

Significance
Gene expression requires accurate copying of the DNA template into messenger RNA by RNA polymerases. Errors occurring during this transcription process can lead to the production of nonfunctional proteins, which is likely to be deleterious. Therefore, natural selection is expected to enhance the fidelity of transcription. However, very little is known about the transcription error rates of different organisms. Here we present a unique method for the detection of transcription errors by replicated high-throughput sequencing of cDNA libraries. Applying this method to the model organism Caenorhabditis elegans, we report a large-scale analysis of transcription errors. Future applications of this method should allow a rapid increase in our knowledge of evolutionary forces acting on transcription fidelity.
Keywords: evolution, base substitution, RNA polymerase fidelity, C. elegans
Abstract
Accurate transmission and expression of genetic information are crucial for the survival of all living organisms. Recently, the coupling of mutation accumulation experiments and next-generation sequencing has greatly expanded our knowledge of the genomic mutation rate in both prokaryotes and eukaryotes. However, because of their transient nature, transcription errors have proven extremely difficult to quantify, and current estimates of transcription fidelity are derived from artificial constructs applied to just a few organisms. Here we report a unique cDNA library preparation technique that allows error detection in natural transcripts at the transcriptome-wide level. Application of this method to the model organism Caenorhabditis elegans revealed a base misincorporation rate in mRNAs of ∼4 × 10−6 per site, with a very biased molecular spectrum. Because the proposed method is readily applicable to other organisms, this innovation provides unique opportunities for studying the incidence of transcription errors across the tree of life.
Errors in biological processes are at the very heart of the evolution of life. Indeed, mutations caused by DNA replication errors are ultimately essential for species adaptation in the face of changing environments. Despite the important role of adaptive mutation for evolution, most mutations are deleterious, especially when they affect protein-coding sequences (1). As a consequence, selection is expected to enhance the fidelity of replication (2–4). However, because errors can occur at any step of the protein synthesis process, even nonmutated sequences can produce nonfunctional proteins. Indeed, misincorporations by RNA polymerase (transcription errors) and erroneous tRNA recruitment (translation errors) may often lead to the synthesis of misfolded, nonfunctional proteins with potentially harmful consequences (5, 6). Therefore, selection is expected to enhance the fidelity of each of these processes. However, the error rates that can be tolerated by different organisms remain unclear, and several hypotheses have been proposed for the limits to the fidelity of replication and transcription (7–10).
Two fundamental differences with DNA mutations may reduce the strength of selection against transcription and translation errors. First, unlike DNA mutations, the latter are not permanently transmitted to daughter cells. Second, individual loci generally produce multiple transcripts with relatively short half-lives (11), so that each error is present in only a fraction of the proteins produced. Therefore, it has been suggested that the strength of selection against transcription and translation errors might be less intense than that operating at the level of genome replication (9).
With the recent improvement in sequencing techniques, detection of mutations is now commonly achieved by next-generation sequencing of mutation accumulation lines (12–15), providing ample opportunities for developing and testing theories on the evolution of mutation rates. However, because of their transient nature, transcription and translation errors have remained difficult to detect. The few attempts to measure transcription error rates have relied on indirect techniques involving reporter constructs and/or in vitro template copying (16–20). Reporter constructs measure transcription errors at only a small number of sites and are often convoluted with translation errors (18, 19), and in vitro methods use experimental conditions that may be quite different from the intracellular environment. Thus, it is not surprising that previous estimates of transcription error rates vary by orders of magnitudes even within the same organism (16–20), although a rough overall average value of 10−5 per nucleotide has been suggested (21). Likewise, measurements of translation error rates are still sparse and can be hard to disentangle from transcription errors (18, 19, 22–24).
Although large-scale analysis of translation errors might require a breakthrough in mass spectrometry techniques, one can imagine that the large amount of RNA-sequencing (RNA-seq) data now routinely obtained by next-generation sequencing could help in detecting transcription errors. Indeed, after mapping RNA-seq reads to a reference genome, transcription errors will appear as mismatches between mRNA reads and the reference genome. Therefore, the billions of RNA-seq reads deposited in public databases probably contain thousands of transcription errors within their sequences. Unfortunately, such a naive approach cannot be used with traditional RNA-seq data, because mismatches caused by transcription errors are not accurately distinguishable from the potentially much more numerous sequencing errors, not to mention errors introduced by reverse transcription during cDNA synthesis [RT (reverse transcription) errors]. In principle, bar-coding of nucleic acid molecules before sequencing can facilitate the discrimination of sequencing errors from real mutations (25). Here we describe a unique method for identifying transcription errors by sequencing multiple cDNAs originating from the same mRNA molecule, using a bar-coding strategy to trace back the origin of individual cDNAs.
Results
A Unique Library Preparation Technique.
To accurately identify transcription errors in RNA-seq data, we developed a unique cDNA library preparation technique. We start by tagging fragmented mRNAs at their 5′ ends with bar codes made of random 8-mers (Fig. 1A). The tagged RNA fragments are then attached to beads and reverse transcribed three times (Fig. 1B). After each round of reverse transcription, the newly generated cDNAs are washed away (Fig. 1C) and characterized by Illumina paired-end sequencing. To simplify the following discussion, we denote a series of reads originating from a unique molecule of fragmented mRNA as a family. Two reads are considered as belonging to the same family if they share the same bar code and have identical 5′ and 3′ breakpoints introduced during the process of mRNA fragmentation.
Fig. 1.

Overview of the method. (A) Fragmented mRNAs are tagged by attaching random 8-mers (bar codes) to their 5′ ends. In this example, one of the four mRNAs contains a transcription error (red star). (B) The tagged RNA fragments are attached to a bead and reverse transcribed. An error introduced by the reverse transcriptase is represented with a green star in one of the cDNAs. (C) The newly generated cDNAs are washed away. (D) After repeating the steps in B and C two more times and sequencing the cDNAs produced after each of the three rounds of reverse transcription, the RNA-seq reads are aligned to the genome and grouped into families according to the combined information of their bar code and breakpoint mapping position. The transcription error (red star, leftmost family) is shared among all members of the family and therefore is easily distinguished from the occasional RT (green star) and sequencing (blue stars) errors.
The rationale for identifying transcription errors in such samples is as follows. Errors that are already present in the fragmented mRNAs (i.e., transcription errors) will be copied by the reverse transcriptase and incorporated into each newly generated cDNA, therefore appearing as a mismatch in every read of a family (Fig. 1D, leftmost family). In contrast, individual reads in the family may contain unique RT and sequencing errors, which will generally occur as singletons (Fig. 1D, blue and green stars). Provided that the sequencing and RT error rates are low enough, the probability of observing the same sequencing or RT error in all three reads would be negligible, and only errors present in the original mRNA molecule could produce the pattern shown in the leftmost family of Fig. 1D. Ideally, every single cDNA generated should be sequenced, so that each family contains three pairs of reads (size 3 family). However, this would require extremely deep sequencing, and in practice, only a fraction of the cDNA library is sequenced (see below), so that most families contain reads from only one or two cDNAs (size 1 and size 2 families). Although size 1 families cannot be used to disentangle transcription errors from other types of errors, we show that size 2 families are sufficient to accurately call transcription errors and that the number of size 2 and size 3 families obtained from a single run of HiSeq sequencing is sufficient to find dozens of transcription errors in Caenorhabditis elegans.
Estimating Sequencing Error Rates.
Every base call in an Illumina sequencing run is given a quality score, which is an indication of the probability that the base call is erroneous. In principle, the product of these probabilities at a given position within a family can be used as an estimation of the probability of observing a false positive caused by multiple sequencing errors. Assuming that all three possible erroneous base calls at a given position are equiprobable, the probability of observing the same erroneous base call in two different reads would be 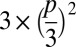 , where p is the probability of an erroneous base call. By setting a threshold on the quality scores (p) of the sites analyzed, we can control the rate at which sequencing errors introduce false positives into our analysis.
, where p is the probability of an erroneous base call. By setting a threshold on the quality scores (p) of the sites analyzed, we can control the rate at which sequencing errors introduce false positives into our analysis.
To evaluate whether the Illumina quality scores do indeed yield correct estimates of the probability of erroneous base calls, we developed our own method of estimating sequencing error rates. With paired-end sequencing of short mRNA fragments, most pairs of reads contain a region of overlap that is sequenced from both ends. Within this region, sequencing errors are revealed as different base calls between the left and right reads. Assuming that the erroneous read is the one containing a different base call than that in the reference genome, we can directly discriminate erroneous from correct base calls and therefore estimate the sequencing error rate for the different values of quality scores. This analysis clearly showed that the Illumina quality scores tend to overestimate the probability of erroneous base calls (Table S1) and therefore can safely be used to estimate an upper limit to the number of false positives introduced by sequencing errors.
Estimating the Reverse Transcriptase Error Rate.
Sequencing errors are not the only possible source of false positives. Reverse transcription of mRNA fragments into cDNAs introduces errors into the newly generated cDNAs at a certain rate. In the extreme case in which the same error is introduced at the same position into every cDNA in a family, these parallel RT errors would be mistaken for a transcription error and contribute false positives to our analysis. To estimate the number of false positives contributed by parallel RT errors, we sought to estimate the reverse transcriptase error rate. With the exception of extremely rare cases where the same RT error occurs in multiple cDNAs within a family, RT errors should be characterized by mismatches present in only one cDNA of families of size 2 or 3. To avoid contamination by sequencing errors, we focused on sites for which the probability of an erroneous base call (computed as described above) was less than 1 × 10−7. Note that this number is less than the probability corresponding to the best possible quality score, so that only positions covered by both reads in a pair can be used here. RT error base substitutions were found at a rate of 1.14 × 10−4 (±6.4 × 10−6, 95% confidence interval). For each of the 12 possible base substitutions, we computed the conditional RT error rate and obtained the full molecular spectrum of RT error base substitutions (Fig. S1). The most common type of RT error corresponds to G → A base substitutions, which occurs at a conditional rate of 1.29 × 10−4 (Fig. S1). Therefore, the probability of observing the same G → A RT error in two cDNAs from the same family is 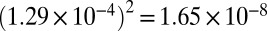 . For all other possible base substitutions, the probability of observing the same error twice in two cDNAs is less than 10−8.
. For all other possible base substitutions, the probability of observing the same error twice in two cDNAs is less than 10−8.
Thus, because the transcription error rate is expected to be on the order of 10−6 to 10−4, it is unlikely that RT errors contribute a significant number of false positives as long as the analysis is restricted to families of size 2 and more. This is confirmed by our observation that within 17,300 size 3 families, we never observed a case where two cDNAs contained a base substitution while the third one contained the same base call as in the reference genome, which would be the signature of parallel RT errors.
Transcription Error Rate and Spectrum of Caenorhabditis elegans.
To demonstrate the feasibility of the proposed technique, we applied it to the transcriptome of C. elegans. We obtained cDNAs from both wild-type (N2) nematodes and an RNA-editing deficient strain (RB886) to disentangle transcription errors from RNA-editing events (26). Because some transcription errors might result in degradation of the corresponding mRNAs by nonsense-mediated mRNA decay (NMD) (27), we also obtained cDNAs from a NMD-deficient strain (VC1305). The three corresponding cDNA libraries were prepared according to the method described in Fig. 1 and sequenced on an Illumina HiSeq 2000, producing a total of 766,868,847 101-nt-long paired reads. Reads were aligned to the reference genome, carefully filtered for mismapping to paralogous regions (Materials and Methods), and grouped into families as defined previously. Reads that mapped to the mitochondrial genome were discarded to avoid confusing transcription errors from faithful transcripts at any potential heteroplasmic sites. This yielded a total of 38,411,057 size 1, 168,094 size 2, and 14,905 size 3 families (Table S2). Informative families (i.e., size 2 and size 3) represent only about 0.5% of all families, which probably reflects the fact that only a fraction of the total cDNAs generated were sequenced. Although deeper sequencing would certainly help increase the fraction of informative families, we show here that the transcription error rate can still be estimated without the need of extremely deep sequencing.
For every family, we built a consensus sequence by selecting only the aligned positions that fulfilled three criteria: (i) in a family of size 2 or 3, (ii) the same base call for all reads, and (iii) a probability of all base calls to be erroneous of <10−8 (computed from the quality score). After filtering for potentially polymorphic sites (see below), consensus sites with a base call different from the reference genome were considered as transcription errors. We found a total of 6, 25, and 52 transcription errors in the wild-type, RNA editing-deficient, and NMD-deficient strains, respectively, yielding base substitional transcription error rates of 2.2 × 10−6, 3.3 × 10−6, and 5.2 × 10−6 per site, respectively (Tables S3 and S4). These three rates are not significantly different from each other (P > 0.1 for all two-by-two comparisons, χ2 test), indicating that the transcription errors recovered in this analysis are likely not to be by-products of RNA-editing processes and that the removal of error-containing transcripts by NMD, if any, is below our statistical power of detection. Because only the small fraction of base substitutions that produce a premature stop codon can be detected by NMD, the expected increased error rate in the NMD-deficient strain is likely to be on the order of only a few percent and will require a much deeper analysis to be detected. Although, we found that 2 out of the 52 base substitutions in the NMD-deficient strain produced a premature termination codon (PTC), this was not significantly different from the 1 out of 31 PTC-creating errors observed in the combined data from the two NMD-capable strains (P > 0.1, Fisher’s exact test).
Although further in-depth analysis may reveal significant differences between these three strains, our results indicate that their transcription error rates are similar enough that the combined data from all three strains should be representative of the overall C. elegans transcription error rate. This leads to an overall transcription error rate estimate of 4.1 × 10−6 (±8.8 × 10−7, 95% confidence interval) per site, which is about one order of magnitude lower than most previously reported transcription error rates (21) but close to the lower estimates obtained from in vitro analysis of wheat germ RNA polymerase II (17). The numbers of transcription errors found in coding (n = 65) and UTR (n = 18) parts of transcripts were not significantly different from a random distribution (P = 0.2, χ2 test). In addition, within coding regions, transcription errors were observed at the first, second, and third positions of codons at frequencies not significantly different from the random expectation of one-third at each position (21, 21, and 23 errors at positions 1, 2, and 3, respectively; P = 0.9, χ2 test).
An important advantage of our method over reporter constructs and in vitro assays is the specific identification of the types of errors that occur, which reveals the full molecular spectrum of transcription errors. We found no significant difference between the three molecular spectra corresponding to the three strains analyzed (Table S4; P > 0.1 for all two-by-two comparisons, log-likelihood ratio test). Although deeper analysis with more statistical power may uncover significant differences, we reasoned that these three spectra were close enough that the combined spectrum should be representative of the C. elegans transcription base substitution spectrum (Fig. 2). There is a wide range of variation in the error rate across the 12 possible base substitutions, the most common one (C → U) being more than 10× more abundant than the least frequent A → C (P < 0.05, Fisher’s exact test), whereas U → A was never observed among the 83 transcription errors found in this analysis (Fig. 2 and Table S4). Transitions are more frequent than transversions, suggesting that RNA polymerases, like DNA polymerases, tend to favor substitutions of bases from the same structural class (28–30). We also noticed that the observed spectrum of transcription errors is relatively close to the DNA mutation spectrum of C. elegans (Fig. 3), indicating that replication and transcription polymerases tend to make the same errors.
Fig. 2.

Molecular spectrum of transcription errors. The values shown in this graph are conditional error rates (i.e., A → U gives the probability of a U to be inserted at a position where an error-free mRNA should contain an A) for all (n = 96) transcription errors detected. Error bars represent the 95% confidence interval of the error rate.
Fig. 3.

Comparison of the genomic and transcription base substitution spectra. The genomic base substitution data are from ref. 41. The total numbers of base substitutions are n = 448 for genomic mutations and n = 96 for transcription errors. Because genomic mutations are not polarized (an A-to-G mutation is equivalent to a T-to-C mutation), transcription errors are merged into groups of complementary mutations to compare them to genomic mutations. This graph shows, for all possible types of transcription and genomic base substitutions, the fraction of base substitutions of a given type. For example, the blue bar A:T → G:C shows that ∼24% of all transcription errors are A → G or T (U) → C errors.
Insertions and Deletions.
In addition to generating base substitution errors, DNA-dependent RNA polymerases can also erroneously skip (deletion) or add (insertion) extra nucleotides in the nascent transcripts. As above, before measuring the RNA polymerase insertion/deletion (indel) error rate, we first need to control the rate of false-positive indels introduced by multiple sequencing or RT errors. We again used the overlapping regions of paired-end reads to detect sequencing indel errors. Using the same method as applied to base substitution errors, we estimated the sequencing indel error rate to be 1.7 × 10−5 (±1.4 × 10−6, 95% confidence interval). This indicates that sequencing indels introduce false positives at a rate of 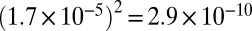 for families with two informative reads and less than 10−10 for families with more than two informative reads.
for families with two informative reads and less than 10−10 for families with more than two informative reads.
Indels introduced by the reverse transcriptase (RT indels) at the same position in two cDNAs also have the potential to create false positives. We searched for RT indels, reasoning that indels present in both reads of a pair but not in the other reads from the same family are caused by RT errors. The resulting RT indel error rate is 8.8 × 10−6 (±1.4 × 10−6, 95% confidence interval). Therefore, assuming that RT indels are randomly distributed, the probability of observing the same RT indel in two reads of a family is 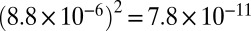 . Given these observations, indels present in all reads of size 2 and size 3 families were considered as being present in the mRNA and therefore corresponding to transcription indels. We found a total of 26 indels (18 insertions and 8 deletions; Table S5), yielding a transcription indel rate of 1.2 × 10−6 (±1.6 × 10−6, 95% confidence interval). One potential consequence of indels in mRNAs is creation of a frameshift in the ORF, leading to the appearance of premature stop codons in the transcript. Because at least some transcripts containing premature termination codons are expected to be degraded by NMD, the indel error rate estimate that we provide is likely biased downward, except in the NMD-deficient strain. However, although the indel error rate measured in the NMD-deficient strain is slightly higher than the average from the two other strains, this difference was not statistically significant (1.5 × 10−6 vs. 1.0 × 10−6 for NMD-deficient and NMD-capable strains, respectively; P = 0.5, χ2 test). All of the transcription indels reported here are one nucleotide in length (see Table S6 for a list of transcription indels). We also observed that transcription indels tend to occur in homopolymeric nucleotide runs (Table S6). It has been previously reported that genomic indels in C. elegans are dominated by insertions and tend to occur in homopolymeric nucleotide runs (31, 32), again suggesting that strong parallels exist in the types of errors generated by DNA and RNA polymerases.
. Given these observations, indels present in all reads of size 2 and size 3 families were considered as being present in the mRNA and therefore corresponding to transcription indels. We found a total of 26 indels (18 insertions and 8 deletions; Table S5), yielding a transcription indel rate of 1.2 × 10−6 (±1.6 × 10−6, 95% confidence interval). One potential consequence of indels in mRNAs is creation of a frameshift in the ORF, leading to the appearance of premature stop codons in the transcript. Because at least some transcripts containing premature termination codons are expected to be degraded by NMD, the indel error rate estimate that we provide is likely biased downward, except in the NMD-deficient strain. However, although the indel error rate measured in the NMD-deficient strain is slightly higher than the average from the two other strains, this difference was not statistically significant (1.5 × 10−6 vs. 1.0 × 10−6 for NMD-deficient and NMD-capable strains, respectively; P = 0.5, χ2 test). All of the transcription indels reported here are one nucleotide in length (see Table S6 for a list of transcription indels). We also observed that transcription indels tend to occur in homopolymeric nucleotide runs (Table S6). It has been previously reported that genomic indels in C. elegans are dominated by insertions and tend to occur in homopolymeric nucleotide runs (31, 32), again suggesting that strong parallels exist in the types of errors generated by DNA and RNA polymerases.
Potential Sources of False Positives.
To validate the results presented in this study, artifacts (other than convergent sequencing/RT errors) that might produce the illusion of transcription errors have to be ruled out. Somatic mutations can produce transcripts whose sequences in the affected cell lineages would differ from the reference genome and would be viewed as transcription errors in our analysis. However, in order for 5% of our inferred base substitution transcription errors to actually be somatic mutation-derived false positives, somatic mutations would have to occur at a frequency of ∼2 × 10−7 per site per cell, which would correspond to a rate of ∼2 × 10−8 mutations per site per cell division (assuming an average of 10 cell divisions from the embryo to the adult worm). This is ∼30× the estimated mutation rate per cell division in the C. elegans germ line (9). Based on the observation that somatic mutations rates per cell division are less than 30× that of the germ line in humans (9), we can reasonably infer that a difference of more than 30× in C. elegans is unlikely and therefore that somatic mutations do not contribute more than 5% of false positives to our estimation of the transcription error rate.
Although C. elegans is self-fertilizing, polymorphic sites also have the potential to produce faithful transcripts that would be mistaken for transcription error-containing mRNAs compared with the reference genome. However, such aberrations are easily detected because 100% (50% if heterozygous) of the families covering such positions would show a mismatch. Therefore, we retained only sites covered by at least 20 families and for which more than 95% of the families support the reference genome base call. We also sequenced the genomic DNA from the same worms that were used to search for transcription errors and mapped these genomic reads against the genomic regions surrounding each of the 83 transcription errors found in this study (Materials and Methods). Out of the 3,547 genomic DNA reads mapped, only 2 supported a base call matching the inferred transcription error, which is exactly the number expected simply from sequencing errors in the genomic DNA reads based on our previous estimate of sequencing error rates (Materials and Methods). This strongly suggests that the transcription errors inferred in our study are not false positives caused by genomic mutations.
We also used genomic DNA reads to search for the presence of inferred transcription indels within the DNA of the worms used in this analysis. We found that 5 out of the 531 mapped genomic DNA reads spanning the position of an inferred transcription indel contained the inferred transcription indel. Based on our estimation of the sequencing indel error rate (1.7 × 10−5 per site), we expect to observe zero (531 × 1.7 × 10−5 = 0.01) such indel-containing genomic DNA reads. The excess of indel-containing genomic DNA reads might be partially explained by an elevated sequencing indel error rate within homoplymeric nucleotide runs. Indeed, these five genomic DNA reads map to only two inferred indels, both of them falling within a large homopolymeric run (9 and 11 nt). However, it is also possible that a fraction of the transcription indels inferred in our study are false positives caused by indels present at low frequency in the genomic DNA of the worms sequenced in this analysis. Therefore, although we are confident that our analysis provides an accurate estimation for the upper limit of the transcription indel rate, the actual rate might be lower than 1.2 × 10−6 indels per site.
Finally, RNA editing (26, 33, 34), a site-specific posttranscriptional process that can deaminate mRNA adenosines to inosine, which is then recognized as a guanosine, could be confused with transcription errors. However, if RNA editing was interfering with our analysis, we would observe an abnormally high level of A → G transcription errors, which is not the case (Fig. 2). Also, we did not observe any significant difference in the transcription error rate and pattern in the editing-deficient strain compared with the wild-type and the NMD-deficient strains (Table S4). It is very unlikely that other types of RNA editing exist in C. elegans [with the exception of very rare C → U changes (35)], and even if such a mechanism existed, our requirement that 95% of the sequenced mRNAs show the same base call as in the reference genome would remove all sites that are systematically edited.
Discussion
A Unique Method for Detecting Transcription Errors.
In this study, we have described a unique cDNA library preparation technique and the associated bioinformatic analyses that allow for detection of transcription errors in RNA-seq data. To demonstrate the feasibility of our method, we applied it to the transcriptome of C. elegans and detected dozens of transcription errors, yielding a base substitution transcription error rate of ∼4 × 10−6. One limitation of our method is that it cannot discriminate misincorporations by the RNA polymerase from posttranscriptional RNA modifications. The observation that the most common type of transcription error is a C → U base substitution suggests that a fraction of the errors observed could be due to cytosine deamination, rather than base misincorporation by the RNA polymerase. However, even if this were true, it would still be the case that the total error rate (misincorporation + posttranscriptional modifications) in mRNAs at the time of translation is being correctly quantified, with the unlikely exception of spontaneous cytosine deamination that could occur in vitro (in the absence of deaminases), after mRNA extraction. In this particular analysis, there was a relatively small ratio of informative families over the total number of reads generated. Even among informative families, those of size 2 were far more abundant than size 3 families, so that all transcription errors reported here were found in size 2 families. However, this is not an inherent limitation of the method because deeper sequencing will improve the ratio of informative families and therefore help reveal many more transcription errors.
Evolution of the Fidelity of Transcription.
Our observation that the C. elegans transcription error rate is about 10 times lower than previously reported transcription error rates for prokaryotes and unicellular eukaryotes is somewhat surprising. Indeed, it has been suggested that the fidelity of replication, transcription, and translation is limited by the ability of selection to increase the fidelity of each of these steps and therefore should scale positively with the effective population size of the organisms considered [the drift barrier hypothesis (36)]. Because multicellular organisms typically have lower effective population sizes than unicellular organisms (3), we expect error rates to be generally higher in multicellular organisms. The larger mutational target of transcripts in multicellular eukaryotes compared with microbes (37) could reinforce the selective pressure on the fidelity of transcription in multicellular eukaryotes and therefore explain the contradiction mentioned above. However, one simple explanation for the inconsistency of current observation with the theory is that previous measures of the transcription error rate were upwardly biased, owing to a reliance on reporter constructs and/or in vitro assays. Indeed, reporter construct analyses are generally incapable of disentangling changes in protein sequences caused by transcription errors from the potentially much more common changes caused by translation errors (18, 19). Likewise, in vitro essays might not be informative about the in vivo error rate, because the conditions used in vitro do not always reflect the intracellular environment, and the different conditions used can change the observed error rate by more than an order of magnitude (17). Therefore, although it is possible that the drift-barrier hypothesis does not apply to the evolution of transcription error rates or that C. elegans is an exception to the rule, we suspect that the real transcription error rates of Escherichia coli and Saccharomyces cerevisiae are much lower than what was previously reported. This point can now be readily evaluated with the method presented above, which fully generalizes to any organism.
The method described in this paper should facilitate a rapid increase of our knowledge on the causes and consequences of transcription errors as well as of the evolutionary forces acting on the fidelity of transcription. We predict that although the transcription error rate is orders of magnitude higher than the replication error rate, the scaling of both with the efficiency of natural selection should be similar, in accordance with the drift-barrier hypothesis (36).
Materials and Methods
Library Preparation and Sequencing.
RNA and DNA isolation.
C. elegans strains (wild-type N2, RB886, and VC1305) were cultured on Nematode growth media following standard protocols (38) with E. coli OP50 except that plates were made using agarose. Mixed-stage worms from 12 crowded plates per strain were washed several times in M9 buffer, and the final worm pellet was divided into two batches for total RNA and genomic DNA extraction. Total RNA was isolated using the the Ambion RiboPure Kit (Life Tech). Total DNA was extracted using Qiagen Genomic-Tip protocol (Qiagen). DNA libraries were generated from 1 μg of genomic DNA sheared to ∼300 base pair fragments on an M-series focused ultrasonicator (Covaris) following the standard TruSeq library protocol (Illumina). Sequencing was conducted on a single lane of a HiSeq2000.
Messenger RNA isolation.
For construction of the RNA-seq libraries, 5 μg of total RNA was resuspended in a final volume of 50 μL Rnase-free water, and mRNA isolation was performed using the Dynal oligo dT bead system from (Life Tech). Messenger RNA was eluted in 17 μL of RNase-free water.
Messenger RNA fragmentation.
Messenger RNA was fragmented using the RNA Fragmentase system (Catalog ID: E6146S; New England BioLabs), per manufacturer’s specifications. Fragmented mRNA (in 100 μL water) was ethanol-precipitated with 2 μL of 5 mg/mL Glycogen (Ambion; Life Tech), 10 μL of 5M sodium acetate pH5.3, and 300 μL of absolute ethanol (chilled). Samples were ethanol precipitated at −80 °C for 1 h, followed by spinning at 15,000 × g for 45 min at 4 °C. Pellets were washed twice with chilled 70% ethanol (vol/vol) and resuspended in a final volume of 5 μL RNase-free water.
Sequential adaptor ligation.
The 3′ RNA adaptor used in these libraries is the standard Illumina TruSeq Small RNA 3′ Adaptor. Ligation of the 3′ adaptor was done in accordance with the Illumina TruSeq Small RNA Sample Prep kit (Illumina). Specifically, 5 μL of fragmented mRNA was mixed with 1 μL of the Illumina TruSeq 3′ adapter and incubated at 70 °C for 2 min before being placed on ice. We then added 2 μL of ligation buffer, 1 μL of RNase inhibitor, and 1 μL of T4 RNA ligase 2 truncated (NEB) followed by incubation at 28 °C for 1 h. Reaction was stopped with 1 μL stop solution followed by incubation at 28 °C for 15 min before being placed on ice. The 5′ adaptor is a modified version of the Illumina TruSeq Small RNA 5′ adaptor (Integrated DNA Technology). The modification includes a 5′ Biotin and eight N bases on the 3′ end of the oligonucleotide (5′-Bio-rGrUrUrCrArGrArGrUrUrCrUrArCrArGrUrCrCrGrArCrGrArUrCrNrNrNrNrNrNrNrN-3′). Ligation included 1 μL of the modified RA5 adapter (100 μM stock), 1 μL of ATP as supplied by Illumina, and 1 μL of T4 RNA Ligase 2 (NEB). This reagent mix was added to the 3′ adapted mRNA, and the sample was incubated at 28 °C for 1 h followed by incubation at 4 °C. Sequentially adapted libraries were then purified using RNA MinElute columns (Qiagen) per the manufacturer's protocol and eluted in 10 μL RNase-free water. Samples were stored overnight at −80 °C.
Reverse transcription cycling.
To capture the products of each RT reaction in separate indexed libraries, we used the biotinylated 5′ ends of the constructs to allow for separation of the RT products after each reaction and transfer of template RNA to a subsequent round of RT. For the first round of reverse transcription the sample was mixed with 1 μL of Illumina TruSeq Small RNA primer and incubated at 70 °C for 2 min, followed by placing the samples on ice. After addition of RT reaction mix, samples were incubated at 50 °C for 30 min. During the incubation, 50 μL of Dynal M270 streptavidin beads (Life Tech) were prepared for RNA use as described in the manufacturer’s protocol by washing the beads for 2 min with an equal volume of Diethylpyrocarbonate (DEPC)-treated 0.1 M NaOH and 0.05 M NaCl followed by 2 min with an equal volume DEPC-treated 0.1 M NaCl. The washed beads were then resuspended in 25 μL of 2X Binding Buffer per the manufacturer's protocol. At the end of the first round of RT, the washed streptavidin beads were added to the first-round RT reaction, and the samples were incubated at room temperature for 15 min on the Lab Quake to allow the streptavidin beads to capture the biotinylated RNA template. The samples were placed on a magnet until the solution cleared, after which the supernatant was transferred to a new PCR tube. The beads were washed twice in 1X Binding Buffer and resuspended in 6 μL RNase-free water. One microliter of Illumina TruSeq Small RNA primer was added, and the samples were incubated at 70 °C for 2 min, followed by placing the samples on ice. Fresh RT reaction mix was then added, and the second round of RT was performed. This process was repeated for a total of three rounds of reverse transcription.
RNA-seq library indexing.
Each round of reverse transcription supernatant was used as a template in an amplification reaction described below to generate three Illumina dRNA TruSeq libraries where each round of reverse transcription is labeled with a different index. Amplification reactions were run for 11 cycles using the thermoprofile described in the Illumina TruSeq Small RNA manual. The amplification reactions were purified using an equal ratio of AmpureXP beads (Beckman Coulter) and eluted in 20 μL of EB buffer (Qiagen). Two microliters of each library was assayed on an RNA pico bioanalyzer chip (Agilent Technologies, Inc.). Paired-end (100-base pair) sequencing was performed on a HiSeq2000.
RNA-Seq Read Mapping.
Reads (with their bar code sequences removed) were mapped to the spliced transcripts predicted by Ensembl (release 66) using the program bwa (39) with default parameters, except for the parameter –o, which was set to zero (i.e., no gaps allowed) for the base substitution analysis. The alignments were then converted into genomic coordinates, and all of the reads mapping to more than one genomic region were discarded. To ensure a very strict filtering of mismapping to paralogous regions, these 69,774,961 uniquely mapping pairs of reads (71,747,935 when allowing indels) were then blated against the reference genome, and every read with a significant hit (E value < 10−2) outside of the genomic region previously assigned was discarded. This step led to the removal of 6,612,765 pairs of reads (6,758,497 when allowing indels).
Genomic DNA Read Mapping.
We generated transcription error-containing genomic sequences by extracting from the C. elegans reference genome (Ensembl release 66) 90 nucleotides on both sides of all inferred transcription errors. Genomic DNA reads were mapped against these transcription error-containing genomic sequencing using the program blat (40) with default parameters. Only reads with a match spanning at least 10 nucleotides on both sides of the position occupied by the inferred transcription error were retained. To estimate the number of genomic DNA reads containing the inferred base substitution because of sequencing error, we extracted, for all of the mapped reads, their base call quality score at the position of the inferred base substitution and converted the base quality into the probability that the base call is wrong according to the observed error rate reported in Table S1. We then summed these probabilities for all mapped reads and divided the total number by 3, assuming that all three possible errors at a given position are equiprobable.
Supplementary Material
Acknowledgments
This work was supported by National Science Foundation Grant EF-0827411 (to M.L.) and National Institutes of Health Grant R01-GM036827 (to M.L. and W.K.T.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. L.D.H. is a guest editor invited by the Editorial Board.
Data deposition: The sequences reported in this paper have been deposited in the National Center for Biotechnology Information Short Read Archive (accession no. SRP030526).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1309843110/-/DCSupplemental.
References
- 1.Eyre-Walker A, Keightley PD. The distribution of fitness effects of new mutations. Nat Rev Genet. 2007;8(8):610–618. doi: 10.1038/nrg2146. [DOI] [PubMed] [Google Scholar]
- 2.Drake JW, Charlesworth B, Charlesworth D, Crow JF. Rates of spontaneous mutation. Genetics. 1998;148(4):1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lynch M. The cellular, developmental and population-genetic determinants of mutation-rate evolution. Genetics. 2008;180(2):933–943. doi: 10.1534/genetics.108.090456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sturtevant AH. Essays on evolution. I. On the effects of selection on the mutation rate. Q Rev Biol. 1937;12(4):464–476. [Google Scholar]
- 5.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134(2):341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet. 2009;10(10):715–724. doi: 10.1038/nrg2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.André JB, Godelle B. The evolution of mutation rate in finite asexual populations. Genetics. 2006;172(1):611–626. doi: 10.1534/genetics.105.046680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baer CF, Miyamoto MM, Denver DR. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet. 2007;8(8):619–631. doi: 10.1038/nrg2158. [DOI] [PubMed] [Google Scholar]
- 9.Lynch M. Evolution of the mutation rate. Trends Genet. 2010;26(8):345–352. doi: 10.1016/j.tig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sniegowski PD, Gerrish PJ, Johnson T, Shaver A. The evolution of mutation rates: separating causes from consequences. Bioessays. 2000;22(12):1057–1066. doi: 10.1002/1521-1878(200012)22:12<1057::AID-BIES3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 11.Wilusz CJ, Wormington M, Peltz SW. The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol. 2001;2(4):237–246. doi: 10.1038/35067025. [DOI] [PubMed] [Google Scholar]
- 12.Denver DR, et al. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci USA. 2009;106(38):16310–16314. doi: 10.1073/pnas.0904895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halligan DL, Keightley PD. Spontaneous mutation accumulation studies in evolutionary genetics. Annu Rev Ecol Evol Syst. 2009;40:151–172. [Google Scholar]
- 14.Lynch M, et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci USA. 2008;105(27):9272–9277. doi: 10.1073/pnas.0803466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ossowski S, et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327(5961):92–94. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blank A, Gallant JA, Burgess RR, Loeb LA. An RNA polymerase mutant with reduced accuracy of chain elongation. Biochemistry. 1986;25(20):5920–5928. doi: 10.1021/bi00368a013. [DOI] [PubMed] [Google Scholar]
- 17.de Mercoyrol L, Corda Y, Job C, Job D. Accuracy of wheat-germ RNA polymerase II. General enzymatic properties and effect of template conformational transition from right-handed B-DNA to left-handed Z-DNA. Eur J Biochem. 1992;206(1):49–58. doi: 10.1111/j.1432-1033.1992.tb16900.x. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberger RF, Hilton J. The frequency of transcriptional and translational errors at nonsense codons in the lacZ gene of Escherichia coli. Mol Gen Genet. 1983;191(2):207–212. doi: 10.1007/BF00334815. [DOI] [PubMed] [Google Scholar]
- 19.Shaw RJ, Bonawitz ND, Reines D. Use of an in vivo reporter assay to test for transcriptional and translational fidelity in yeast. J Biol Chem. 2002;277(27):24420–24426. doi: 10.1074/jbc.M202059200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Springgate CF, Loeb LA. On the fidelity of transcription by Escherichia coli ribonucleic acid polymerase. J Mol Biol. 1975;97(4):577–591. doi: 10.1016/s0022-2836(75)80060-x. [DOI] [PubMed] [Google Scholar]
- 21.Ninio J. Transient mutators: A semiquantitative analysis of the influence of translation and transcription errors on mutation rates. Genetics. 1991;129(3):957–962. doi: 10.1093/genetics/129.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kramer EB, Farabaugh PJ. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA. 2007;13(1):87–96. doi: 10.1261/rna.294907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ortego BC, Whittenton JJ, Li H, Tu SC, Willson RC. In vivo translational inaccuracy in Escherichia coli: Missense reporting using extremely low activity mutants of Vibrio harveyi luciferase. Biochemistry. 2007;46(48):13864–13873. doi: 10.1021/bi602660s. [DOI] [PubMed] [Google Scholar]
- 24.Zaher HS, Green R. Fidelity at the molecular level: Lessons from protein synthesis. Cell. 2009;136(4):746–762. doi: 10.1016/j.cell.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitt MW, et al. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci USA. 2012;109(36):14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gray MW. Evolutionary origin of RNA editing. Biochemistry. 2012;51(26):5235–5242. doi: 10.1021/bi300419r. [DOI] [PubMed] [Google Scholar]
- 27.Isken O, Maquat LE. Quality control of eukaryotic mRNA: Safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21(15):1833–1856. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- 28.Fitch WM. Evidence suggesting a non-random character to nucleotide replacements in naturally occurring mutations. J Mol Biol. 1967;26(3):499–507. doi: 10.1016/0022-2836(67)90317-8. [DOI] [PubMed] [Google Scholar]
- 29.Vogel F, Röhrborn G. Amino-acid substitutions in haemoglobins and the mutation process. Nature. 1966;210(5031):116–117. doi: 10.1038/210116a0. [DOI] [PubMed] [Google Scholar]
- 30.Wakeley J. The excess of transitions among nucleotide substitutions: New methods of estimating transition bias underscore its significance. Trends Ecol Evol. 1996;11(4):158–162. doi: 10.1016/0169-5347(96)10009-4. [DOI] [PubMed] [Google Scholar]
- 31.Denver DR, Morris K, Lynch M, Thomas WK. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature. 2004;430(7000):679–682. doi: 10.1038/nature02697. [DOI] [PubMed] [Google Scholar]
- 32.Denver DR, Morris K, Lynch M, Vassilieva LL, Thomas WK. High direct estimate of the mutation rate in the mitochondrial genome of Caenorhabditis elegans. Science. 2000;289(5488):2342–2344. doi: 10.1126/science.289.5488.2342. [DOI] [PubMed] [Google Scholar]
- 33.Benne R, et al. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell. 1986;46(6):819–826. doi: 10.1016/0092-8674(86)90063-2. [DOI] [PubMed] [Google Scholar]
- 34.Shaw JM, Feagin JE, Stuart K, Simpson L. Editing of kinetoplastid mitochondrial mRNAs by uridine addition and deletion generates conserved amino acid sequences and AUG initiation codons. Cell. 1988;53(3):401–411. doi: 10.1016/0092-8674(88)90160-2. [DOI] [PubMed] [Google Scholar]
- 35.Chester A, Scott J, Anant S, Navaratnam N. RNA editing: Cytidine to uridine conversion in apolipoprotein B mRNA. Biochim Biophys Acta. 2000;1494(1–2):1–13. doi: 10.1016/s0167-4781(00)00219-0. [DOI] [PubMed] [Google Scholar]
- 36.Lynch M. The lower bound to the evolution of mutation rates. Genome Biol Evol. 2011;3:1107–1118. doi: 10.1093/gbe/evr066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lynch M. The origins of eukaryotic gene structure. Mol Biol Evol. 2006;23(2):450–468. doi: 10.1093/molbev/msj050. [DOI] [PubMed] [Google Scholar]
- 38.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77(1):71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kent WJ. BLAT—The BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denver DR, et al. Variation in base-substitution mutation in experimental and natural lineages of Caenorhabditis nematodes. Genome Biol Evol. 2012;4(4):513–522. doi: 10.1093/gbe/evs028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.