

Abstract
The proinflammatory cytokine tumor necrosis factor-alpha (TNF-α) is associated with myocardial dysfunction observed in sepsis and septic shock. There are two fundamental components to this dysfunction. (1) systolic dysfunction; and (2) diastolic dysfunction. The aim of these experiments was to determine if any aspect of whole-heart dysfunction could be explained by alterations to global intracellular calcium ([Ca2+]i), contractility, and [Ca2+]i handling, by TNF-α, at the level of the individual rat myocyte. We took an integrative approach to simultaneously measure [Ca2+]i, contractility and sarcolemmal Ca fluxes using the Ca indicator fluo-3, video edge detection, and the perforated patch technique, respectively. All experiments were performed at 37°C. The effects of 50 ng/mL TNF-α were immediate and sustained. The amplitude of systolic [Ca2+]i was reduced by 31% and systolic shortening by 19%. Diastolic [Ca2+]i, myocyte length and relaxation rate were not affected, nor were the activity of the [Ca2+]i removal mechanisms. The reduction in systolic [Ca2+]i was associated with a 14% reduction in sarcoplasmic reticulum (SR) content and a 11% decrease in peak L-type Ca current (ICa-L). Ca influx was decreased by 7% associated with a more rapid ICa-L inactivation. These data show that at the level of the myocyte, TNF-α reduces SR Ca which underlies a reduction in systolic [Ca2+]i and thence shortening. Although these findings correlate well with aspects of systolic myocardial dysfunction seen in sepsis, in this model, acutely, TNF-α does not appear to provide a cellular mechanism for sepsis-related diastolic myocardial dysfunction.
Keywords: Calcium, L-type calcium channel, myocyte, sarcoplasmic reticulum, tumor necrosis factor-alpha
Introduction
Impaired cardiac function is a well-recognized feature of sepsis and septic shock (Kumar et al. 2001a; Court et al. 2002). Manifestations of this dysfunction fall into two broad categories. (1) Systolic dysfunction including impaired contractility and reduced ejection fraction and (2) diastolic dysfunction including ventricular dilation and impaired relaxation. It is well known that proinflammatory cytokines are implicated in the pathogenesis of sepsis, a notable example being tumor necrosis factor-alpha (TNF-α) (Kumar et al. 2001b). With regard to myocardial dysfunction, there is considerable evidence that TNF-α is implicated. Serum concentrations of TNF-α are elevated in sepsis (Waage et al. 1989; Casey et al. 1993), and animal models of sepsis (Michie et al. 1988), and a correlation between plasma concentrations of TNF-α and myocardial dysfunction has been established (Vincent et al. 1992; Herbertson et al. 1995; Kumar et al. 1996; Forfia et al. 1998). Furthermore, in vivo studies in dogs have demonstrated that injection of TNF-α can replicate the profile of systolic and diastolic dysfunction observed in sepsis (Natanson et al. 1989; Eichenholz et al. 1992; Walley et al. 1994; Murray and Freeman 1996). However, as most of these studies involved chronic exposure to TNF-α, it is difficult to elucidate, which, if any of the aspects of myocardial dysfunction are due to the direct action of TNF-α, or secondary to activation of other inflammatory cascades (Halliwell and Gutteridge 1999; Bayir 2005; Brandes and Kreuzer 2005) (e.g., generation of reactive oxygen species or nitric oxide species).
The purpose of this study was two-fold: (1) to determine if any of the above aspects of myocardial dysfunction caused by TNF-α could be explained by alterations to global [Ca2+]i and contractility at the level of the single cardiac myocyte and (2) to explain any changes to global dysfunction by determining the effects of TNF-α on [Ca2+]i handling. Because of the acute nature of TNF-α exposure, any effects observed would most likely be as a result of a direct action of TNF-α.
Others have studied the effects of TNF-α on the individual myocyte, however, the findings are contradictory and incomplete. Most have demonstrated a reduction in systolic [Ca2+]i and shortening (Yokoyama et al. 1993; Goldhaber et al. 1996; Kumar et al. 1996; Amadou et al. 2002; Cailleret et al. 2004), although others have reported no effect on systolic [Ca2+]i (Goldhaber et al. 1996) and even an increase (Amadou et al. 2002). The time frame of effect also varies considerably which has important implications not only in terms of relevance to pathology but also whether the reported effects are direct or indirect. Most report effects occurring progressively, over tens of minutes (Yokoyama et al. 1993; Goldhaber et al. 1996; Kumar et al. 1996; Amadou et al. 2002) though relatively acute effects (1 min) have been reported (Sugishita et al. 1999). Furthermore, some have demonstrated a biphasic effect of TNF-α, where an initial enhancement of systolic [Ca2+]i precedes an ultimate decrease (Amadou et al. 2002; Cailleret et al. 2004). Despite these studies, the effects of TNF-α on the diastolic aspects of global [Ca2+]i and contractility have been largely overlooked, as have the effects on the mechanisms of [Ca2+]i handling.
The lack of consensus regarding the effects and time frame of effects of TNF-α on myocyte function may be as a result of the diverse range of models and techniques used. We present the first integrative approach in that the effects of TNF-α on global [Ca2+]i, contractility and [Ca2+]i handling have been studied simultaneously. This has the advantage that correlations between findings are likely to be more robust.
These data suggest that TNF-α directly affects the L-type calcium channel (LCC) contributing to a reduced sarcoplasmic reticulum (SR) Ca content. This underlies a reduction in systolic [Ca2+]i and thence shortening. However, we observed no effect on any aspect of diastolic function studied suggesting modifications to global [Ca2+]i, contractility and [Ca2+]i handling at the level of the single myocyte by direct action of TNF-α can only be contributory factors to systolic whole-heart dysfunction.
Methods
Unless stated, all chemicals used were obtained from Sigma Aldrich, Dorset, U.K.
Cell isolation
Adult, male Wistar rats were humanely killed by stunning and cervical dislocation in accordance with the U.K. Animals (Scientific Procedures) Act 1986. Hearts were removed and the aorta cannulated for retrograde perfusion with a Ca-free solution containing (in mmol/L) NaCl: 134, HEPES: 10, Glucose: 11.1, NaH2PO4: 1.2, MgSO4: 1.2, KCl: 4, pH 7.34 with NaOH. Following a 10-min wash, Collagenase (Worthington Biochemical Cooperation, NJ) and type XIV protease (Sigma-Aldrich, Dorset, U.K.) were added at typical concentrations of 0.6 and 0.067 mg/mL, respectively, for a digest lasting around 7 min. For a 10-min wash, the solution was then switched to a low Ca solution containing (in mmol/L) NaCl: 115, HEPES: 10, Glucose: 11.1, NaH2PO4: 1.2, MgSO4: 1.2, KCl: 4, Taurine: 50, CaCl2: 0.1, pH 7.34 with NaOH. Cells were stored in this solution until use.
Voltage clamp by perforated patch
Sarcolemmal currents were measured using the perforated patch technique under voltage clamp, using the switch clamp facility of the Axoclamp 2B voltage clamp amplifier (Axon instruments, CA). Microelectrodes with a typical resistance of 5 MΩ were filled with a caesium-based (to control outward currents) pipette solution containing (in mmol/L) CsCl: 20, Cs3CH3O3S: 125, NaCl: 10, HEPES: 10, MgCl2: 5, Cs2EGTA: 0.1, pH 7.2 with CsOH. Electrical access was achieved by addition of amphotericin B (240 μg/mL). ICa-L was activated using a 100-msec duration, 40 mV voltage step, from a holding potential of −40 mV applied at 0.5 Hz. An experimental solution containing (in mmol/L) NaCl2: 134, HEPES: 10, Glucose: 11.1, MgCl2: 1.2, CaCl2: 1, KCl: 4, probenecid; 2, pH 7.34 with NaOH was used for all experiments. Outward currents were inhibited by addition of BaCl2 (0.1 mmol/L) and 4-Aminopyridine (5 mmol/L). All experiments were carried out at 37°C.
Measurement of [Ca2+]i and cell shortening
[Ca2+]i was measured by loading the cells with fluo-3 AM (5 μmol/L) for 10 min (Greensmith et al. 2010). Fluorescence was excited at 488 nm and emission measured at wavelengths greater than 525 nm (Greensmith et al. 2010). Fluorescence signals were calibrated and converted to reflect absolute Ca2+ concentrations using the following equation (Dibb et al. 2004):
Where Kd is the dissociation constant of fluo-3 (864 nmol/L at 37°C), F is fluorescence, and Fmax is maximal fluorescence obtained by damaging the cell at the end of each experiment. Background fluorescence was recorded and subtracted from the total fluorescence to derive F.
Diastolic myocyte length and the degree of systolic shortening were measured using a video edge detection system (Crescent Electronics Ltd, Sandy, Utah), calibrated against a graticule slide (Greensmith et al. 2010).
Quantification of sarcolemmal Ca influx and sarcoplasmic reticulum Ca2+ content
Sarcolemmal Ca influx was quantified by integration of ICa-L activated by the voltage step described above. SR Ca2+ content was determined by integration of the inward sodium current evoked by rapid application of 10 mmol/L caffeine (Varro et al. 1993).
Quantification of the activity of SERCA and NCX
To measure the combined activity of sarco-endoplasmic reticulum calcium ATPase (SERCA) and sodium-calcium exchanger (NCX), the rate of decay of the systolic [Ca2+]i transient (ksys) was determined by fitting a single exponential. The activity of NCX alone was inferred from the rate of decay of the caffeine-evoked [Ca2+]i transient (kNCX). The activity of SERCA alone (kSERCA) was calculated by subtraction of kNCX from ksys (Diaz et al. 2004; Dibb et al. 2004).
Data analysis and statistics
For the experiments studying the chronic effects of TNF-α, we compared two groups; control (cells stored as above) and cells incubated with 25 ng/mL TNF-α for at least 1 h. For each parameter tested, mean data were determined by averaging 10 transients. Statistical significance was evaluated using a t-test (P < 0.05). For the experiments studying the acute effects of 50 ng/mL TNF-α, for each parameter tested, mean data were determined by averaging 10 transients from three experimental periods; control, initial TNF application (the first 20 sec of exposure), and prolonged TNF application (following ∼3 min of exposure). Repeated measures Analysis of variance (ANOVA), repeated measures ANOVA on ranks and paired t-tests were used to determine statistical significance (P < 0.05).
Results
Our first experiments (Fig. 1) were designed to determine if relatively long-term exposure to TNF-α could produce any alterations to [Ca2+]i, or contractility. Cells were incubated with 25 ng/mL TNF-α for at least 1 h. Incubation with TNF-α had no significant effect on the amplitude of systolic [Ca2+]i (Control: 546 ± 92, TNF-α: 477 ± 86 nmol/L, n = 8 and 9, P = 0.59) or the degree of systolic shortening (Control: 4.4 ± 0.8, TNF-α: 4.0 ± 0.9%, n = 8 and 9, P = 0.75). Incubation with TNF-α also produced no effect on any parameter of diastolic function measured, including diastolic [Ca2+]i (Control: 139 ± 24, TNF-α: 129 ± 8 nmol/L, n = 8 and 9, P = 0.47), the rate of systolic [Ca2+]i removal (Control: 7.5 ± 1.1, TNF-α: 7.0 ± 1.0 sec−1, n = 8 and 9, P = 0.77), diastolic cell length (Control: 121 ± 4, TNF-α: 129 ± 6 μm, n = 8 and 9, P = 0.27) or the rate of relaxation (Control: 267 ± 33, TNF-α: 326 ± 72 msec, n = 8 and 9, P = 0.96). Furthermore, we observed no significant effect on peak ICa-L (Control: 39 ± 4, TNF-α: 35 ± 3 pA/pL, n = 8 and 9, P = 0.50), Ca influx (Control: 3.70 ± 0.39, TNF-α: 3.35 ± 0.25 μmol/L, n = 8 and 9, P = 0.46), or SR Ca content (Control: 90 ± 6, TNF-α: 90 ± 9 μmol/L, n = 7 and 9, P = 0.97).
Figure 1.

The chronic effects of 25 ng/mL tumor necrosis factor-alpha (TNF-α). In all panels, two groups are compared; control, and cells incubated with TNF-α for at least 1 h and show mean data for (A) [Ca2+]i transient amplitude, (B) diastolic [Ca2+]i, (C) ksys, (D) systolic contraction, (E) diastolic cell length, (F) relaxation time (90–10% maximal), (G) peak ICa-L (normalized to cell volume), (H) Ca influx, (I) SR Ca content.
Next, we studied the effects of acute exposure to 50 ng/mL TNF-α in order to (1) see if a larger concentration could produce an effect, and to (2) determine the direct effects of TNF-α. The effects of 50 ng/mL TNF-α on systolic [Ca2+]i and myocyte contraction were immediate and sustained (Figs. 2, 3). In most myocytes, the application of TNF-α was associated with an immediate augmentation of the [Ca2+]i transient amplitude, associated with an increase in systolic shortening. However, this lasted only one beat and was followed by a rapid reduction in systolic [Ca2+]i occurring within seconds of exposure. Following a typical exposure of 3 min, systolic [Ca2+]i was reduced by 31% (Control: 462 ± 48, TNF-α: 321 ± 32 nmol/L, n = 8, P < 0.005). This was associated with a 19% reduction in systolic shortening (Control: 4.3 ± 0.6, TNF-α: 3.5 ± 0.6%, n = 9, P < 0.05). Diastolic [Ca2+]i (Control: 126 ± 14, TNF-α: 125 ± 13 nmol/L, n = 8, P = 0.7), myocyte length (Control: 116 ± 3, TNF-α: 116 ± 3 μm, n = 9, P = 0.6) and relaxation time (90–10% of maximal shortening: Control: 326 ± 46, TNF-α: 354 ± 79 msec, n = 9, P = 0.6) were unaffected by exposure to TNF-α. After TNF-α exposure, we attempted a wash period to study the reversibility of the effects. However application of caffeine to quantify SR Ca following TNF-α usually resulted in cell death. Consequently, the reversibility of the above effects could not be studied. In time-dependent controls (data not shown), no effects were observed over equivalent time periods to those of TNF-α exposure.
Figure 2.
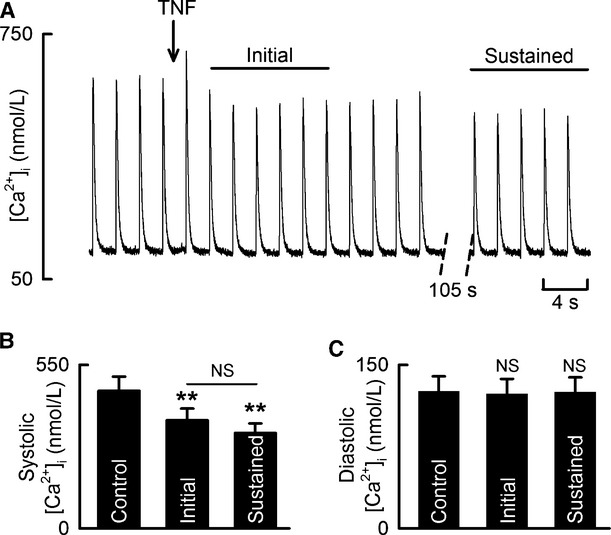
The effects of 50 ng/mL TNF-α on [Ca2+]i. (A) specimen record showing [Ca+]i transients. (B and C), respectively; mean [Ca2+]i transient amplitude and diastolic [Ca2+]i. In all panels, the periods compared are control, initial TNF-α application (within 20 sec), and sustained TNF-α application (∼3 min).
Figure 3.

The effects of 50 ng/mL TNF-α on contractility. (A) Specimen record showing contractility transients. (B) Mean systolic shortening. (C) Mean diastolic cell length. (D) Mean relaxation time (90–10% of maximal systolic shortening). In all panels, the periods compared are control, initial TNF-α application (within 20 sec), and sustained TNF-α application (∼3 min).
We next investigated whether changes to [Ca2+]i handling played a role in the decrease in systolic [Ca2+]i, starting with the LCC. These data were recorded simultaneously with those above. Figure 4 summarizes the effects of TNF-α on ICa-L. Peak ICa-L was reduced by 11% (Control: 0.65 ± 0.06, TNF-α: 0.58 ± 0.05 nA, n = 9, P < 0.05). Ca influx via the LCC was reduced by 7% (Control: 3.64 ± 0.28, TNF-α: 3.38 ± 0.32 μmol/L, n = 9, P < 0.05). LCC inactivation time (as determined by the time taken for ICa-L to decay from 90% to 10% of maximal) was reduced by ∼6% (Control: 46.45 ± 2.55, TNF-α: 43.89 ± 2.35 msec, n = 9, P < 0.05) suggesting a direct inhibition of the LCC.
Figure 4.

The effects of 50 ng/mL TNF-α on the LCC and ICa-L. (A) Specimen record showing the time course of effect on peak ICa-L. (B) Specimen current traces from the periods highlighted in A. (C) Mean peak ICa-L. (D) Mean Ca influx on the ICa-L. (E) Mean LCC inactivation time (90–10% of peak ICa-L). In all panels, the periods compared are control, initial TNF-α application (within 20 sec), and sustained TNF-α application (∼3 min).
To determine if a decrease in SR Ca played a role in the decrease in systolic [Ca2+]i, SR Ca content was quantified by integration of caffeine-evoked inward sodium current (Fig. 5). After ∼3 min exposure to TNF-α, mean SR Ca2+ content was reduced by 14% (Control: 79 ± 5; TNF-α: 68 ± 5 μmol/L, n = 7, P < 0.05).
Figure 5.

The effects of 50 ng/mL TNF-α on SR Ca. (A) Specimen records showing caffeine-evoked inward sodium currents (generated by the NCX-mediated removal of cytoplasmic Ca) (top) and the integral of those currents (below). B (inset); mean SR Ca content. The periods compared are control and sustained TNF-α application (∼3 min).
To further investigate diastolic function in the presence of TNF-α, the activity of the [Ca2+]i removal mechanisms were quantified (Fig. 6). TNF-α had no effect on the activity of the combined [Ca2+]i removal mechanisms (ksys – Control; 6.62 ± 0.56, TNF-α; 6.29 ± 0.42 sec−1, n = 8, P = 0.1), NCX (kNCX – Control; 1.60 ± 0.23, TNF-α; 1.65 ± 0.20 sec−1, n = 5, P = 0.4) or SERCA (kSERCA – Control; 4.18 ± 0.35, TNF-α; 4.10 ± 0.32 sec−1, n = 5, P = 0.5).
Figure 6.

The effects of 50 ng/mL TNF-α on the [Ca2+]i removal mechanisms. (A) Specimen records showing normalized systolic [Ca2+]i transients. (B) Specimen records showing normalized caffeine-evoked [Ca2+]i transients. (C) Mean ksys. (D) Mean kNCX. (E) mean kSERCA.
Discussion
The effects of chronic TNF-α exposure
In our hands, incubation with 25 ng/mL TNF-α produced no significant effect on any measured parameter of global systolic or diastolic function (Fig. 1). Nor did we observe any effect on the major [Ca2+]i handling mechanisms. Subsequently, we studied the effects of acute exposure to TNF-α. Our aim was to (1) determine if a larger concentration of TNF-α could produce an effect, and (2) determine the direct effects of TNF-α. With regard to (1), based on the lack of effect described above, we increased the TNF-α concentration to 50 ng/mL.
The plasma concentration of TNF-α reported in patients with sepsis is around 1 ng/mL. However, the action of cytokines such as TNF-α can be paracrine in nature so plasma concentrations underestimate the relevant concentrations at the site of action. Also, the use of 50 ng/mL TNF-α is typical of other experimenters in the field (Kumar et al. 2001a; Court et al. 2002).
The acute effects of TNF-α on systolic [Ca2+]i and contractility
Our data demonstrating the effects of 50 ng/mL TNF-α confirm previously reported findings that a reduction in cellular systolic [Ca2+]i may, at least partially, contribute to TNF-α-induced reduction in myocardial contractility (Eichenholz et al. 1992; Walley et al. 1994). In most cells (Figs. 2, 3), we observed an initial augmentation of systolic [Ca2+]i and contraction. Although technically this agrees with the biphasic effect of TNF-α seen by others (Cailleret et al. 2004), in our hands this effect lasted only one beat before rapidly developing into a reduced systolic [Ca2+]i and % shortening. Although agreeing qualitatively with the ultimate effect seen in most previous studies (Yokoyama et al. 1993; Goldhaber et al. 1996; Sugishita et al. 1999; Amadou et al. 2002; Cailleret et al. 2004), the onset of the reduction in systolic [Ca2+]i observed by us is the most rapid reported, possibly as a result at working at physiologically relevant temperatures.
The acute effects of TNF-α on diastolic [Ca2+]i and relaxation
Although some studies have shown that TNF-α has no effect on global diastolic function (Yokoyama et al. 1993; Goldhaber et al. 1996), the effects on other diastolic functions such as the [Ca2+]i removal mechanisms, have been largely overlooked. This study has added to the current body of knowledge by demonstrating that, in our hands acute exposure to 50 ng/mL TNF-α has no effect on both myocyte relaxation and the activity of the [Ca2+]i removal mechanisms (Figs. 3, 6). The absence of direct effects on myocyte relaxation or the [Ca2+]i removal mechanisms is in fact surprising given the fact that TNF-α has been shown to inhibit phosphorylation of phospholamban (Yokoyama et al. 1999). Also, the fact we observe no changes to resting myocyte length at constant diastolic [Ca2+]i suggests a lack of effect on myofilament sensitivity, a phenomenon which has been reported by others (Goldhaber et al. 1996). Furthermore, this finding suggests that the reduction in systolic shortening occurs directly as a result of reduced systolic [Ca2+]i rather than as a result of or in combination with a direct effect on the myofilaments.
The acute effects of TNF-α on the L-type calcium channel
There is currently little consensus as to the effects of TNF-α on the LCC and thus ICa-L (Yokoyama et al. 1993; Krown et al. 1995; Sugishita et al. 1999). The integrative approach used in this study is advantageous as it has allowed us to study the effects of TNF-α on the ICa-L simultaneously with global myocyte function. We suggest therefore that correlations between observed effects are more robust. We observed a small, but significant reduction in peak ICa-L (Fig. 4) which itself may contribute to the reduction in systolic [Ca2+]i (Eisner et al. 2000). Under physiological conditions, one may expect a reduction in systolic [Ca2+]i to be associated with delayed LCC inactivation and thence enhanced Ca influx (Trafford et al. 1997). Following TNF-α exposure we observed a small but significant increase in the rate of LCC inactivation as indicated by the reduction in the time taken for ICa-L to decay from 90% to 10% of peak. This actually results in a 7% decrease in Ca influx. This suggests a direct action of TNF-α on the LCC.
What underlies the reduction in systolic [Ca2+]i?
Others have suggested that the effects of TNF-α on the LCC are directly responsible for the reduction in systolic [Ca2+]i (Krown et al. 1995). However, in our hands, given the relatively small effect of TNF-α on peak ICa-L¸ it is unlikely that this alone can account for the reduction in systolic [Ca2+]i (Bassani et al. 1995). To seek an alternative explanation, we quantified the effect of TNF-α on SR Ca content (Fig. 5). The amplitude of systolic [Ca2+]i is proportional to the third power of SR Ca content (Trafford et al. 2000). Following TNF-α exposure, systolic [Ca2+]i was seen to fall to 69% of control values. The cube root of this decrease predicts that SR Ca content would need to fall by 12% to account for the observed decrease in systolic [Ca2+]i. We observe a 14% decrease in SR Ca which suggests that this alone can account for the reduction in systolic [Ca2+]i.
What mechanisms reduce sarcoplasmic reticulum Ca content?
TNF-α had no effect on the activity of SERCA (Fig. 6), which can, to some extent, modulate SR Ca (Bode et al. 2011). Can the reduced Ca influx on ICa-L be responsible for the decreased SR Ca content? First, we must consider that the ICa-L plays two roles in Ca handling: (1) it triggers SR Ca release in a graded fashion such that the greater the peak ICa-L the greater fractional release and thence systolic [Ca2+]i (Bassani et al. 1995), and (2) it loads the cytoplasm with Ca, increasing Ca availability to SERCA facilitating SR loading (Fabiato 1985). Trafford et al. (2001) have shown that increasing ICa-L (both in terms of peak current and influx) increases systolic [Ca2+]i but has no effect on SR Ca (Trafford et al. 2001). This is because although an increase in Ca influx facilitates Ca loading thus increasing SR Ca content, the potentiation of systolic [Ca2+]i by the increased trigger leads to enhanced efflux decreasing SR content (Trafford et al. 2000; Eisner et al. 2013). The converse is true if ICa-L is decreased. The effect of the roles explained above effectively balance out and SR Ca remains constant.
However, in our hands, although TNF-α reduces both peak ICa-L and influx, the effect on peak ICa-L is small. Under these conditions, the effects of reduced loading may be greater than the effect of the reduction in trigger-dependent systolic [Ca2+]i leading to a decrease in SR Ca. Can the 7% decrease in Ca influx therefore account for the 14% reduction in SR Ca? If we assume the decrease in systolic [Ca2+]i is entirely due to a decrease in SR Ca caused by reduced Ca influx, and, we assume the reduction in systolic [Ca2+]i is proportional to the decrease in Ca influx and thence efflux, a 31% decrease in Ca influx would be required. Therefore, although the reduction in Ca influx by TNF-α may contribute to the reduced SR Ca2+ content, it cannot fully explain it.
What other mechanism could contribute to SR Ca loss? Duncan et al. (2010) reported that TNF-α increases the open probability of the ryanodine receptor (RyR); which is known to contribute to SR Ca loss (Zima et al. 2010). The first beat augmentation of systolic [Ca2+]i seen in this study is consistent with an increased RyR open probability (Trafford et al. 2000).
Summary and clinical relevance
We have shown that, by a reduction in SR Ca, TNF-α reduces systolic [Ca2+]i and shortening at the level of the individual myocyte. Although inhibition of the LCC may contribute directly to this, we suggest it is the subsequent depletion of SR Ca that is the primary mechanism. In this study, TNF-α had no effect on any measured parameter of diastolic function. We suggest therefore that the direct effects of TNF-α on cell [Ca2+]i and contractility could contribute to whole-heart systolic dysfunction observed in sepsis. However, these data suggest that the direct effects of TNF-α on cell [Ca2+]i and contractility do not contribute whole-heart diastolic dysfunction.
Acknowledgments
At the time this study was carried out, David James Greensmith was supported by the Manchester University Strategic Studentship Scheme.
Conflict of Interest
None declared.
References
- Amadou A, Nawrocki A, Best-Belpomme M, Pavoine C, Pecker F. Arachidonic acid mediates dual effect of TNF-alpha on Ca2+ transients and contraction of adult rat cardiomyocytes. Am. J. Physiol. 2002;282:C1339–C1347. doi: 10.1152/ajpcell.00471.2001. [DOI] [PubMed] [Google Scholar]
- Bassani JWM, Yuan W, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 1995;268:C1313–C1329. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- Bayir H. Reactive oxygen species. Crit. Care Med. 2005;33(12 Suppl):S498–S501. doi: 10.1097/01.ccm.0000186787.64500.12. [DOI] [PubMed] [Google Scholar]
- Bode EF, Briston SJ, Overend CL, O'Neill SC, Trafford AW, Eisner DA. Changes of SERCA activity have only modest effects on sarcoplasmic reticulum Ca2+ content in rat ventricular myocytes. J. Physiol. 2011;589(Pt 19):4723–4729. doi: 10.1113/jphysiol.2011.211052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc. Res. 2005;65:16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Cailleret M, Amadou A, Andrieu-Abadie N, Nawrocki A, Adamy C, Ait-Mamar B, et al. N-acetylcysteine prevents the deleterious effect of tumor necrosis factor-(alpha) on calcium transients and contraction in adult rat cardiomyocytes. Circulation. 2004;109:406–411. doi: 10.1161/01.CIR.0000109499.00587.FF. [DOI] [PubMed] [Google Scholar]
- Casey LC, Balk RA, Bone RC. Plasma cytokine and endotoxin levels correlate with survival in patients with the sepsis syndrome. Ann. Intern. Med. 1993;119:771–778. doi: 10.7326/0003-4819-119-8-199310150-00001. [DOI] [PubMed] [Google Scholar]
- Court O, Kumar A, Parrillo JE, Kumar A. Clinical review: myocardial depression in sepsis and septic shock. Crit. Care. 2002;6:500–508. doi: 10.1186/cc1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz ME, Graham HK, Trafford AW. Enhanced sarcolemmal Ca2+ efflux reduces sarcoplasmic reticulum Ca2+ content and systolic Ca2+ in cardiac hypertrophy. Cardiovasc. Res. 2004;62:538–547. doi: 10.1016/j.cardiores.2004.01.038. [DOI] [PubMed] [Google Scholar]
- Dibb KM, Rueckschloss U, Eisner DA, Isenberg G, Trafford AW. Mechanisms underlying enhanced cardiac excitation contraction coupling observed in the senescent sheep myocardium. J. Mol. Cell. Cardiol. 2004;37:1171–1181. doi: 10.1016/j.yjmcc.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Duncan DJ, Yang Z, Hopkins PM, Steele DS, Harrison SM. TNF-alpha and IL-1beta increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium. 2010;47:378–386. doi: 10.1016/j.ceca.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenholz PW, Eichacker PQ, Hoffman WD, Banks SM, Parrillo JE, Danner RL, et al. Tumor necrosis factor challenges in canines: patterns of cardiovascular dysfunction. Am. J. Physiol. 1992;263(3 Pt 2):H668–H675. doi: 10.1152/ajpheart.1992.263.3.H668. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Choi HS, Díaz ME, O'Neill SC, Trafford AW. Integrative analysis of calcium cycling in cardiac muscle. Circ. Res. 2000;87:1087–1094. doi: 10.1161/01.res.87.12.1087. [DOI] [PubMed] [Google Scholar]
- Eisner D, Bode E, Venetucci L, Trafford A. Calcium flux balance in the heart. J. Mol. Cell. Cardiol. 2013;58:110–117. doi: 10.1016/j.yjmcc.2012.11.017. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Simulated calcium current can both cause calcium loading in and trigger calcium release from the sarcoplasmic reticulum of a skinned canine cardiac purkinje cell. J. Gen. Physiol. 1985;85:291–320. doi: 10.1085/jgp.85.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forfia PR, Zhang X, Ochoa F, Ochoa M, Xu X, Bernstein R, et al. Relationship between plasma NOx and cardiac and vascular dysfunction after LPS injection in anesthetized dogs. Am. J. Physiol. 1998;274(1 Pt 2):H193–H201. doi: 10.1152/ajpheart.1998.274.1.H193. [DOI] [PubMed] [Google Scholar]
- Goldhaber JI, Kim KH, Natterson PD, Lawrence T, Yang P, Weiss JN. Effects of TNF-alpha on [Ca2+]i and contractility in isolated adult rabbit ventricular myocytes. Am. J. Physiol. 1996;271:H1449–H1455. doi: 10.1152/ajpheart.1996.271.4.H1449. [DOI] [PubMed] [Google Scholar]
- Greensmith DJ, Eisner DA, Nirmalan M. The effects of hydrogen peroxide on intracellular calcium handling and contractility in the rat ventricular myocyte. Cell Calcium. 2010;48:341–351. doi: 10.1016/j.ceca.2010.10.007. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. 3rd ed. Oxford: Oxford Univ. Press; 1999. [Google Scholar]
- Herbertson MJ, Werner HA, Goddard CM, Russell JA, Wheeler A, Coxon R, et al. Anti-tumor necrosis factor-alpha prevents decreased ventricular contractility in endotoxemic pigs. Am. J. Respir. Crit. Care Med. 1995;152:480–488. doi: 10.1164/ajrccm.152.2.7633696. [DOI] [PubMed] [Google Scholar]
- Krown KA, Yasui K, Brooker MJ, Dubin AE, Nguyen C, Harris GL, et al. TNF alpha receptor expression in rat cardiac myocytes: TNF alpha inhibition of L-type Ca2+ current and Ca2+ transients. FEBS Lett. 1995;376:24–30. doi: 10.1016/0014-5793(95)01238-5. [DOI] [PubMed] [Google Scholar]
- Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J. Exp. Med. 1996;183:949–958. doi: 10.1084/jem.183.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Haery C, Parrillo JE. Myocardial dysfunction in septic shock: Part I. Clinical manifestation of cardiovascular dysfunction. J. Cardiothorac. Vasc. Anesth. 2001a;15:364–376. doi: 10.1053/jcan.2001.22317. [DOI] [PubMed] [Google Scholar]
- Kumar A, Krieger A, Symeoneides S, Kumar A, Parrillo JE. Myocardial dysfunction in septic shock: Part II. Role of cytokines and nitric oxide. J. Cardiothorac. Vasc. Anesth. 2001b;15:485–511. doi: 10.1053/jcan.2001.25003. [DOI] [PubMed] [Google Scholar]
- Michie HR, Manogue KR, Spriggs DR, Revhaug A, O'Dwyer S, Dinarello CA, et al. Detection of circulating tumor necrosis factor after endotoxin administration. N. Engl. J. Med. 1988;318:1481–1486. doi: 10.1056/NEJM198806093182301. [DOI] [PubMed] [Google Scholar]
- Murray DR, Freeman GL. Tumor necrosis factor-alpha induces a biphasic effect on myocardial contractility in conscious dogs. Circ. Res. 1996;78:154–160. doi: 10.1161/01.res.78.1.154. [DOI] [PubMed] [Google Scholar]
- Natanson C, Eichenholz PW, Danner RL, Eichacker PQ, Hoffman WD, Kuo GC, et al. Endotoxin and tumor necrosis factor challenges in dogs simulate the cardiovascular profile of human septic shock. J. Exp. Med. 1989;169:823–832. doi: 10.1084/jem.169.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugishita K, Kinugawa K-I, Shimizu T, Harada K, Matsui H, Takahashi T, et al. Cellular basis for the acute inhibitory effects of IL-6 and TNF- [alpha] on excitation-contraction coupling. J. Mol. Cell. Cardiol. 1999;31:1457–1467. doi: 10.1006/jmcc.1999.0989. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Díaz ME, Negretti N, Eisner DA. Enhanced calcium current and decreased calcium efflux restore sarcoplasmic reticulum Ca content following depletion. Circ. Res. 1997;81:477–484. doi: 10.1161/01.res.81.4.477. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Díaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J. Physiol. 2000;522:259–270. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafford AW, Díaz ME, Eisner DA. Coordinated control of cell Ca2+ loading and triggered release from the sarcoplasmic reticulum underlies the rapid inotropic response to increased L-type Ca2+ current. Circ. Res. 2001;88:195–201. doi: 10.1161/01.res.88.2.195. [DOI] [PubMed] [Google Scholar]
- Varro A, Negretti N, Hester SB, Eisner DA. An estimate of the calcium content of the sarcoplasmic reticulum in rat ventricular myocytes. Pflügers Archiv. 1993;423:158–160. doi: 10.1007/BF00374975. [DOI] [PubMed] [Google Scholar]
- Vincent JL, Bakker J, Marecaux G, Schandene L, Kahn RJ, Dupont E. Administration of anti-TNF antibody improves left ventricular function in septic shock patients. Results of a pilot study. Chest. 1992;101:810–815. doi: 10.1378/chest.101.3.810. [DOI] [PubMed] [Google Scholar]
- Waage A, Brandtzaeg P, Halstensen A, Kierulf P, Espevik T. The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J. Exp. Med. 1989;169:333–338. doi: 10.1084/jem.169.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walley KR, Hebert PC, Wakai Y, Wilcox PG, Road JD, Cooper DJ. Decrease in left ventricular contractility after tumor necrosis factor-alpha infusion in dogs. J. Appl. Physiol. 1994;76:1060–1067. doi: 10.1152/jappl.1994.76.3.1060. [DOI] [PubMed] [Google Scholar]
- Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest. 1993;92:2303–2312. doi: 10.1172/JCI116834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama T, Arai M, Sekiguchi K, Tanaka T, Kanda T, Suzuki T, et al. Tumor necrosis factor-alpha decreases the phosphorylation levels of phospholamban and troponin I in spontaneously beating rat neonatal cardiac myocytes. J. Mol. Cell. Cardiol. 1999;31:261–273. doi: 10.1006/jmcc.1998.0863. [DOI] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Bers DM, Blatter LA. Ca(2)+ spark-dependent and -independent sarcoplasmic reticulum Ca(2)+ leak in normal and failing rabbit ventricular myocytes. J. Physiol. 2010;588(Pt 23):4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]