

Significance
In this paper, both oxygen and carbon K-edge spectra of CO2 polymorphs at high pressure and room temperature were reported. The electronic structure of CO2 shows remarkable change from molecular to nonmolecular transition. Furthermore, a 532-eV feature was observed only at a limited pressure range from ∼37 to 53 GPa, suggesting the presence of a transient species in the nonmolecular phase.
Keywords: mineral physics, diamond anvil cell, inelastic x-ray scattering
Abstract
Knowledge of the high-pressure behavior of carbon dioxide (CO2), an important planetary material found in Venus, Earth, and Mars, is vital to the study of the evolution and dynamics of the planetary interiors as well as to the fundamental understanding of the C–O bonding and interaction between the molecules. Recent studies have revealed a number of crystalline polymorphs (CO2-I to -VII) and an amorphous phase under high pressure–temperature conditions. Nevertheless, the reported phase stability field and transition pressures at room temperature are poorly defined, especially for the amorphous phase. Here we shed light on the successive pressure-induced local structural changes and the molecular-to-nonmolecular transition of CO2 at room temperature by performing an in situ study of the local electronic structure using X-ray Raman scattering, aided by first-principle exciton calculations. We show that the transition from CO2-I to CO2-III was initiated at around 7.4 GPa, and completed at about 17 GPa. The present study also shows that at ∼37 GPa, molecular CO2 starts to polymerize to an extended structure with fourfold coordinated carbon and minor CO3 and CO-like species. The observed pressure is more than 10 GPa below previously reported. The disappearance of the minority species at 63(±3) GPa suggests that a previously unknown phase transition within the nonmolecular phase of CO2 has occurred.
Molecular compounds such as N2 and H2O have been known to acquire a nonmolecular structure under compression and ultimately transform into highly disordered and/or amorphous phases (1–4). Other solids, such as group IV oxides SiO2 (5) and GeO2 (6), are prone to amorphize under pressure despite the covalent framework structure. However, it was not until recently that CO2, both a molecular compound and group IV oxide, was also reported to form several polymorphs and a pressure-induced amorphous phase (7–23). Previous experimental evidence on the formation of nonmolecular phases of N2 and CO2 was mainly based on the loss of optical vibrons (1) and/or the emergence of a broad IR or Raman band in the stretching mode region. Unfortunately, these new features are often very weak and accurate measurement was hindered by significant noises (17, 22, 23), making it difficult to extract useful information on the local coordination in the amorphous phase. This difficulty is illustrated by studies of the high-pressure high-temperature amorphous a-CO2 phase in which the polymeric local structure was claimed to be a mixture of tetrahedral and octahedral coordinated carbon based on Raman spectroscopy (17), but to a mixture of tetrahedral and threefold coordinated polymeric CO2 from a combined theoretical and infrared spectroscopy study (20). Moreover, knowledge of the phase diagram and kinetics for the various phase transitions in solid CO2 are obscured by significant disparities in the estimated phase transition pressures from different studies and techniques. An example is the pressures reported for the molecular to nonmolecular phase transformation ranging from 48 to 65 GPa (17, 22, 23).
The discrepancies between different experiments and ambiguities in the transition pressures in solid CO2 motivate a thorough investigation of the pressure dependence of the local structural and electronic environment. Here we report a survey of the evolution of the chemical bonding of CO2 under pressure, using X-ray Raman scattering (XRS)—a nonresonant, hard X-ray photon-in photon-out technique at both the carbon (C) and oxygen (O) K edges. XRS provides a convenient way to measure absorption spectra of low Z materials in high-pressure diamond anvil cell. From analysis of the spectral features, insights into the effects of pressure on the unoccupied π* and σ* orbitals of C and O aided with first-principles calculations have been obtained. The present results provide a framework to further the understanding of the local mechanism of polymerization of CO2 under pressure.
Results and Discussion
The O K-edge XRS spectrum obtained for the molecular phase CO2-I at 2 GPa (Fig. 1A) is characterized by an intense π* resonance peak at 534.6 eV and very weak σ* features located between 540 and 550 eV. In comparison, the spectrum of gaseous CO2 shows a single, relatively stronger π* peak and a σ* peak of moderate intensity (24). The C K-edge XRS spectrum of CO2-I shares similarities with the O K-edge spectrum with an intense π* resonance peak at 291 eV and a very broad and weak σ* peak at about 313.2 eV (Fig. 1B). The sensitivity of the XRS spectrum to local chemical bonding is exhibited by the stark contrast in the spectral features for the transitions to π* at the C K edge between CO2 (sp hybridization), and graphite (sp2 hybridization) and diamond (sp3 hybridization) (Fig. 2) in which graphite shows both π* and σ* features whereas there is only a broad σ* band in the spectrum of diamond.
Fig. 1.

Representative X-ray Raman spectra. (A) Oxygen and (B) carbon K edge of CO2 at pressures up to 63(±3) GPa. The ambient pressure spectra of oxygen and carbon K edge were obtained at low temperature in vacuum condition. Evolution of the π* and σ* peaks between CO2-III and the amorphous phase, showing (C) at the O K edge the decrease of π* from CO2-III at 534.6 eV and the appearance and then weakening of π* of the i-CO2 phase at about 532 eV, and (D) at the C K edge the decrease of the π* and increase of the σ* peaks between CO2-III and the amorphous phase. The pressure is labeled along with the spectra.
Fig. 2.

Comparison of the C K edge for the graphite, diamond, and CO2-I. The single crystalline graphite was probed along [001] with a reflection geometry and along [100] with a transmission geometry, and the diamond was probed along [111] with a reflection geometry.
In solid CO2 the O K-edge spectral profile is observed to undergo gradual changes upon compression at pressure above 7.4 GPa (Fig. 3A). The two σ* peaks at 542.3 and 546.7 eV become more distinct but the intensities remain rather weak. At about 10.7 GPa, the σ* peak of the O K edge at 542.3 eV has disappeared and concomitantly a new peak starts to emerge at 537.4 eV. At the same time, the σ* peak at 546.7 eV becomes stronger with the intensity increases up to ∼17 GPa. The progressive spectral changes (i.e., simultaneous diminishing π* and increasing σ* excitations) point to a gradual transition of CO2 from a linear molecule with planar π bonding to a distorted 3D network structure. We note that throughout the transition, the variation of energy and width of the π* peak at both edges is negligibly small, suggesting that the π* orbitals are not significantly affected by the structural change.
Fig. 3.

CO2 phase I–III transitions observed in this study. (A) XRS spectra of the O K edge of CO2 between 2.0 and 17 GPa (circles) and their best fit (solid line) using a weighted sum of the spectra at 2.0 and 17 GPa, considered respectively as the spectrum of pure phase I and pure phase III. (B) The fraction of the CO2 phase as a function of pressure shows a mixture of CO2-I and -III in the 7–15-GPa range. The error bars for the fraction of phase are estimated about ±5%. The error bars for the pressure below 20 GPa are smaller than the symbols.
Previous studies have shown that the transformation from CO2-I to -III is sluggish (8, 9, 13) as both phase I and III were found to coexist in an extended pressure range. To extract structural information from within the transition region, we fit the O K-edge spectra measured between 7.4 and 12.8 GPa with a weighted sum of the reference spectra for CO2-I at 2.0 GPa and CO2-III at ∼17 GPa. The results, summarized in Fig. 3B, show that CO2-III begins to appear as low as ∼7.4 GPa and coexists with CO2-I up to ∼17 GPa. In the earlier studies, the transition pressures have been estimated to be between 10 and 20 GPa (8, 9, 16). The present result imposes a constraint on the transition pressure range. The lower onset pressure of the transition observed here can be attributed to the local character of XRS, which is sensitive to the first coordination shells of the excited atom in contrast to X-ray diffraction or Raman and infrared spectroscopy. We note that pressure gradient measurements on the sample detected no gradient up to 10 GPa and only a small pressure gradient (0.017 GPa/μm) from 10 to 17 GPa (25).
Upon further compression to about 37 GPa, we observe dramatic changes in both O and C K-edge XRS patterns (Fig. 1 A–D). Note that the pressure distribution within the CO2 sample was 35–40 GPa before the XRS measurement, whereas after the XRS measurement, the sample pressure was found to be about 37 GPa throughout the path of the X-ray beam. In addition, the CO2 samples at P > 30 GPa were all freshly prepared except for the 53(±3) GPa spectrum, which was obtained by further compressing the sample after measuring the 30-GPa spectrum. In the O K-edge spectra, the π* peak of CO2-III at 535 eV has nearly disappeared, and concurrently broad, salient features emerge in the 537∼547 eV range, together with a weak peak at ∼532 eV. We ascribe the appearance of the broad features to the formation of extended σ* band structure, showing that CO2 has lost its molecular character and has started to polymerize. The appearance of a small peak at lower energy side of the π* excitation between ∼37 and 53(±3) GPa (Fig. 1A) suggests the formation of a nonmolecular phase, and will be discussed later. Similarly, a broad σ* band also starts to appear at 296 eV in the C K-edge XRS spectra. This feature has no analog in the graphite (sp2) or diamond (sp3) spectra (Fig. 2).
The evolution of the O and C K-edge XRS line shapes with pressure is summarized in Fig. 1 C and D, respectively. The appearance of the prominent σ* features and ensuing decrease of the π* peaks at both O and C K edges offers compelling evidence that CO2 has almost completely transformed into a nonmolecular polymeric phase at ∼37 GPa. This result is significant, as it shows the transformation into a nonmolecular phase occurs at a substantially lower pressure than the previously reported values of 48–65 GPa using Raman and infrared spectroscopy (17, 21, 22), thereby delineating new stability fields for the intermediate bonding (CO2-III) and amorphous phases. This highlights the advantage of using a spectroscopic technique sensitive to the local bonding environment, such as XRS, to monitor the formation of a continuous network of bonds between molecular entities.
Above 53(±3) GPa the broad σ* feature at the O K edge is unaffected by further pressure increase while the π* peak of CO2-III vanished (Fig. 1C). In addition, the π* peak at ∼532 eV weakened and eventually disappeared at 63(±3) GPa. This observation indicates that another structural transition has occurred. The intensity of the π* peak at the C K edge also decreases with respect to the σ* peak. Unfortunately, above 53(±3) GPa the XRS intensity at the C K edge was too weak and contaminated with C signals from the diamond anvils and no reliable signal could be measured. It is important to note that the 532 eV feature was observed only in a limited pressure range from ∼37 to 53 GPa and, therefore, it is unlikely that this feature is an indication of X-ray irradiation damage.
To better understand the XRS spectra, first-principles calculations of both C and O K-edge X-ray absorption spectra (XAS) were performed (Fig. 4 A and B). The structural models were selected from several reported high-pressure structures. We found that calculations using the independent particle approximation (i.e., full core, half core, and screened core hole models) failed to reproduce even the qualitative features of both the carbon and oxygen XRS spectra (Fig. S1). The use of the more accurate Bethe–Salpeter equation (BSE) method, which explicitly takes into account the electron correlation and excitonic effects, was found to be essential. These calculations are computationally very demanding and can only be applied to systems with a relatively small number of atoms. As seen in Fig. 4 A and B, apart from a shift in the absolute energy scale, there is a close agreement between the theoretical and experimental π* and σ* excitation profiles in the C and O K-edges for the phase CO2-I (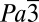 ) and -III (Cmca). Note that the experimental resolution and the core-hole lifetime broadening were not included in the calculations, which explains why the theoretical spectra are sharper.
) and -III (Cmca). Note that the experimental resolution and the core-hole lifetime broadening were not included in the calculations, which explains why the theoretical spectra are sharper.
Fig. 4.

XAS spectra calculated from BSE method for different phases of CO2 at (A) the O K edge and (B) C K edge.
The spectra calculated for the nonmolecular polymeric phase V (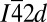 ) are strikingly different from that of the molecular phases. The absence of a strong π* excitation at both C and O K edges is expected because the carbon atom in the nonmolecular phase has a fourfold coordination. Instead, transitions to C and O σ* orbitals located respectively 6.6 and 4 eV above the π* excitation in CO2-III became more prominent. The theoretical result is in excellent agreement with the experimental C and O K-edge spectra measured above 37 GPa. Therefore, the observations of prominent, higher-energy σ* peaks can be attributed unequivocally to the formation of a nonmolecular structure with fourfold coordinated carbon atoms.
) are strikingly different from that of the molecular phases. The absence of a strong π* excitation at both C and O K edges is expected because the carbon atom in the nonmolecular phase has a fourfold coordination. Instead, transitions to C and O σ* orbitals located respectively 6.6 and 4 eV above the π* excitation in CO2-III became more prominent. The theoretical result is in excellent agreement with the experimental C and O K-edge spectra measured above 37 GPa. Therefore, the observations of prominent, higher-energy σ* peaks can be attributed unequivocally to the formation of a nonmolecular structure with fourfold coordinated carbon atoms.
Theoretical calculations also suggest that the observed low-energy peak, which emerges around 532 eV at the O K edge at ∼37–53(±3) GPa and then disappears at 63(±3) GPa, may due to minor phases with residual C = O π bonds. The π* peak calculated for the O K edge of the P42/mnm phase coincides in energy with the π* peak of Cmca (Fig. 4A) in accord with the experimental spectra, which show that the 532 eV peak is ∼3 eV below the π* peak of Cmca (Fig. 1A). We can therefore rule out P42/mnm structure as the minority phase. To search for an alternate explanation, it is noted that at 63(±3) GPa and 2,000 K solid CO2 was reported to dissociate into an ionic phase i-CO2 consisting of CO32- and CO2+ ion pairs in the postaragonite structure (21). To evaluate whether the low-energy peak at the O K edge may be attributed to either CO32- or CO2+ like species, C and O K-edge XAS spectra of i-CO2 were calculated (26). The O K-edge XAS spectra of the distinct oxygen atoms in i-CO2 are found to be overall similar to each other, showing a broad distribution of π* excitations centered around 4–5 eV below the σ* feature of CO2-V (Fig. 4A). This is in agreement with the experimental observation where the 532-eV π* peak is located ∼5 eV below the σ* feature. At the C K edge in Fig. 4B, we observe two main peaks separated by 2 eV, the lower energy peak corresponding to the CO moiety and the higher energy peak to CO3. These two peaks are located slightly above the π* peak of CO2-III, although in the experiment the π* peak is at 290 eV for both CO2-III and the minority phase. In conclusion, despite slightly higher calculated energies for both C and O π* excitations in CO and CO3, the qualitative similarity with the experimental features at 532 and 290 eV, respectively, for the O K and C K edges, suggests that the minority phase may consist of dissociated CO moieties and/or CO3. Because the 532-eV peak in the O K-edge spectrum disappeared at 63(±3) GPa, we speculate that the minority phase is metastable in the 37–63(±3) GPa range.
To gain further insight into the local coordination of the amorphous phase at 63(±3) GPa, we have performed molecular dynamics calculations starting with the crystalline i-CO2 structure (26). We found that i-CO2 is unstable and collapsed into a disordered structure consisting of mixed threefold (CO3) and fourfold (CO4) coordination. The predicted structure is in agreement with the proposal of a mixed coordinated phase from a previous report (20). This result, combined with our unraveling of the metastable minority phase, marks a major step in the understanding of the nonmolecular mix-coordinated phase, by suggesting a route of formation involving intermediate CO-like and CO3-like species. It is not computationally feasible to calculate the XAS of the disordered structure using the BSE method. Our theoretical results, however, are consistent with the experimental observations of the appearance of the broad core → σ* band at the expense of the C = O π* excitation.
From in situ measurements of the electronic structure of CO2 under pressure, we have obtained information of crucial importance to the understanding of the phase transitions from the local point of view. Our results show that the successive I–III–amorphous phase boundaries are at significantly lower pressures than previous reports. The present results cast new light on the phase diagram of CO2, and highlight the continuous character of these transitions. Our study also reveals that the local coordination of the carbon atoms in the amorphous phase is fourfold, with a minor metastable phase of mixed CO and CO3 species. The present study demonstrates that site-specific XRS is a powerful and unambiguous probe for the study of molecular-to-nonmolecular transitions at high pressure.
Methods
Experiment.
CO2 sample with 99.995% purity was cryogenically loaded in a beryllium gasket chamber with a hole of 70–250 µm in a diamond anvil cell. A small ruby sphere was placed at the edge of the hole, serving as a pressure calibrant. In addition, the first-order Raman peak of the diamond was also used as a pressure reference in some cases for pressure above 30 GPa. The high-pressure inelastic X-ray scattering measurements of CO2 were conducted at the Taiwan beamline BL12XU, SPring-8. A monochromatic beam with incident energy fixed at Ei = 9,887 eV with a beam size of 13–20 (horizontal) by 22–30 (vertical) μm2 was focused onto the sample. A small pinhole (∼10 µm in diameter) was installed in front of the diamond cell for the collection of higher pressure data. The wavelengths of scattered photons were selected using three Si(555) spherically bent analyzers in near-backscattering conditions and counted with a Si solid-state detector. The scattering angle was ∼35°. A total energy resolution of ∼1.4 eV was achieved with this setup. Depending on the pressure, the measurement time for one spectrum ranged between 8 and 20 h. The oxygen and carbon K-edge absorption spectra were obtained by scanning the incident energy around Ei + EC-K and Ei + EO-K, respectively.
Computation.
The XAS were calculated with the BSE method, using the Exciting code, which is in the frame of all-electron density-functional theory based on the linearized augmented plane-wave method. Calculations using the full and half core-hole approximations were performed with the gauge included projected augment wave code from Quantum Espresso (http://www.pwscf.org) (27, 28). The BSE calculations were performed using all electron full potential linearized augmented plane wave code EXCITING (http://exciting-code.org) (29). The BSE method is found to be necessary in CO2 XAS calculation due to the electron–hole interaction during the excitation process. The spectra calculated using core-hole method showed poor agreement with the experiment (Fig. S1). Convergence with respect to k point and q point sampling was tested, and convergence was obtained with a 3 × 3 × 3 mesh on the CO2-I phase and a 4 × 4 × 4 mesh on the other crystal structures. Up to 115 and 90 empty states were included for the calculation of screening and excitation, respectively. For the high-pressure calculations, we optimized the structure at the appropriate pressures using the quantum Espresso code.
Supplementary Material
Acknowledgments
We thank Yun-Mei Yiu for help in calculations at initial work and Cheng-Chi Chen for technical support at Taiwan beamline BL12XU, SPring-8. This work is financially supported by Natural Sciences and Engineering Research Council. The synchrotron radiation experiments were performed at the BL12XU of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI). Key Basic Research Program of China (2012CB825700), National Natural Science Foundation of China (Grants 10979002 and 51371157), and the Fundamental Research Funds for the Central Universities are gratefully acknowledged. M.W. thanks the Chinese Government for a scholarship to study at University of Saskatchewan, Canada. I.J. and Y.Q.C. are supported by the U.S. Department of Energy, Office Basic Energy Science, under Contract DE-AC02-98CH10886.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305116110/-/DCSupplemental.
References
- 1.Goncharov AF, Gregoryanz E, Mao Hk, Liu Z, Hemley RJ. Optical evidence for a nonmolecular phase of nitrogen above 150 GPa. Phys Rev Lett. 2000;85(6):1262–1265. doi: 10.1103/PhysRevLett.85.1262. [DOI] [PubMed] [Google Scholar]
- 2.Gregoryanz E, Goncharov AF, Hemley RJ, Mao HK. High pressure amorphous nitrogen. Phys Rev B. 2001;64(5):052103. [Google Scholar]
- 3.Eremets MI, Hemley RJ, Mao Hk, Gregoryanz E. Semiconducting non-molecular nitrogen up to 240 GPa and its low-pressure stability. Nature. 2001;411(6834):170–174. doi: 10.1038/35075531. [DOI] [PubMed] [Google Scholar]
- 4.Mishima O, Calvert LD, Whalley E. ‘Melting ice’ I at 77 K and 10 kbar: A new method of making amorphous solids. Nature. 1984;310:393–395. [Google Scholar]
- 5.Hemley RJ, Jephcoat AP, Mao HK, Ming LC, Manghnani MH. Pressure-induced amorphization of crystalline silica. Nature. 1988;334:52–54. [Google Scholar]
- 6.Prakapenka VP, Shen G, Dubrovinsky LS, Rivers ML, Sutton SR. High pressure induced phase transformation of SiO2 and GeO2: Difference and similarity. J Phys Chem Solids. 2004;65(8-9):1537–1545. [Google Scholar]
- 7.Aoki K, Yamawaki H, Sakashita M, Gotoh Y, Takemura K. Crystal structure of the high-pressure phase of solid CO2. Science. 1994;263(5145):356–358. doi: 10.1126/science.263.5145.356. [DOI] [PubMed] [Google Scholar]
- 8.Olijnyk H, Jephcoat AP. Vibrational studies on CO2 up to 40 GPa by Raman spectroscopy at room temperature. Phys Rev B. 1998;57(2):879–888. [Google Scholar]
- 9.Yoo CS, et al. Crystal structure of carbon dioxide at high pressure: “Superhard” polymeric carbon dioxide. Phys Rev Lett. 1999;83(26):5527–5530. [Google Scholar]
- 10.Iota V, Yoo CS, Cynn H. Quartzlike carbon dioxide: An optically nonlinear extended solid at high pressures and temperatures. Science. 1999;283(5407):1510–1513. doi: 10.1126/science.283.5407.1510. [DOI] [PubMed] [Google Scholar]
- 11.Serra S, Cavazzoni C, Chiarotti GL, Scandolo S, Tosatti E. Pressure-induced solid carbonates from molecular CO2 by computer simulation. Science. 1999;284(5415):788–790. doi: 10.1126/science.284.5415.788. [DOI] [PubMed] [Google Scholar]
- 12.Yoo CS, Iota V, Cynn H. Nonlinear carbon dioxide at high pressures and temperatures. Phys Rev Lett. 2001;86(3):444–447. doi: 10.1103/PhysRevLett.86.444. [DOI] [PubMed] [Google Scholar]
- 13.Iota V, Yoo CS. Phase diagram of carbon dioxide: Evidence for a new associated phase. Phys Rev Lett. 2001;86(26 Pt 1):5922–5925. doi: 10.1103/PhysRevLett.86.5922. [DOI] [PubMed] [Google Scholar]
- 14.Yoo CS, et al. Crystal structure of pseudo-six-fold carbon dioxide phase II at high pressures and temperatures. Phys Rev B. 2002;65(10):104103. [Google Scholar]
- 15.Bonev SA, Gygi F, Ogitsu T, Galli G. High-pressure molecular phases of solid carbon dioxide. Phys Rev Lett. 2003;91(6):065501. doi: 10.1103/PhysRevLett.91.065501. [DOI] [PubMed] [Google Scholar]
- 16.Lu R, Hofmeister AM. Infrared fundamentals and phase transitions in CO2 up to 50 GPa. Phys Rev B Condens Matter. 1995;52(6):3985–3992. doi: 10.1103/physrevb.52.3985. [DOI] [PubMed] [Google Scholar]
- 17.Santoro M, et al. Amorphous silica-like carbon dioxide. Nature. 2006;441(7095):857–860. doi: 10.1038/nature04879. [DOI] [PubMed] [Google Scholar]
- 18.Iota V, et al. Six-fold coordinated carbon dioxide VI. Nat Mater. 2007;6(1):34–38. doi: 10.1038/nmat1800. [DOI] [PubMed] [Google Scholar]
- 19.Giordano VM, Datchi F. Molecular carbon dioxide at high pressure and high temperature. Europhys Lett. 2007;77(4):46002. [Google Scholar]
- 20.Montoya JA, Rousseau R, Santoro M, Gorelli F, Scandolo S. Mixed threefold and fourfold carbon coordination in compressed CO2. Phys Rev Lett. 2008;100(16):163002. doi: 10.1103/PhysRevLett.100.163002. [DOI] [PubMed] [Google Scholar]
- 21.Sengupa A, Yoo CS. Raman study of molecular-to-nonmolecular transitions in carbon dioxide at high pressure. Phys Rev B. 2009;80(1):014118. [Google Scholar]
- 22.Kume T, Ohya Y, Nagata M, Sasaki S, Shimizu H. A transformation of carbon dioxide to nonmolecular solid at room temperature and high pressure. J Appl Phys. 2007;102:053501. [Google Scholar]
- 23.Santoro M, Gorelli FA. Constraints on the phase diagram of nonmolecular CO2 imposed by infrared spectroscopy. Phys Rev B. 2009;80(18):184109. [Google Scholar]
- 24.Yang BX, Kirz J, Sham TK. Oxygen K-edge absorption spectra of O2, CO and CO2. Phys Lett. 1985;110A(6):301–304. [Google Scholar]
- 25. Kaci L (2011) Strength and elasticity of CO2 under high pressure. MS thesis (Univ of Western Ontario, London, Canada)
- 26.Yoo CS, Sengupta A, Kim M. Carbon dioxide carbonates in the earth’s mantle: Implications to the deep carbon cycle. Angew Chem Int Ed Engl. 2011;50(47):11219–11222. doi: 10.1002/anie.201104689. [DOI] [PubMed] [Google Scholar]
- 27.Giannozzi P, et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J Phys Condens Matter. 2009;21(39):395502. doi: 10.1088/0953-8984/21/39/395502. [DOI] [PubMed] [Google Scholar]
- 28.Taillefumier M, Cabaret D, Flank AM, Mauri F. X-ray absorption near-edge structure calculations with the pseudopotentials: Application to the K edge in diamond and α-quartz. Phys Rev B. 2002;66(19):195107. [Google Scholar]
- 29.Sagmeister S, Ambrosch-Draxl C. Time-dependent density functional theory versus Bethe-Salpeter equation: An all-electron study. Phys Chem Chem Phys. 2009;11(22):4451–4457. doi: 10.1039/b903676h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.