman Coulter), and data were analyzed with Summit 4.3 software.
ROS Induction and Detection
Primary mouse pancreatic cancer cell line 9805 was treated with 600 μM H2O2 for 1h and the presence of ROS was detected using CellROX Green reagent (C10444, Invitrogen), a fluorogenic probe, according to the manufacturer’s instructions. Briefly, cells were then incubated with 10 μM CellROX Green in RPMI with 10% FBS for 30 min at 37°C. Cells were then washed in PBS and imaged on a Olympus IX71 inverted microscope. Signal intensity was analyzed using Image-Pro Plus software.
In Vivo Anti-IL6 Treatment
Pdx1-Cre;KrasLSL-G12D/+;p53fl/+;Rosa26LSL-YFP/+ mice aged 10 weeks were randomized to two treatment arms: anti-IL6 antibody treatment (25 mg/kg; clone MP5-20F3, Bioxcell, West Lebanon, NH) or Rat IgG1 control (25 mg/kg; BE0088, BioXCell, West Lebanon, NH). Mice (3 for each group) received treatments on days 0, 2, 4, and 6 intraperitoneally then sacrificed on day 7 for histologic analysis. PanIN lesions in 3 sections per mouse were quantified by grade in a blinded manner. Data expressed as number of ADMs or PanINs of each grade per medium (10X) powered field. No discernible side effects were noted in either of the treatment groups.
Statistical Analysis
All data were presented as means ± SEM. Intergroup comparisons were performed using the Student’s t test. Prism 6 was used for all statistical analyses, and P<0.05 was considered statistically significant.
Detailed procedures and standard procedures are included in the Supplemental Methods; detailed antibody information and primer sequences are listed in Table S1 and S2.
Results
IL6 is expressed by several cell types within the pancreatic cancer microenvironment
Analysis of human and mouse pancreatic cancer samples revealed IL6 immuno-staining in several cell compartments, including epithelial cells, Smooth Muscle Actin (SMA)-positive fibroblasts and immune cells (Figure S1A and S1B). Interestingly, IL6 was expressed in mouse primary pancreatic fibroblasts only upon incubation with conditioned medium from pancreatic cancer cells (Figure S1C). Since previous studies had described expression restricted to immune cells (18), we sought to confirm the immunostaining results by quantitative RT-PCR, and observed that primary mouse pancreatic fibroblasts in culture expressed Il6 mRNA when exposed to conditioned medium from pancreatic cancer cells (Figure S1D). Moreover, one of two primary pancreatic cancer cells and three commercially available pancreatic cancer cells lines tested expressed IL6 (Figure S1E). Thus, multiple sources of IL6 are present within the pancreatic cancer microenvironment.
IL6 is required for PanIN formation in iKras* mice with embryonic Kras activation
In iKras* mice the expression of oncogenic Kras can be timed at will by adding or removing doxycycline (doxy) from the animal’s food or water (21). We previously reported that activation of KrasG12D in adult animals leads to PanIN formation with low penetrance and long latency (21). In contrast, embryonic activation of KrasG12D (Figure S1F) resulted in PanIN formation in all the animals by 6 weeks of age (Figure S1I). In order to determine the effect of IL6 inactivation on PanIN formation in iKras* mice we generated iKras*;IL6−/− mice (Figure 1A). iKras*;IL6−/− and iKras* littermates were sacrificed at 6 weeks of age and their pancreata were harvested (Figure S1F). The expression of the KrasG12D transgene was comparable in iKras* and iKras*;IL6−/− mice, and undetectable in wild-type controls. Il6 mRNA was elevated in iKras* pancreata compared to control, and, as expected, undetectable in iKras*;IL6−/− mice (Figure S1G). Histological examination of iKras* pancreata revealed PanIN lesions surrounded by extensive fibro-inflammatory stroma (Figure S1I). We detected IL6 expression, as well as activation of the downstream effector Stat3, both in the lesions and in the surrounding stroma. Moreover, we detected elevated expression of p-ERK1/2, as readout of MAPK pathway activity. Similar findings were obtained in iKras*;IL6+/− mice. In iKras*;IL6−/− pancreata we observed a majority of normal acini with infrequent areas of acinar-ductal metaplasia (ADM) and rare PanINs (Figure S1I and histopathological analysis in Figure S1H). As expected, IL6 expression was completely abrogated in these tissues. Interestingly, the residual lesions in iKras*;IL6−/− mice had reduced p-ERK1/2 and p-Stat3 compared to lesions in iKras* mice. Thus, in the iKras* model, IL6 is important for the onset of PanINs. These findings are consistent with the observation that IL6 is important for pancreatic cancer initiation (18).
Figure 1. IL6 expression is dispensable for pancreatitis-induced PanIN formation.
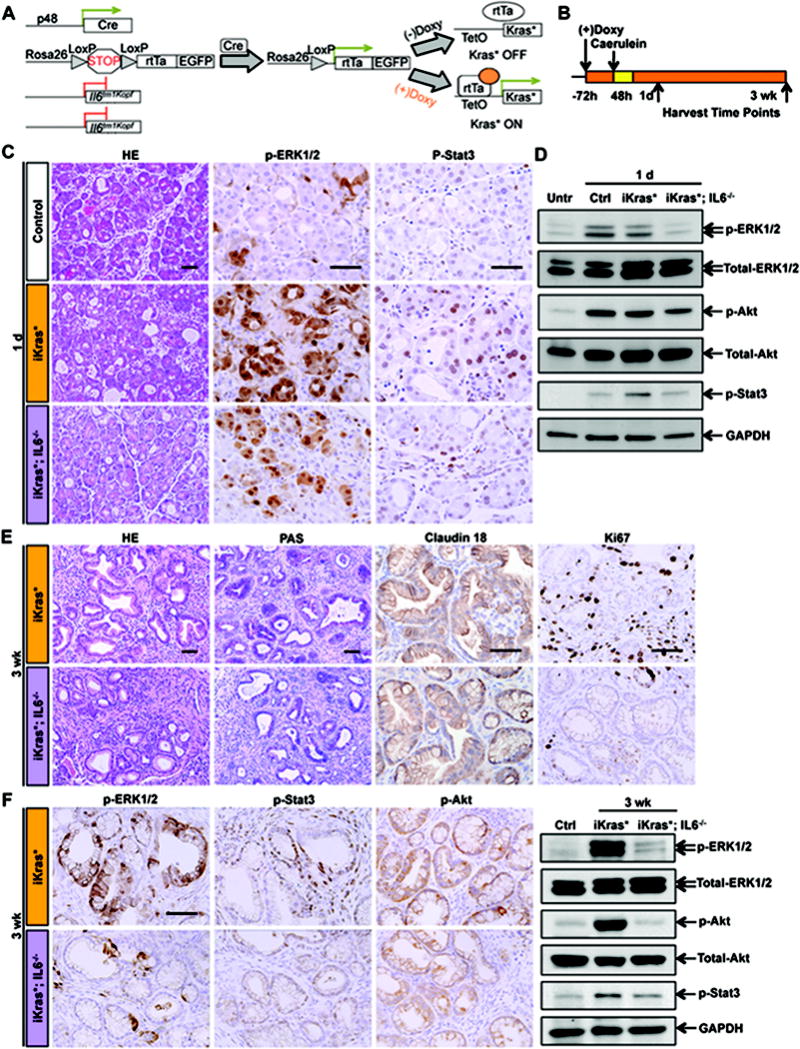
(A) Genetic makeup of the iKras*;IL6−/− mouse model. (B) Experimental design, n=4~7. (C) H&E and immunohistochemistry staining for p-ERK1/2 and p-Stat3 in control, iKras* and iKras*;IL6−/− mice pancreata 1 day post pancreatitis induction. Scale bar 50μm. (D) Western blot for p-ERK1/2, total-ERK1/2, p-Akt, total-Akt, p-Stat3 and GAPDH in untreated, control, iKras* and iKras*;IL6−/− mice pancreata 1 day post pancreatitis induction. (E) H&E, PAS and immunohistochemistry staining for Claudin 18 and Ki67 in iKras* and iKras*;IL6−/− mice pancreata 3 weeks post pancreatitis. Scale bar 50μm. (F) Immunohistochemistry staining and western blot for p-ERK1/2, p-Stat3 and p-Akt in iKras* and iKras*;IL6−/− mice pancreata 3 weeks post pancreatitis. Scale bar 50μm.
Pancreatitis-driven PanINs in IL6 deficient mice have altered signaling and reduced proliferation
In a next set of experiments we let iKras* and iKras;IL6−/− mice reach adulthood in absence of doxy, thus maintaining Kras* expression off. Doxy was then administered when the animals reached 4–6 weeks of age, to induce Kras* expression, and pancreatitis was induced by two series of caerulein injections, for two consecutive days, starting 72 hours after doxy administration, as previously described (12, 21) (Figure 1B). One day later, pancreatitis was evident in control, iKras* and iKras*;IL6−/− pancreata, with characteristic acinar damage, acinar-ductal metaplasia, edema and inflammatory infiltrates, as well as increased expression of p-ERK1/2 and p-Stat3 (Figure 1C). Flow cytometry showed the number of infiltrating CD45+ immune cells was similar in all the experimental groups; although we observed a trend towards reduced infiltration of T cells, and a significant increase in macrophages (but not in the related myeloid-derived suppressor cells) in iKras*;IL6−/− mice (Figure S2A). The acute response to pancreatitis is accompanied by up-regulation of the MAPK pathway (25, 26), the PI3K/Akt pathway (27) and by activation of Stat3 (19). In iKras*;IL6−/− mice the levels of p-ERK1/2, p-Stat3 and, to a lesser extent, p-AKT were reduced (Figure 1D). Thus, while the absence of IL6 did not prevent induction of pancreatitis, it qualitatively changed the response to the inflammatory stimulus.
In order to determine whether IL6 deficiency altered PanIN formation, we dissected tissues 3 weeks post-pancreatitis, when full recovery is expected in control animals, and extensive PanIN lesions are expected in iKras* mice (21). Upon histopathological analysis, both in iKras* and in iKras*;IL6−/− mice we observed pancreas-wide PanIN lesions with accumulation of fibro-inflammatory stroma, characteristic PAS staining, and expression of the PanIN marker Claudin 18 (Figure 1E). Quantification of the type and extent of lesions revealed no significant changes in iKras*;IL6−/− compared to iKras* mice (Figure 2E, 3 week time point). Thus, PanIN lesions formed even in absence of IL6, upon induction of pancreatitis.
Figure 2. IL6 is necessary for PanIN progression and maintenance.
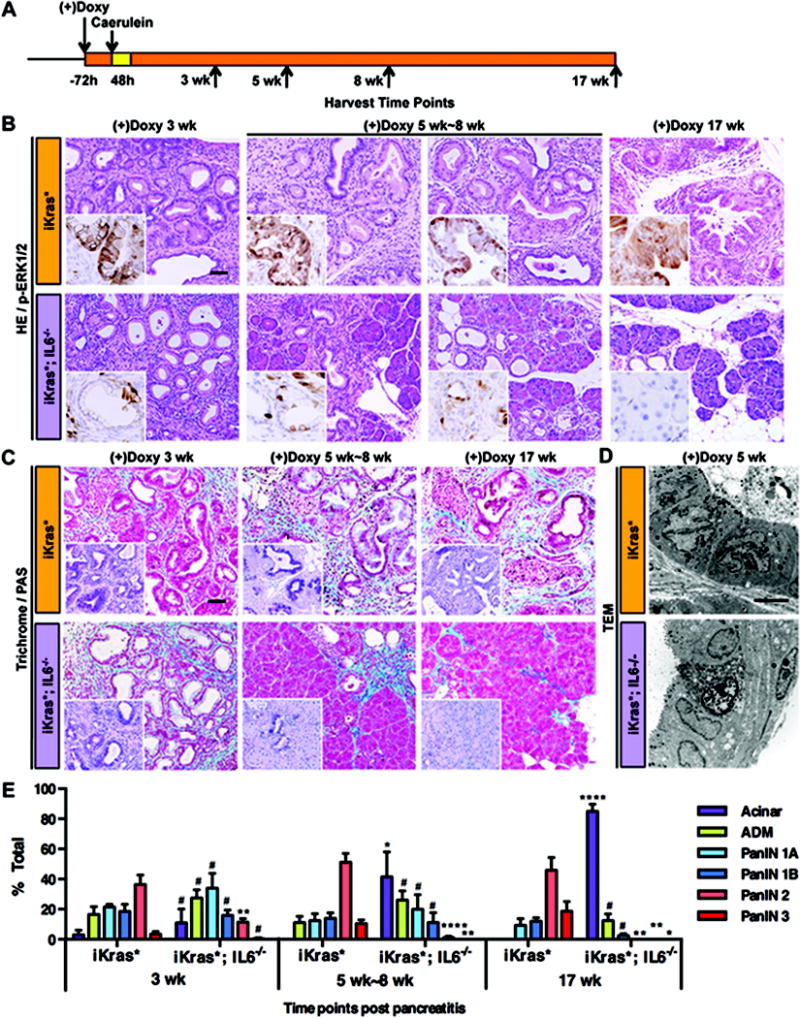
(A) Experimental design, n=4~7. (B) H&E and p-ERK1/2 staining (insets) of iKras* and iKras*;IL6−/− mice pancreata 3 weeks, 5 weeks~8 weeks and 17 weeks following pancreatitis. Scale bar 50μm. (C) Gomori Trichrome staining and Periodic Acid Schiff staining (insets) of iKras* and iKras*;IL6−/− mice pancreata. Scale bar 50μm. (D) Transmission electron microscope image of iKras* and iKras*;IL6−/− mice pancreata 5 weeks following pancreatitis. Scale bar 10μm. (E) Pathological analysis. Data represent mean ± SEM, n=3~5. The statistical difference between iKras* and iKras*;IL6−/− mice at the same time point per lesion type was determined by two-sided Student’s t-test. *p<0.05, **p<0.01, ****p<0.0001, #not significant.
Further characterization of the lesions in iKras* and iKras*;IL6−/− animals revealed striking differences. First, the proliferation index was dramatically reduced in absence of IL6, both within the lesions and in the surrounding stroma (Figure 1E, Ki67 immunostaining). Second, the levels of p-ERK1/2, p-Akt and p-Stat3 were reduced in IL6-deficient pancreata (Figure 1F).
In mice bearing mutant Kras*, PanIN formation is associated with persistence of inflammatory infiltrates (28). In order to determine whether the absence of IL6 altered the prevalence, or nature of the inflammatory infiltrates we performed flow cytometry for components of both the innate and adaptive immune systems in iKras* and iKras*;IL6−/− pancreata. Our analysis revealed a higher number of inflammatory cells in iKras* pancreata compared to control 3 weeks post-pancreatitis – consistent with the completed repair of the pancreatic tissue in control mice and with the accumulation of a fibro-inflammatory stroma in iKras* animals. When we compared iKras* with iKras*;IL6−/− pancreata we did not observe a change in the total immune component, or in the number and nature of most infiltrating T cell subsets (Figure S2A). However, we observed a reduction in several immune subtypes that have been associated with tumor progression, such as macrophages, myeloid-derived monocyte suppressor cells, and a trend towards a reduction in the number of other myeloid-derived suppressor cell subsets and regulatory T cells (Figure S2A). We also observed a dramatic decrease in infiltrating mast cells (Figure S2B), a cell type that has been linked to pancreatic carcinogenesis (29); however, whether the lack of mast cells was the cause or effect of the lack of progression remains to be established.
Taken together, our data showed that pancreatitis-driven PanIN formation was independent of IL6, possibly mediated by other cytokines that are released during the induction of pancreatitis. However, lesions formed in absence of IL6 were qualitatively distinct. In fact, IL6 was required for the activation of the MAPK signaling pathway, and, to a lesser extent, for the activation of Akt and Stat3. Moreover, IL6 deficient PanINs had a low proliferation index. Thus, while histologically similar, PanINs in iKras and iKras;IL6−/− mice had significant molecular differences in the activation of signaling pathways underlying pancreatic cancer progression.
IL6 is necessary for PanIN maintenance and progression
We next compared the kinetics of PanIN progression between iKras* and iKras*;IL6−/− mice. We aged cohorts of each genotype (n=4~7/genotype/time point) for 3, 5, 8 or 17 weeks following induction of pancreatitis (see scheme in Figure 2A). As previously described, at the 3 weeks time-point histopathological analysis revealed no significant differences between iKras* and iKras*;IL6−/− tissues. Over time, however, iKras* mice developed high-grade PanIN lesions with sustained MAPK activity (Figure 2B). We observed PAS-positive epithelial cells and expansion of a collagen-rich stroma (Figure 2C). In contrast, in iKras*;IL6−/− tissues we observed a progressive decrease in the prevalence of PanINs and stroma and conversely an increase of acinar clusters (Figure 2B and 2C). PanIN lesions are characterized by distinct microscopic features, that can be highlighted by Transmission Electron Microscopy (TEM): multilobular nuclei, lack of large secretory granules (which are found in acinar cell), microvilli on the luminal surface and large, irregular ductal lumen (Figure 2D); PanINs in iKras*;IL6−/− samples had similar features, although the nuclear shape was less irregular. In addition, by 5 weeks after the induction of pancreatitis, apoptotic cells were detected within the ductal structures (Figure 2D). Within the residual lesions, MAPK activation was low, as determined by p-ERK1/2 immunostaining, and was confined to a small subset of cells (Figure 2B, insets). In addition, we observed reduction in p-Stat3 and p-Akt levels and, as expected, lack of IL6 expression (S3A–C). By 17 weeks, the pancreata of iKras*;IL6−/− mice were populated by normal acini for over 80% of the tissue, with rare ADM and sporadic PanIN1A, as confirmed by histopathological analysis of de-identified samples (Figure 2E). Since no lesions were observed at this time point, it was not surprising to observe limited p-ERK1/2 and p-Akt staining; occasional p-Stat3 was observed in acini and ducts of normal appearance, possibly indicating a residual inflammatory response in the pancreas (Figure S3C). Contemporary to the changes in the epithelium, the stroma surrounding the PanIN lesions also underwent major remodeling, resulting in very little residual fibrosis and conversely in accumulation of adipose tissue (Figure 2B and 2C). This process correlated with the up-regulation of MMP7, MT1-MMP and, to a lesser extent, MMP9 (Figure S4A). These proteases might play a role in the remodeling of the tissue, but future experimental work, beyond the scope of the current study, will be needed to address this possibility. Interesting, the expression of SMA, a fibroblast activation marker, decreased prior to the remodeling of the stroma in iKras*;IL6−/− mice (Figure S3D). Thus, remodeling of the stroma was preceded by the return of fibroblasts to a non-active state, a similar finding to what we previously observed upon inactivation of Kras* expression in the pancreas (21). We then investigated whether IL6 influenced the expression of other inflammatory cytokines in tissues harvested 3, 5–8 or 17 weeks after pancreatitis. We did not observe changes in several cytokines and inflammation-related genes such as Il2, Il4, Il10, Il11, Il17, Cox2, Tnfα and Ifnγ (Figure S4B). However, in addition to confirming the lack of Il6 expression, we detected lower levels of Il1β (a proinflammatory cytokine), Slfn4 (a myeloid activation marker (30)) and GM-CSF (a cytokine that is required for pancreatic cancer growth (31, 32)) in iKras*;IL6−/− mice compared with iKras* (Figure 3A).
Figure 3. IL6 regulates proliferation and survival of PanIN cells.
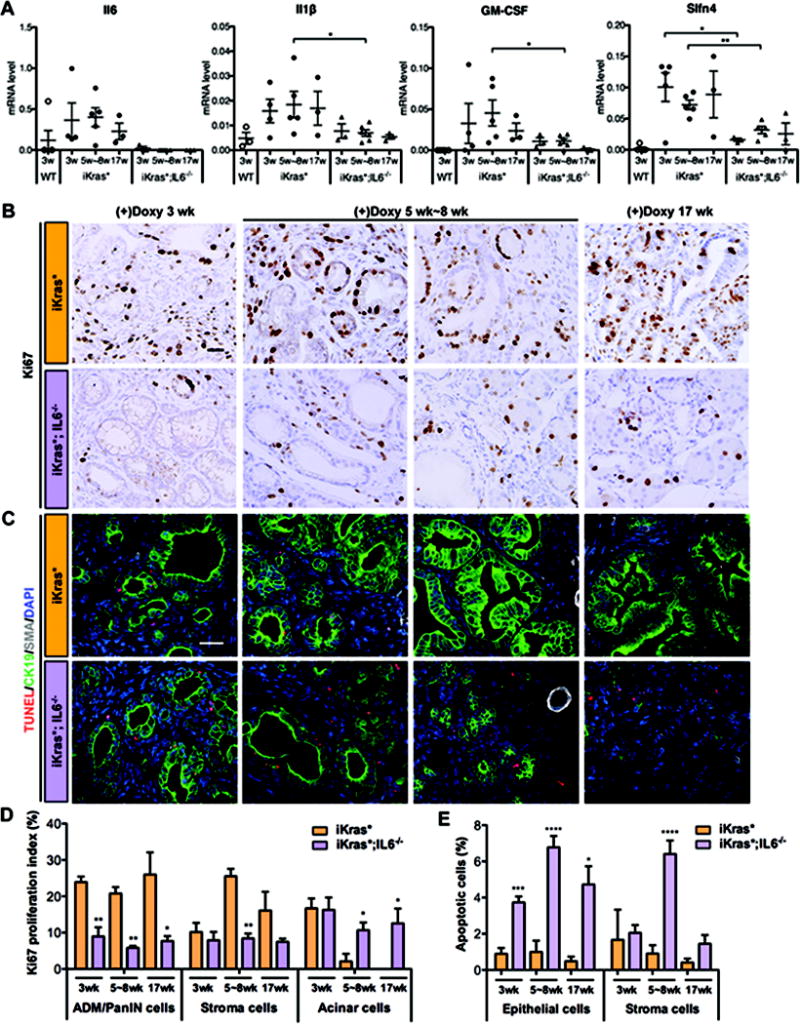
(A) RT-qPCR for Il6, Il1β, GM-CSF and Slfn4 expression in WT, iKras* and iKras*;IL6−/− mice pancreata. (B) Ki67 immunohistochemistry staining of iKras* and iKras*;IL6−/− mice pancreata 3 weeks, 5 weeks~8 weeks and 17 weeks following pancreatitis. Scale bar 25μm. (C) TUNEL (red) and co-immunofluorescence staining for CK19 (green), SMA (grey) and DAPI (blue). Scale bar 25μm. (D) Ki67 proliferation index in ADM/PanIN, stroma and acinar cells and (E) quantification of apoptotic cells in iKras* and iKras*;IL6−/− mice pancreata. Data represent mean ± SEM, n=3. The statistical difference was determined by two-sided Student’s t-test, comparing the different genotype mice at each time point for each category considered (acini, stroma, ADM/PanIN). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
In summary, while pancreatitis-induced lesions in iKras* mice progressed over time to high-grade PanINs surrounded by abundant stroma, neoplastic lesions in iKras*;IL6−/− mice regressed, the stroma was remodeled, eventually resulting in a normal pancreatic parenchyma, albeit with some adipose tissue infiltration. The remodeling process in iKras*;IL6−/− pancreata resembled the effect of Kras* inactivation in PanIN bearing iKras* mice (21); indicating that IL6 is a key player in Kras*-driven carcinogenesis.
IL6 regulates proliferation, survival and differentiation status of PanIN cells
Since expression of IL6 was associated with enhanced proliferation of PanIN lesions (Figure 3B), we quantified the proliferation index for the different cell populations present in the tissues, namely ADM/PanIN cells, components of the stroma and acinar cells. In iKras* mice the PanIN cells had a high proliferation index that was maintained over time, and proliferation in the stroma reached a peak at 5–8 weeks. Proliferation in both compartments was significantly reduced in iKras*;IL6−/− lesions. Interestingly, when we compared the proliferation of the acinar compartment between the two genotypes we observed a significant increase (p<0.05) in iKras*;IL6−/− compared to iKras* at the 5–8 weeks and at the 17 weeks time points (Figure 3B and 3D). Taken together, our data indicate that IL6 promotes proliferation of the metaplastic/dysplastic cells, and possibly prevents acinar proliferation, a normal aspect of the tissue recovery following pancreatitis.
In iKras*;IL6−/− mice, this decrease in proliferation potentially explains the lack of progression in the lesions, but not the progressive elimination of the lesions. To investigate this aspect, we investigated two possible mechanisms of regression: apoptosis of PanIN cells, as well as re-differentiation of PanIN cells into normal acini. First, we determined that PanIN cells have severely compromised survival in absence of IL6. Apoptotic cells (as determined by TUNEL or cleaved caspase 3 staining) were occasionally present in iKras* tissues; the overall level of apoptosis was low (less than 1% of the epithelial cells), and it decreased over time (Figure 3C, 3E, S4C and S5A). In contrast, iKras*;IL6−/− PanINs had elevated levels of apoptosis both within the epithelium and in the surrounding stroma (Figure 3C, 3E, S4C and S5A). In addition to the immunostaining analysis, we performed RT-qPCR to determine the relative ratio between the mediators of apoptosis Bak and Bax and the anti-apoptotic genes Bcl-2 and Bcl-x. At the 5–8 week time point, when most of the changes within the tissue were taking place, iKras*;IL6−/− tissues, compared with iKras*, had a higher pro-apoptotic ratio, for Bak/Bcl-x and for Bax/Bcl-x (Figure S5B). In a second set of experiments, we performed co-immunofluorescent staining with CK19, a ductal and PanIN marker, and amylase, an acinar marker. In iKras* tissues, as expected, we observed almost exclusive expression of CK19 in epithelial cells (Figure 4A and 4B). In iKras*;IL6−/− tissues, CK19 was prevalent at 3 weeks post-pancreatitis, but by 5–8 weeks post pancreatitis up to a third of the epithelial cells co-expressed CK19 and amylase, and a significant amylase positive population was observed (Figure 4A and 4B). By 17 weeks, CK19-only cells were rare, while amylase positive cells represented the majority of the epithelial population, and some cells co-expressing CK19 and amylase were still present (Figure 4A and 4B). The transcription factor Mist1 is a key regulator of acinar cell differentiation (33), and its expression is lost during PanIN formation. Importantly, inactivation of Mist1 in the context of oncogenic Kras accelerates pancreatic carcinogenesis (34), while conversely forced expression of Mist1 prevents Kras-driven carcinogenesis (35). As expected, we did not observe any Mist1 expression in the lesions of iKras* mice (Figure 4C). Similarly, in iKras*;IL6−/− lesions at 3 weeks post-pancreatitis Mist1 expression was absent. However, by 5 weeks post-pancreatitis, Mist1 was expressed in the residual ductal/PanIN structures thus confirming their mixed differentiation status (Figure 4C). At later time points, Mist1 expression was confined to acini and excluded from the ducts, as in the normal pancreas.
Figure 4. IL6 inhibition results in ductal-to-acinar re-differentiation of PanIN cells.
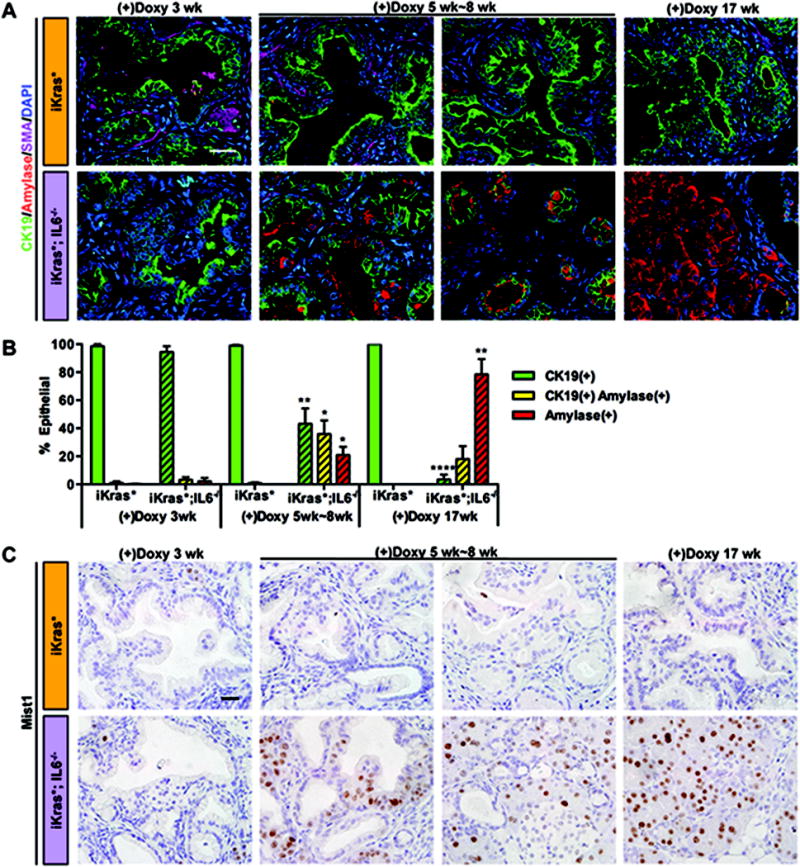
(A) Co-immunofluorescence staining for CK19 (green), amylase (red), SMA (magenta) and DAPI (blue). Scale bar 25μm. (B) Quantification of CK19 and amylase single or double positive epithelial cells in iKras* and iKras*;IL6−/− mice pancreata. Data represent mean ± SEM, n=3. The statistical difference was determined by two-sided Student’s t-test. For each time point, the number of single or double-positive cells was compared between genotypes. *p<0.05, **p<0.01, ****p<0.0001. (C) Mist1 immunohistochemistry staining. Scale bar 25μm.
Taken together, our data indicate that PanIN elimination is underscored by a combination of apoptotic cell death, re-differentiation of cells towards the acinar lineage and increased proliferation of acinar cells, mechanisms of pancreas repair that we have previously observed upon inactivation of oncogenic Kras in PanIN bearing mice (21).
IL6 is required for Nrf2 up-regulation and oxidative stress resistance
The onset of PanIN formation is accompanied by the activation of a Reactive Oxygen Species (ROS) detoxification program (36). PanINs that form in mice deficient for Nrf2, a key factor in the ROS detoxification pathway, are non-proliferative (36). Expression of Nrf2 has been shown to be activated downstream of oncogenic Kras (37) via the MAPK/ERK signaling pathway (36, 38). Given that iKras*;IL6−/− mice had a defect in MAPK/ERK activation, we decided to investigate whether Nrf2 expression was defective in these animals. Indeed, we found robust expression of Nrf2 in PanINs of iKras* mice at all the time points studied, but drastically reduced Nrf2 expression in iKras*;IL6−/− lesions at those time points when lesions could still be detected (Figure 5A). Thus, the presence of oncogenic Kras* was not sufficient to activate the ROS detoxification program, and additional inflammatory signals were required.
Figure 5. IL6 inhibits ROS induction in pancreatic cancer cells.
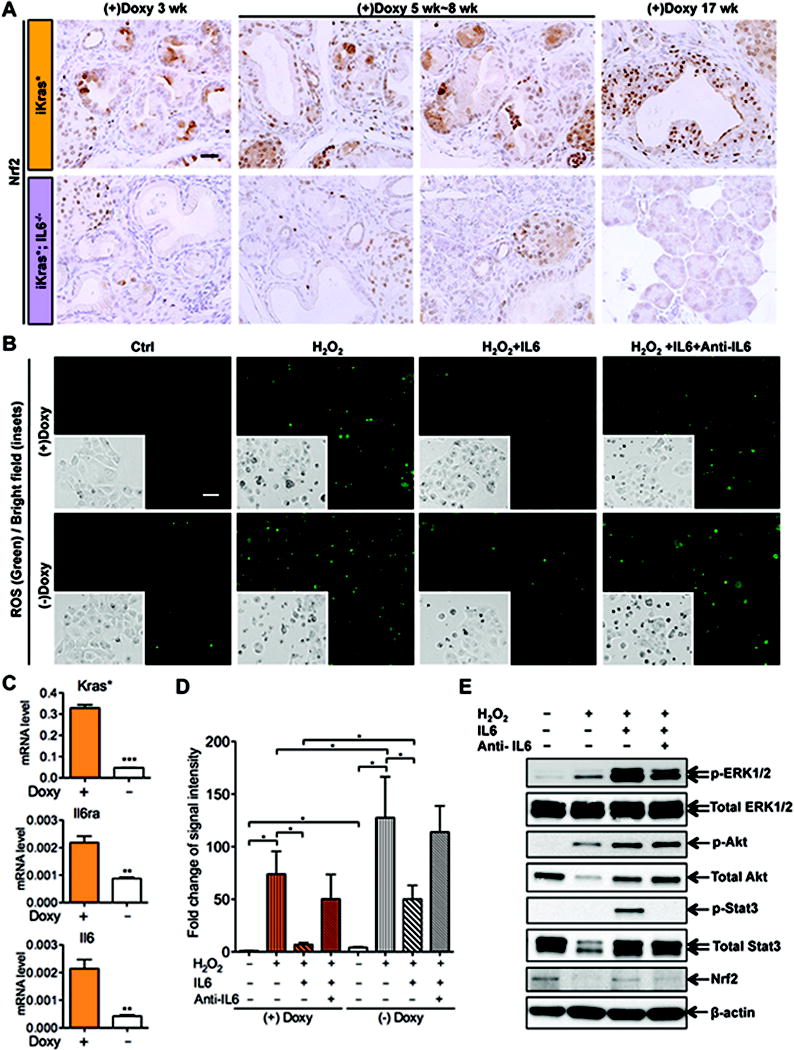
(A) Nrf2 immunohistochemistry staining of iKras* and iKras*;IL6−/− mice pancreata 3 weeks, 5 weeks~8 weeks and 17 weeks following pancreatitis. Scale bar 25μm. (B) ROS staining and bright field images of primary mouse pancreatic cancer cell line 9805 treated with 600 μM H2O2 alone, or in the presence of IL6 (50ng/ml), or IL6 pre-incubated with anti-IL6 (8μg/ml), for 1h. Scale bar 50μm. (C) RT-qPCR of Kras*, Il6ra and Il6 expression in 9805 cells with or without Doxy treatment. Data represent mean ± SEM, n=3. **p<0.01; ***p<0.001. (D) Quantification of ROS signal intensity. Data represent mean ± SEM, n=3. *p<0.05. Each category was compared between the two genotypes for each time point. (E) Western blot for p-ERK1/2, total-ERK1/2, p-Akt, total-Akt, p-Stat3, total Stat3, Nrf2 and β-actin in 9805 cells 30 minutes following ROS induction.
To investigate in further detail the effect of IL6 depletion on the ability of tumor cells to cope with oxidative damage, we used the primary mouse pancreatic cancer cell line, 9805, derived from iKras*;p53* mice (24). The in vitro approach allowed us to investigate the effect of IL6 on the tumor cells, independently from possible indirect effects mediated by components of the stroma. Of note, 9805 cells express the Il6 receptor (Il6r) in a Kras*-dependent manner, and are thus able to respond to IL6 signaling (Figure 5C). The cancer cells were seeded to chamber slides at 50~60% confluency and cultured in complete medium containing doxy at 1ug/ml overnight. Then, the cells were subdivided into 4 groups: control, treated with H2O2 alone to induce intracellular ROS accumulation, treated with H2O2 in the presence of recombinant IL6, or IL6 pre-incubated with an IL6 blocking antibody - as these cells express low levels of IL6 in a Kras-dependent manner (Figure 5C). The experiment was performed in presence of doxy (thus, with expression of oncogenic Kras*) or without doxy (thus, with no oncogenic Kras). First, we confirmed the notion that oncogenic Kras* protects the cells from ROS accumulation, as both ROS staining and the number of dead cells were higher in absence of doxy. Second, we determined that treatment with IL6 had a protective effect both on ROS accumulation and on cell death; this effect was observed independently from Kras* expression, and was abrogated by the anti-IL6 antibody (Figure 5B, quantification in 5D and Figure S5C). In order to determine whether the MAPK pathway was affected by induction of oxidative stress we performed western blot analysis for p-ERK1/2 in our samples. Of note, even though 9805 cells expressed some IL6, exogenous IL6 was effective in increasing the activation of downstream pathways such as MAPK, PI3K and p-Stat3. We observed that treatment with H2O2 caused a modest increase in p-ERK1/2, even in absence of exogenous IL6. However, there was a strong increase of p-ERK1/2 in cells treated with H2O2 in the presence of IL6, which was reversed by the anti-IL6 antibody. A similar effect was observed with p-Akt and Nrf2, while p-Stat3 did not seem to be affected by the induction of oxidative stress (Figure 5E). Thus, our data indicate that IL6 provides protection from oxidative stress.
In order to determine whether the failure to activate the ROS detoxification program in iKras*;IL6−/− mice could explain the lack of PanIN progression, we used a ROS scavenger, N-Acetyl-L-Cysteine (NAC). In brief, iKras*;IL6−/− mice were treated with NAC starting 2 weeks after induction of pancreatitis, and then for 3 weeks (Figure S5D). The mice were harvested at 5 weeks post-pancreatitis. Compared with untreated iKras*;IL6−/− mice (Figure 2B), NAC-treated iKras*;IL6−/− mice revealed partial rescue of carcinogenesis. Their pancreata had extensive PanIN lesions with elevated p-ERK1/2 and p-Stat3 levels, and rescue of proliferation both within the epithelial and the stroma compartments (Figure S5E). In conclusion the requirement for IL6 during pancreatic carcinogenesis might be explained, at least in part, by preventing ROS accumulation.
IL6 prevents tissue repair following Kras inactivation
The iKras* mouse allows us to turn off Kras* expression at will, thus it is a suitable model to study pancreatic repair following inactivation of the Kras oncogene. In order to study the role of IL6 during pancreatic repair, we performed the following experiment: adult mice were kept on doxy for 3 weeks following pancreatitis induction; then some of the animals were harvested, and the others were placed on doxy-free chow and water. 4–5 mice per genotype were then harvested 1 day, 3 days and 2 weeks later (Figure 6A), based on our previous characterization of the dynamics of pancreas repair in this model (21). In iKras* mice, IL6 was expressed through most of the repair process, though with decreasing expression levels over time (Figure S6B and S6D). Whole tissue analysis by western blot indicated that inactivation of oncogenic Kras* lead to rapid down-regulation of p-ERK1/2 and p-Akt levels within 1 day, and to a transient up-regulation of cleaved caspase 3 levels, indicating cell death. However p-Stat3 remained high initially, possibly as a result of its expression in the inflammatory cells that infiltrate the tissue during the repair process (Figure S6C). In fact, immunohistochemistry revealed abundant p-Stat3 positive cells within the stroma compartments (Figure S6E). iKras*;IL6−/− mice had lower levels of p-ERK1/2, p-Akt and p-Stat3 than iKras* mice; these pathways were further down-regulated upon Kras* inactivation. In contrast, these mice had a higher basal level of cleaved caspase 3, and it further increased in a transient manner following Kras* inactivation. However, cleaved caspase 3 became undetectable as early as 3 days following Kras* inactivation (Figure S6C).
Figure 6. IL6 prevents tissue repair following Kras* inactivation.
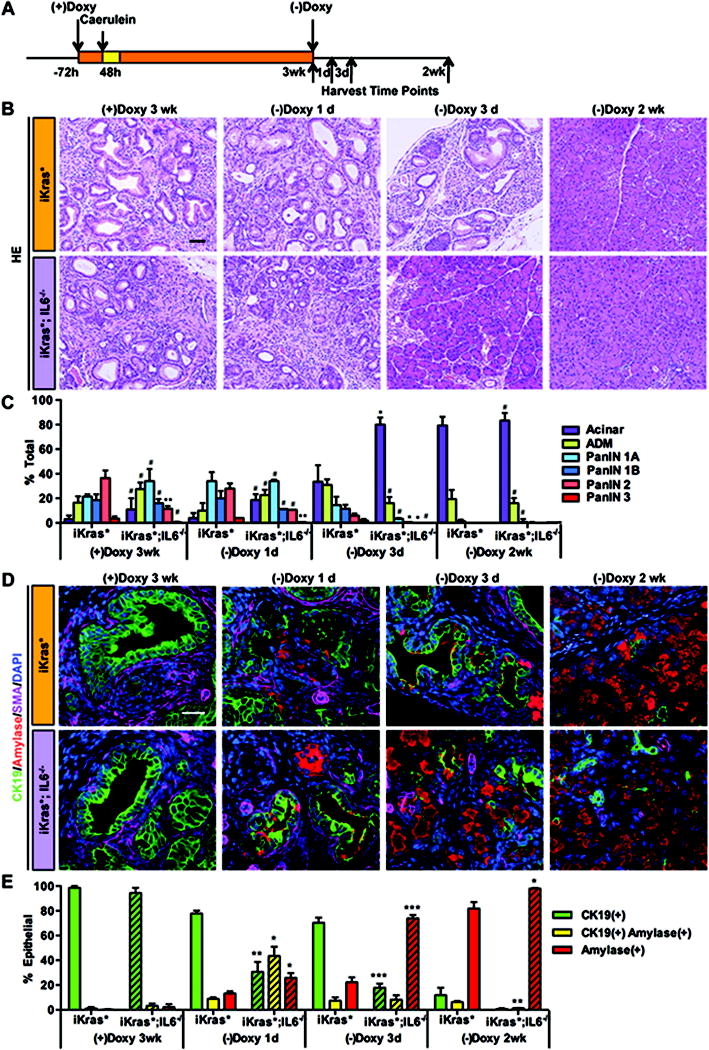
(A) Experimental design, n=4~5. (B) H&E staining of iKras* and iKras*;IL6−/− mice pancreata 3 weeks post pancreatitis and 1 day, 3days and 2 weeks following Kras* inactivation. Scale bar 50μm. (C) Pathological analysis. Data represent mean ± SEM, n=3~4. The statistical difference was determined by two-sided Student’s t-test. *p<0.05, **p<0.01, #not significant. (D) Co-immunofluorescence staining for CK19 (green), amylase (red), SMA (magenta) and DAPI (blue). Scale bar 25μm. (E) Quantification of CK19 and amylase single or double positive epithelial cells in iKras* and iKras*;IL6−/− mice pancreata. Data represent mean ± SEM, n=3. The statistical difference was determined by two-sided Student’s t-test. *p<0.05, **p<0.01, ***p<0.001.
In iKras* mice, the repair process was complete by 2 weeks (Figure 6B and quantification in 6C). Consistently, PAS positivity was still abundant 1 day after Kras* inactivation, while by 3 days the dysplastic ducts were mostly lined by PAS negative cells (Figure S7A). Co-immunostaining for CK19, amylase and SMA highlighted some limited expression of amylase already 1 day after Kras* inactivation, and clusters of CK19/amylase positive cells with ductal morphology 3 days after Kras* inactivation (Figure 6D and quantification in 6E). The stroma was still SMA positive 3 days after Kras* inactivation (Figure 6D, top row, magenta staining, and Figure S7B), and trichrome staining for collagen deposition was still abundant (Figure S7C). iKras*;IL6−/− mice had an accelerated repair process, with several acinar clusters already evident 1 day after Kras* inactivation, and almost complete recovery as early as 3 days after doxy removal (Figure 6B and 6C). Consistently, mixed differentiation acinar-ductal cells were common 1 day after Kras* inactivation, and a majority of normal acini, amylase positive and CK19 negative, populated the tissue by 3 days after Kras* inactivation (Figure 6D and 6E). The appearance of mixed acinar-ductal differentiation cells is preceded by expression of the transcription factor Mist1. Interestingly, we observed earlier re-expression of Mist1 in iKras*;IL6−/− pancreata (1 day after inactivation of Kras* compared to 3 days after inactivation in iKras* mice), possibly explaining the accelerated recovery of the pancreas parenchyma (Figure S8A). Remodeling of the stroma also occurred in an accelerated manner: both SMA and trichrome staining were clearly down-regulated already 1 day after Kras inactivation, and then abrogated in large areas of the pancreas by 3 days (Figure 6D, bottom row, magenta staining, and Figure S7B and S7C).
In pancreata that have been exposed to Kras* activity for a limited period of time, Kras inactivation results in re-differentiation of PanINs to acinar cells and proliferation of the newly formed acini (21). Upon Kras* inactivation, there is a decrease in proliferation both in the CK19(+) epithelial cell compartment and in the stroma; in contrast, CK19(−) epithelial cells, corresponding to the newly formed acinar cells, enter the cell cycle, as part of the repair process. In iKras*;IL6−/− pancreata, acinar cells were formed and entered the cell cycle sooner upon Kras inactivation, possibly contributing to the faster recovery process (Figure S8B and S8C, quantification in S8D). Thus, while IL6 was required for proliferation of PanIN cells and stroma, it was dispensable for – and possibly inhibited – proliferation of normal acinar cells. Our data indicate that IL6 has a negative effect on pancreatic tissue repair, possibly inhibiting pancreatic acinar differentiation and acinar cell proliferation.
Therapeutic inhibition of IL6 causes PanIN regression
In a final set of experiments we investigated the effect of blocking IL6 signaling in Pdx1-Cre;KrasLSL-G12D/+;p53fl/+;Rosa26LSL-YFP/+ (KPCY) (22, 39) mice using an anti-IL6 blocking antibody, a strategy that has therapeutic relevance. The KPCY model was chosen because of its proven relevance to clinical response in patients (40), and for its predictable disease progression. Our goal was to treat KPCY mice after PanIN lesions had developed, in order to determine whether IL6 was required for PanIN maintenance. Ten-week old KPCY mice were randomized to two treatment arms, and administered respectively anti-IL6 antibody or isotype control. The animals were sacrificed 7 days after the beginning of the treatment (see scheme in Figure 7A). The tissue was then prepared for histopathological examination. In the control group (n=3), mice presented with PanINs interspersed within normal exocrine tissue and areas of ADM, as expected based on the disease progression in this model (Figure 7C). While the majority of PanINs were of grade 1A-2, PanIN3 lesions were also observed (Figure 7B). In contrast, in the anti-IL6 treated group, the tissue was largely populated by normal acini, with occasional areas of ADM and virtually no PanINs (Figure 7D and quantification in Figure 7B). Thus, these results indicate that IL6 signaling is required for PanIN maintenance once initiated, mimicking the findings obtained in iKras* mice using genetic inactivation of IL6. Notably, the requirement for IL6 was maintained even in presence of a loss-of function allele of the tumor suppressor p53.
Figure 7. In vivo anti-IL6 treatment reverses PanIN lesions.
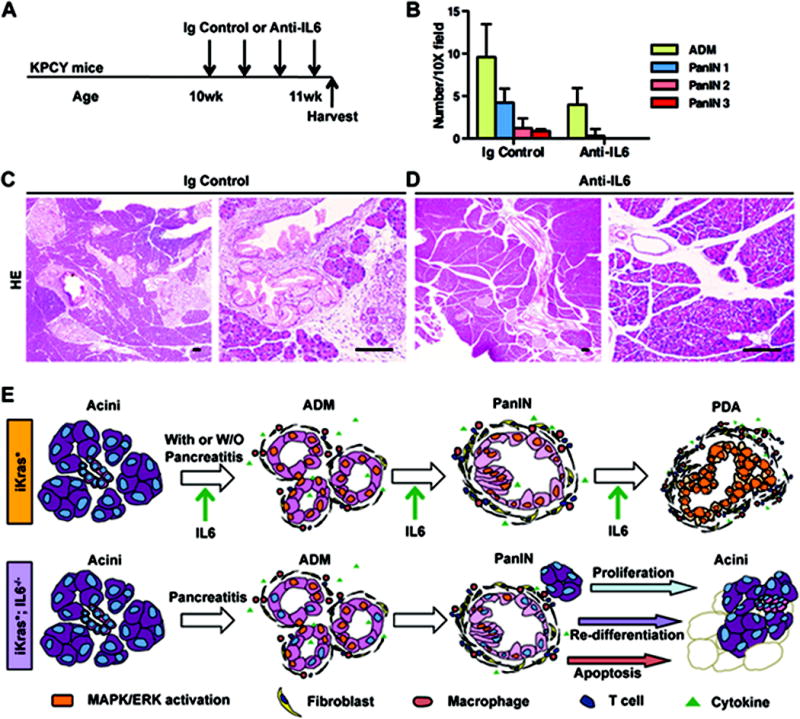
(A) Experimental design, n=3. (B) Pathological analysis. Data represent mean ± SEM, n=3. (C, D) H&E staining of KPCY mice pancreata treated with isotype control (C) or anti-IL6 antibody (D) 1 week following the beginning of treatment. Scale bar 100μm. (E) Working model indicating the requirement of IL6 during the maintenance and progression of pancreatic cancer in mice.
Discussion
The formation of PanINs, the most common precursor lesions of pancreatic cancer, is accompanied by the accumulation of a desmoplastic stroma with abundant immune infiltrates (28) and by expression of several inflammatory cytokines, including IL6 (18). It has been suggested that inflammatory environment is an important component of pancreatic cancer (41); however, our mechanistic understanding of the effect inflammation during cancer progression is incomplete. Recent evidence indicates that, at least in mice, the most common cell of origin for pancreatic cancer is an acinar cell that has de-differentiated to a duct-like cell, often in response to local tissue injury (42, 43). The process of acinar-ductal metaplasia occurs in the pancreas in response to damage such as the induction of acute or chronic pancreatitis. However, the wild-type pancreas is able to undergo tissue repair rapidly and effectively within a short time from the cessation of the injury stimulus. In contrast, pancreata bearing an oncogenic form of Kras are unable to undergo tissue repair, and get “locked” in the acinar-ductal metaplasia status, and progress to PanIN lesions (12). We have previously shown that inactivation of oncogenic Kras at early stages of carcinogenesis allows tissue repair, by allowing re-differentiation of duct-like cells into acinar cells (21). We have also shown that IL6 is expressed in a Kras-dependent manner in the iKras* pancreas. Ras-dependent expression of IL6 was previously described in multiple cell types (44). Progression towards cancer is accompanied by high levels of IL6 while the repair process correlates with repression of IL6 expression (21).
Given the important role IL6 plays during the initiation of pancreatic cancer (18), we decided to investigate whether it constitutes a key player in modulating the balance between tissue repair and carcinogenesis. Our findings showed that IL6 was dispensable for pancreatitis-driven PanIN formation, but necessary for the maintenance and progression of the lesions. In fact, in absence of IL6, the lesions were progressively cleared from the tissue, by a combination of cell-intrinsic mechanisms within the epithelial cells, and extrinsic mechanisms involving epithelial-mesenchymal interactions (see scheme in Figure 7E).
Within the epithelial cells, we observed extensive apoptotic cell death in absence of IL6, consistent with previous reports associating IL6 with cell survival (15), as well as re-differentiation of PanIN cells into normal acini. Moreover, PanIN lesions that formed in absence of IL6 failed to activate the Kras effector pathways MAPK, PI3K/Akt and had reduced level of p-Stat3. The failure to activate the MAPK and PI3K pathways was somewhat surprising, since it could be expected that the presence of constitutively active, oncogenic Kras* would bypass any upstream signal. However, our finding is consistent with several recent reports indicating the need for a positive feedback loop to ensure full activation of oncogenic Kras* (45–47). Importantly, activation of the MAPK pathway is both sufficient and necessary for pancreatic carcinogenesis in mice (45, 48). An important target of the MAPK signaling pathway in pancreatic cancer is Nrf2 (36), a transcription factor that drives the reactive oxygen species detoxification program (49). ROS have been associated with carcinogenesis both as a tumor promoter, given the connection between ROS and genome instability (50), and as a barrier to carcinogenesis as ROS accumulation is toxic for the cells (51). Nrf2 deletion in mouse models of pancreatic cancer prevents tumor progression, indicating the ROS detoxification is an essential step during tumorigenesis (36). Here, we show that, even in presence of mutant Kras, IL6 signaling was essential to up-regulate Nrf2 expression in PanIN lesions. We then showed, using a primary mouse pancreatic cancer cell line, that failure to up-regulate Nrf2 caused oxidative damage and led to tumor cell death. In fact, the failure of iKras*;IL6−/− lesions to progress could be rescued, at least partially, by treating the mice with the ROS scavenger NAC. Thus, our results indicate that IL6 signaling is essential to activate the ROS detoxification program in pancreatic cancer.
In addition to the epithelial changes, important differences also occur within the stroma. Lack of IL6 affected both the fibroblasts within the stroma, which became unable to maintain their activation status, and the inflammatory microenvironment. In particular, the cytokines IL1β and GM-CSF were expressed at lower levels, and fewer myeloid-derived suppressor cells accumulated within IL6-deficient tissues. IL1β can directly drive oncogenesis and suppress cancer immunosurveillance mechanisms via mobilizing myeloid derived suppressor cells (52). GM-CSF is overexpressed by pancreatic cancer cells and promotes tumor growth by creating a permissive immune environment (31, 32). Thus IL6 emerges as a key regulator of multiple immune signals that are important for tumor growth.
Our working model is that IL6 promotes proliferation and survival of neoplastic cells while inhibiting proliferation of acinar cells, as evidenced by our tissue repair experiments. The iKras* mouse model has the unique feature of allowing inactivation of oncogenic Kras* at will (21). Mice lacking IL6 expression had accelerated recovery of acinar cells and increased acinar cell proliferation. Thus, IL6 switches the balance between tissue-repair and carcinogenesis in the pancreas.
Our results have potential therapeutic implications by providing a rationale for the use of IL6 inhibitors in pancreatic cancer, at a time when several inhibitors are reaching the clinic (for review, see (53)). In fact, our proof of principle test in KPCY mice demonstrated that IL6 blocked PanIN progression in this model. Expression patterns of IL6 in human tissues and blood should be explored in a large panel of human cancers, as the current published findings relied on a relatively small sample set (14), and compared to the conclusions made in mouse models. Further work will also be needed to determine the best potential use of IL6 inhibitors in this disease, either for treatment or to prevent progression of latent lesions in high-risk individuals or to prevent relapse in patients following resection, and possibly as an adjuvant combined with standard of care chemotherapy.
Supplementary Material
Significance
Our studies show that IL6 is indispensable for the progression of pancreatic cancer precursor lesions and for tumor cells survival. Therefore, we provide a rationale for targeting of IL6 in pancreatic cancer, a disease in dire need of new therapeutic options.
Acknowledgments
We thank Esha Mathew, Arthur L. Brannon III and Dr. Jörg Zeller for scientific discussion and critical reading of the manuscript. We thank Marsha M. Thomas for lab support. The p48-Cre mouse was a generous gift from Dr Chris Wright (Vanderbilt University). We thank Dr Stephen Konieczny (Purdue University) for providing the Mist1 antibody and Dr Diane M. Simeone for providing human pancreatic cancer samples and primary cell lines. The CK19 antibody (Troma III) was obtained from the Iowa Developmental Hybridoma Bank.
Financial Support: This project was supported by the University of Michigan Biological Scholar Program, the University of Michigan Cancer Center and NCI-1R01CA151588-01. ADR was supported by NIDDK (K08DK088945) and utilized the Morphology and Molecular Biology Cores of the Penn Center for Molecular Studies in Digestive and Liver Diseases (P30-DK050306). MAC was supported by a University of Michigan Program in Cellular and Molecular Biology training grant (NIH T32 GM07315) and by a University of Michigan Center for Organogenesis Training Grant (5-T32-HD007515). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists.
References
- 1.Katz MH, Wang H, Fleming JB, Sun CC, Hwang RF, Wolff RA, et al. Long-term survival after multidisciplinary management of resected pancreatic adenocarcinoma. Ann Surg Oncol. 2009;16:836–47. doi: 10.1245/s10434-008-0295-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.National Cancer Institute. Surveillance, Epidemiology and End Results (SEER) Program. Available at http://seer.cancer.gov.
- 3.American Cancer Society. Cancer Facts and Figures. Available at http://www.cancer.org/Research/CancerFactsFigures.
- 4.Biankin AV, Kench JG, Dijkman FP, Biankin SA, Henshall SM. Molecular pathogenesis of precursor lesions of pancreatic ductal adenocarcinoma. Pathology. 2003;35:14–24. [PubMed] [Google Scholar]
- 5.Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–82. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–54. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 7.Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, et al. Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. Gastroenterology. 2012 doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan L, McFaul C, Howes N, Leslie J, Lancaster G, Wong T, et al. Molecular analysis to detect pancreatic ductal adenocarcinoma in high-risk groups. Gastroenterology. 2005;128:2124–30. doi: 10.1053/j.gastro.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Parsons BL, Meng F. K-RAS mutation in the screening, prognosis and treatment of cancer. Biomark Med. 2009;3:757–69. doi: 10.2217/bmm.09.95. [DOI] [PubMed] [Google Scholar]
- 10.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–7. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 11.Lankisch PG, Breuer N, Bruns A, Weber-Dany B, Lowenfels AB, Maisonneuve P. Natural history of acute pancreatitis: a long-term population-based study. Am J Gastroenterol. 2009;104:2797–805. doi: 10.1038/ajg.2009.405. quiz 806. [DOI] [PubMed] [Google Scholar]
- 12.Morris JPt, Cano DA, Sekine S, Wang SC, Hebrok M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120:508–20. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–39. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talar-Wojnarowska R, Gasiorowska A, Smolarz B, Romanowicz-Makowska H, Kulig A, Malecka-Panas E. Clinical significance of interleukin-6 (IL-6) gene polymorphism and IL-6 serum level in pancreatic adenocarcinoma and chronic pancreatitis. Dig Dis Sci. 2009;54:683–9. doi: 10.1007/s10620-008-0390-z. [DOI] [PubMed] [Google Scholar]
- 15.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naugler WE, Karin M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14:109–19. doi: 10.1016/j.molmed.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Howlett M, Giraud AS, Lescesen H, Jackson CB, Kalantzis A, Van Driel IR, et al. The interleukin-6 family cytokine interleukin-11 regulates homeostatic epithelial cell turnover and promotes gastric tumor development. Gastroenterology. 2009;136:967–77. doi: 10.1053/j.gastro.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–69. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 19.Fukuda A, Wang SC, Morris JPt, Folias AE, Liou A, Kim GE, et al. Stat3 and MMP7 Contribute to Pancreatic Ductal Adenocarcinoma Initiation and Progression. Cancer Cell. 2011;19:441–55. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020–9. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–53. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–61. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 24.Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, et al. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One. 2012;7:e49707. doi: 10.1371/journal.pone.0049707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan RD, Zheng CF, Guan KL, Williams JA. Activation of MAP kinase kinase (MEK) and Ras by cholecystokinin in rat pancreatic acini. Am J Physiol. 1995;268:G1060–5. doi: 10.1152/ajpgi.1995.268.6.G1060. [DOI] [PubMed] [Google Scholar]
- 26.Duan RD, Williams JA. Cholecystokinin rapidly activates mitogen-activated protein kinase in rat pancreatic acini. Am J Physiol. 1994;267:G401–8. doi: 10.1152/ajpgi.1994.267.3.G401. [DOI] [PubMed] [Google Scholar]
- 27.Lupia E, Goffi A, De Giuli P, Azzolino O, Bosco O, Patrucco E, et al. Ablation of phosphoinositide 3-kinase-gamma reduces the severity of acute pancreatitis. Am J Pathol. 2004;165:2003–11. doi: 10.1016/s0002-9440(10)63251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–27. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 29.Chang DZ, Ma Y, Ji B, Wang H, Deng D, Liu Y, et al. Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2011;17:7015–23. doi: 10.1158/1078-0432.CCR-11-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Zuylen WJ, Garceau V, Idris A, Schroder K, Irvine KM, Lattin JE, et al. Macrophage activation and differentiation signals regulate schlafen-4 gene expression: evidence for Schlafen-4 as a modulator of myelopoiesis. PLoS One. 2011;6:e15723. doi: 10.1371/journal.pone.0015723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–47. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Direnzo D, Hess DA, Damsz B, Hallett JE, Marshall B, Goswami C, et al. Induced Mist1 expression promotes remodeling of mouse pancreatic acinar cells. Gastroenterology. 2012;143:469–80. doi: 10.1053/j.gastro.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi G, Zhu L, Sun Y, Bettencourt R, Damsz B, Hruban RH, et al. Loss of the acinar-restricted transcription factor Mist1 accelerates Kras-induced pancreatic intraepithelial neoplasia. Gastroenterology. 2009;136:1368–78. doi: 10.1053/j.gastro.2008.12.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi G, Direnzo D, Qu C, Barney D, Miley D, Konieczny SF. Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene. 2013;32:1950–8. doi: 10.1038/onc.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Recktenwald CV, Kellner R, Lichtenfels R, Seliger B. Altered detoxification status and increased resistance to oxidative stress by K-ras transformation. Cancer Res. 2008;68:10086–93. doi: 10.1158/0008-5472.CAN-08-0360. [DOI] [PubMed] [Google Scholar]
- 38.Papaiahgari S, Kleeberger SR, Cho HY, Kalvakolanu DV, Reddy SP. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J Biol Chem. 2004;279:42302–12. doi: 10.1074/jbc.M408275200. [DOI] [PubMed] [Google Scholar]
- 39.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 40.Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. 2010;28:585–93. doi: 10.1038/nbt.1640. [DOI] [PubMed] [Google Scholar]
- 41.Clark CE, Beatty GL, Vonderheide RH. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Lett. 2009;279:1–7. doi: 10.1016/j.canlet.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 42.Morris JPt, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10:683–95. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JPt, et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2012;22:737–50. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–9. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, et al. EGF Receptor Is Required for KRAS-Induced Pancreatic Tumorigenesis. Cancer Cell. 2012;22:304–17. doi: 10.1016/j.ccr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–30. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daniluk J, Liu Y, Deng D, Chu J, Huang H, Gaiser S, et al. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest. 2012;122:1519–28. doi: 10.1172/JCI59743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, et al. A Central Role for RAF->MEK->ERK Signaling in the Genesis of Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2012 doi: 10.1158/2159-8290.CD-11-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–56. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 50.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–4. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 51.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–88. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 52.Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–19. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012;38:904–10. doi: 10.1016/j.ctrv.2012.04.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.