

Abstract
Oligomerization of G protein-coupled receptors is a recognized mode of regulation of receptor activities, with alternate oligomeric states resulting in different signaling functions. The CXCR4 chemokine receptor is a G protein-coupled receptor that is post-translationally modified by tyrosine sulfation at three sites on its N-terminus (Y7, Y12, Y21), leading to enhanced affinity for its ligand, stromal cell derived factor (SDF-1, also called CXCL12). The complex has been implicated in cancer metastasis and is a therapeutic target in cancer treatment. Using molecular dynamics simulation of NMR-derived structures of the CXCR4 N-terminus in complex with SDF-1, and calculations of electrostatic binding energies for these complexes, we address the role of tyrosine sulfation in this complex. Our results show that sulfation stabilizes the dimeric state of the CXCR4:SDF-1 complex through hydrogen bonding across the dimer interface, conformational changes in residues at the dimer interface, and an enhancement in electrostatic binding energies associated with dimerization. These findings suggest a mechanism through which post-translational modifications such as tyrosine sulfation might regulate downstream function through modulation of the oligomeric state of the modified system.
Keywords: post-translational modification, sulfotyrosine, molecular dynamics, chemokine receptor, dimerization
Introduction
CXCR4, a member of the G protein-coupled receptor family, is the primary receptor for stromal cell derived factor (SDF-1, also called CXCL12). The complex plays a role in normal and pathological cell migration processes including embryonic development, leukocyte trafficking, and cancer mestastasis.1,2 Over 23 cancers express elevated levels of CXCR4, which assists in metastasis of cancer cells to distant tissues which express SDF-1.3 Cell culture studies have shown that dimeric SDF-1 fails to induce cell migration due to the activation of “cellular idling signaling pathways.”4 Notably, silencing of the SDF-1 gene in tumor cells enhances metastasis while the presence of re-expressed or exogenously administered SDF-1, in its monomeric or dimeric form, inhibits metastasis.4
Many GPCRs undergo oligomerization either in the presence or absence of their ligands, and oligomerization has been shown to expand the diversity of signaling activities.5 CXCR4 forms dimers in living cells2,6–9 which may be further stabilized by the presence of SDF-1.9 The recently solved crystal structures of complexes of CXCR4 reveal a dimeric arrangement with either a 1:2 or 2:2 ligand:receptor complex; it has been suggested that double occupancy of the dimeric receptor by SDF-1 may account for the inhibition of cellular migration at high chemokine concentrations.1
SDF-1 has a typical chemokine fold characterized by a flexible N-terminus connected by the N loop to a three stranded antiparallel beta sheet and a C terminal alpha helix. It has been shown to exist in a monomer–dimer equilibrium with the dimer stabilized by electrostatic interactions between the first beta sheet on each monomer.10 Veldkamp et al. engineered an SDF-1 dimer stabilized by disulfide bonds between SDF-1 units to allow for NMR structure determination of a dimeric CXCR4 N-terminus:SDF-1 complex.11
High affinity binding between CXCR4 and SDF-1 requires that the N-terminus of CXCR4 be post-translationally modified by sulfation at three tyrosine residues (Y7, Y12, Y21).11–14 This post-translational modification is not unusual in this setting, as tyrosine sulfation has been predicted or observed in the N-terminal extracellular domain of most chemokine receptors.15 A tyrosine corresponding to Y21 exists in almost all CXC family receptors, and residues lining the Y21 binding pocket are conserved in at least half of all chemokines.11 Although neither Y7 nor Y12 nor their chemokine binding sites are conserved among receptor-chemokine complexes, sulfation at these sites is shown to enhance the affinity of the CXCR4 N-terminus for SDF-1 by approximately twenty times while sulfation at Y21 alone provides only a three-fold enhancement.14 The NMR structures show that in the dimeric complex, the sulfated Y7 and Y12 residues interact with the SDF-1 on the opposite side of the dimer.11 This suggests that sulfation helps stabilize the dimeric complex by promoting interactions between each CXCR4 N-terminus and both molecules of the SDF-1 dimer.
Our work addresses the structural effects of sulfation in the CXCR4:SDF-1 complex using molecular dynamics simulations of CXCR4 N-terminus:SDF-1 NMR structures that are unsulfated (PDB: 2k04), partially sulfated (PDB: 2k03; sulfated at only Y21) or fully sulfated (PDB: 2k05; sulfated at Y7, Y12, and Y21). Simulations were conducted on the dimeric complexes, and monomers taken from these complexes, to investigate the effects of sulfation on dimerization. To validate the use of our force field parameters for the simulation of sulfated protein systems, we asked whether sulfating the unsulfated NMR conformers would recover the most favorable interactions seen in the sulfated conformers. Toward this end, we conducted a control study in which we added sulfate groups to Y7, Y12, and Y21 in the unsulfated conformers, and compared simulations of this system (which we refer to as 2k04_S) with simulations of the originally sulfated systems.
We address the impact of sulfation on CXCR4:SDF-1 complex stabilization using calculations of electrostatic binding energies for the monomeric and dimeric complexes in different sulfation states. Complexes were taken from the originally sulfated conformers (PDB: 2k05), and these same conformers sulfated only at Y21, or completely unsulfated. Electrostatic interactions have been shown to play an important role in protein–ligand binding and specificity, through a balance of favorable Coulombic interaction energies and desolvation penalties incurred by removing charged groups from the surrounding solvent.16–20
Although the CXCR4:SDF-1 complex has been well characterized with respect to structure,1,11–13 oligomerization state2,6–10,21 and physiological function3,4,8,15,22,23 no study to date has focused specifically on how sulfation impacts the dynamics and electrostatic stabilization of this complex. Our results support a role for tyrosine sulfation, in particular at sites Y7 and Y12, in the dimerization of the CXCR4:SDF-1 complex through hydrogen bonding across the dimer interface, dynamical changes at the dimer interface, and an enhancement in the electrostatic contribution to binding energy for the dimeric complex.
Results and Discussion
Validation of simulation trajectories
As a starting point for our analysis, we validate our simulation protocol by comparing inter-atomic distances and NOE distance restraint violations with those in the NMR-derived PDB entries. Table I lists the percentage of NOE violations in a given range for simulation of the first six conformers of 2k03, 2k04, and 2k05. The table also lists violations from the set of twenty NMR generated conformers in the original PDB file. Results for a combined trajectory over six simulations show only a small increase in violations above 1 Å for the simulated conformers as compared to the experimental conformers, and less than 5% of violations above 2 Å. We note that simulations were run on only the first six conformers in each entry; had simulations been run starting from all twenty conformers, then agreement with the NOE violations would most likely have been equivalent to the agreement shown by the PDB ensemble. Agreement with NOE restraints for both the unsulfated and sulfated simulations demonstrates that the force fields can be used to simulate the behavior of systems that undergo tyrosine sulfation.
Table I.
NOE Violations for Simulation of Six Conformers of Dimeric CXCR4:SDF-1, for 2k03 (Sulfated at Y21), 2k04 (Unsulfated), and 2k05 (Fully Sulfated)
| 2k03 (Y21) | 2k04 (unsulfated) | 2k05 (Y7,Y12,Y21) | ||||
|---|---|---|---|---|---|---|
| Violation range | MD | PDB | MD | PDB | MD | PDB |
| Viol=0 | 70.3 (60.2 ± 0.5) | 74.8 | 71.0 (59.4 ± 0.3) | 78.1 | 70.4 (59.1 ± 0.3) | 74.3 |
| 0<Viol≤1 | 18.9 (23.7 ± 0.4) | 16.9 | 18.7 (22.8 ± 0.3) | 17.2 | 18.9 (23.9 ± 0.3) | 19.2 |
| 1<Viol≤2 | 7.1 (9.1 ± 0.2) | 6.6 | 7.0 (8.9 ± 0.3) | 3.6 | 5.7 (8.8 ± 0.1) | 5.0 |
| 2<Viol≤3 | 2.6 (3.9 ± 0.1) | 1.5 | 2.2 (5.1 ± 0.3) | 1.1 | 3.5 (4.4 ± 0.1) | 1.4 |
| Viol>3 | 1.2 (3.2 ± 0.2) | 0.2 | 1.1 (3.8 ± 0.3) | 0.1 | 1.5 (3.9 ± 0.2) | 0.2 |
Columns list the violation range in Angstroms and the percentage of violations in a given range, for the simulations (MD) and the native ensembles (PDB). Values are reported for a combined trajectory from simulation of six conformers, and as an average with standard errors over the separate simulations of each of the six conformers (in parenthesis).
Plots of RMSD versus time for all simulations (Fig. 1) show that the 20 ns simulation time is sufficient for equilibration of all systems, and that comparable positional deviations are observed among all monomeric and dimeric complexes. The overall RMSD of the complexes is higher than might be expected from molecular dynamics simulation of X-ray structures due to the flexibility of the CXCR4 N-termini which are for the most part not constrained by secondary structure.
Figure 1.
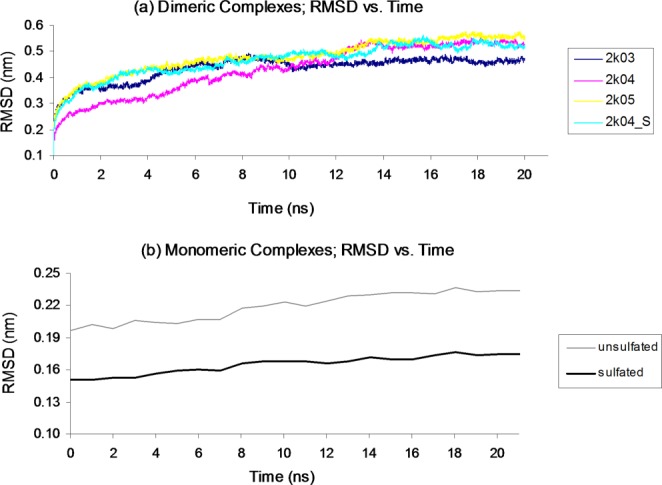
Simulations show equilibration within 20 ns, and comparable positional deviations among all (a) dimeric and (b) monomeric systems. RMSD values for each system are averaged over simulation of six conformers. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Sulfation impacts the structure and dynamics of dimeric CXCR4:SDF-1 complexes
Though residues Y7 and Y12 on the CXCR4 N-terminus are not conserved among chemokine receptors, binding between CXCR4 and SDF-1 is enhanced by sulfation at these sites. NMR structures show that the sulfotyrosine residues at Y7 and Y12 form interactions across the dimer interface, that is, between each CXCR4 N-terminus and the SDF-1 molecule on the other side of the dimer.11 Thus we propose a role for tyrosine sulfation at Y7 and Y12 in stabilizing the dimeric state of the CXCR4:SDF-1 complex, thereby initiating functional consequences that depend on the dimeric state of the chemokine, its receptor, or the complex as a whole.
We compare hydrogen bonding between the unsulfated (2k04) and sulfated (2k03 and 2k05) simulations to assess to what extent hydrogen bonding is enhanced by sulfation, and to identify interactions occurring across the dimer interface. Tables II and III list percentage occupancies for hydrogen bonds between side chains of Y7, Y12, and Y21 on the CXCR4 N-termini, and residues of SDF-1, for the 2k03, 2k04, 2k04_S, and 2k05 simulations. The unsulfated conformers (2k04) show limited hydrogen bonding for Y21, while the conformers sulfated at Y21 (2k03) show hydrogen bonding with residues in either the N loop (N22) or the 40's loop (N44, N46, R47) of the neighboring SDF-1 molecule [Fig. 2(a)]. These SDF-1 residues are thought to form a conserved basic pocket in chemokines of the CXC family.11
Table II.
Hydrogen Bonds to Y7, Y12, and Y21 Side Chains for Simulation of 2k04, Unsulfated
| Tyr | SDF-1 residue | Frames (%) |
|---|---|---|
| 7 | A:Ile28 | 7.7 ± 3.7 |
| 7 | C:Leu26 | 3.6 ± 2.4 |
| 7 | C:Trp57 | 2.6 ± 1.8 |
| 12 | A:Ile28 | 2.0 ± 1.9 |
| 21 | A:Glu15 | 4.3 ± 2.1 |
Table III.
Hydrogen Bonds to Y7, Y12, and Y21 Side Chains for Simulation of 2k05 (Fully Sulfated), 2k04_S (Fully Sulfated), and 2k03 (Sulfated at Y21)
| 2k05 | 2k04 Sulfated | 2k03 | ||
|---|---|---|---|---|
| sTyr | SDF-1 Residue | %Frames (%Bidenate) | % Frames (%Bidentate) | %Frames (%Bidenate) |
| 7 | A:Asn30 | 19.8 ± 5.8 | 13.1 ± 3.9 | – |
| 7 | C:Arg20 | 18.9 ± 5.2(2.8 ± 1.7) | 17.6 ± 6.4(5.3 ± 2.4) | – |
| 7 | C:His25 | 3.9 ± 2.2 | 6.3 ± 3.4 | – |
| 7 | C:Leu26 | 7.7 ± 4.3 | 27.1 ± 5.9 | – |
| 7 | C:Tyr61 | 15.1 ± 5.0 | 38.1 ± 6.8 | – |
| 7 | C:Lys64 | 4.7 ± 2.5 | 0.4 ± 0.4 | – |
| 12 | A:Lys27 | 11.3 ± 3.0 | 24.9 ± 5.2 | – |
| 12 | A:Arg41 | 14.7 ± 5.0(3.0 ± 1.9) | 5.0 ± 3.5(0.0 ± 0.0) | – |
| 12 | A:Gln48 | 3.3 ± 1.3 | 13.2 ± 4.0 | – |
| 12 | C:Lys1 | 3.5 ± 2.2 | 0.0 ± 0.0 | – |
| 12 | C:Arg20 | 7.0 ± 4.8(4.0 ± 2.7) | 0.0 ± 0.0(0.0 ± 0.0) | – |
| 12 | C:Lys24 | 6.4 ± 2.9 | 2.2 ± 1.4 | – |
| 12 | C:His25 | 2.3 ± 1.2 | 1.2 ± 0.7 | – |
| 12 | C:Arg41 | 3.3 ± 1.9(0.0 ± 0.0) | 12.5 ± 4.7(0.2 ± 0.2) | – |
| 21 | A:Asn22 | 3.1 ± 1.5 | 11.5 ± 3.6 | 3.5 ± 1.7 |
| 21 | A:Asn44 | 0.4 ± 0.4 | 4.4 ± 3.3 | 5.8 ± 2.9 |
| 21 | A:Asn45 | 2.1 ± 1.4 | 1.9 ± 0.8 | 0.7 ± 0.5 |
| 21 | A:Asn46 | 4.2 ± 2.4 | 0.7 ± 0.7 | 7.9 ± 3.4 |
| 21 | A:Arg47 | 19.9 ± 5.5 | 3.3 ± 1.6 | 9.8 ± 3.7(0.1 ± 0.0) |
Values in parenthesis represent percentages of bidentate interactions.
Figure 2.
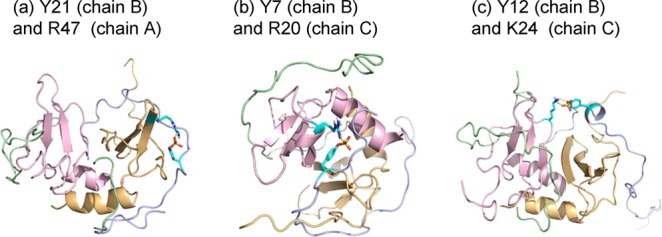
Sulfation at CXCR4 sites Y7 and Y12 results in hydrogen bonding across the dimer interface. CXCR4 N-terminal chains shown in blue and green; SDF-1 molecules shown in yellow and pink. Representative hydrogen bonds in simulation snapshots are shown for sulfation sites on one CXCR4 N-terminus as follows: (a) between Y21, and SDF-1 residue R47 on the same side of the dimer interface, (b) between Y7, and SDF-1 residue R20 on the opposite side of the dimer interface, and (c) between Y12, and SDF-1 residue K24 on the opposite side of the dimer interface. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
For Y7 and Y12, the unsulfated conformers show limited hydrogen bonding, including interactions between Y7 and residues of both molecules of the SDF-1 dimer. Hydrogen bonding is significantly enhanced by sulfation, with percentage occupancies up to 20% for the fully sulfated (2k05) simulation. Y7 and Y12 form hydrogen bonds with residues on both SDF-1 molecules; Figure 2(b,c) shows simulation snapshots highlighting hydrogen bonds that cross the dimer interface (between Y7 and SDF-1 residue R20, and Y12 and SDF-1 residue K24). For interactions with arginine, Table III also lists the percentage of frames with bidentate interactions; these interactions are commonly observed for phosphorylated residues and are also observed for sulfated residues, albeit with less frequency.24 Our results show that sulfation of the CXCR4 N-terminus increases hydrogen bonding with SDF-1, and plays a role in CXCR4:SDF-1 complex dimerization through extensive hydrogen bonding interactions across the dimer interface.
We used Kullback–Leibler (K–L) divergence to identify main-chain and side-chain dihedral population shifts that are a result of sulfation, by a comparison of torsions in the unsulfated, partially sulfated, and fully sulfated simulations. Our results show that sulfation impacts residues near the sulfation site, and notably, also at a remote location at the interface between SDF-1 chains. Figure 3(a) shows K–L divergence between the unsulfated (2k04) simulation and the partially sulfated (2k03) simulation (left), and between the unsulfated (2k04) simulation and the fully sulfated (2k05) simulation (right). Both partially sulfated and fully sulfated systems show some divergence due to sulfation in residues K24 and H25 of the first beta sheet of SDF-1, which is positioned at the dimer interface. The 2k05 simulation shows more extensive divergence at the dimer interface, including residues K27, I28, and L29. Velkamp et al.10 found that acidic pH promotes the monomeric state of SDF-1 by disrupting electrostatic interactions between the charged K27 residue and a neutral H25 residue at the dimer interface, presumably through protonation of H25, highlighting the role of these residues in SDF-1 dimer stabilization. Our K–L divergence analysis shows that sulfation results in structural and dynamical changes in residues that play a role in dimer stabilization, and that these changes are enhanced by sulfation at sites Y7 and Y12.
Figure 3.
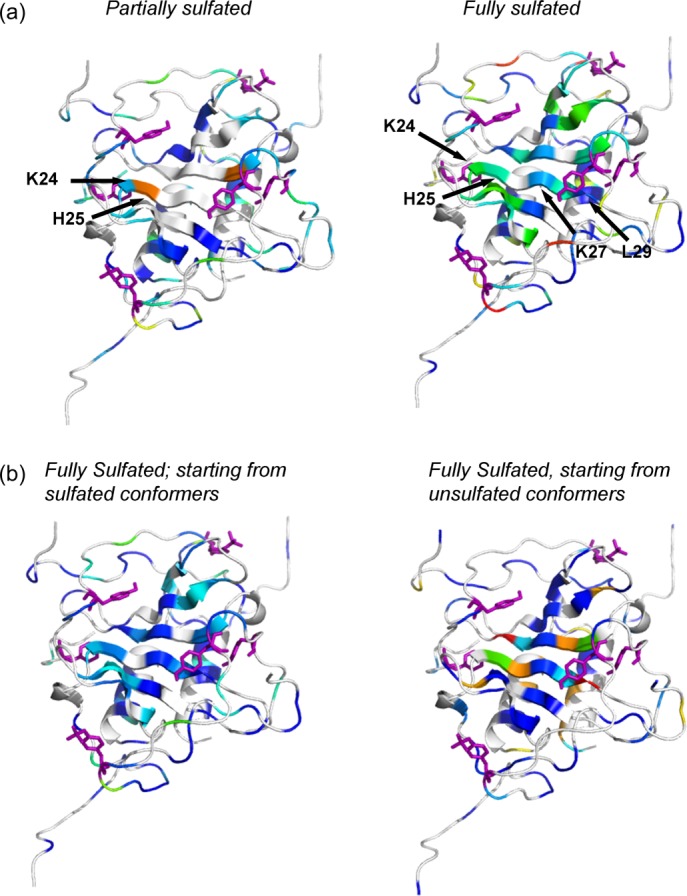
K–L Divergences between the unsulfated and sulfated CXCR4:SDF-1 complexes show significant dihedral population shifts in residues at the dimer interface for only the fully sulfated systems. (a) Divergences between unsulfated (2k04) and partially sulfated (2k03) conformers (left), and between unsulfated (2k04) and fully sulfated (2k05) conformers (right). (b) Divergences between unsulfated (2k04) and fully sulfated (2k05) conformers (left), and between unsulfated (2k04) and fully sulfated (2k04_S) conformers sulfated in PRIME (right). Divergent residues at the dimer interface are labeled in (a) for one half of the dimer and Y7, Y12, and Y21 are shown as purple sticks. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
As a further validation of the effectiveness of our utilized force fields and simulation protocols to reliably model the effects of sulfation, we added sulfate groups to Y7, Y12, and Y21 in conformers from the unsulfated NMR ensemble (2k04), subjected them to molecular dynamics simulation, and compared these simulations (2k04_S) with the originally sulfated (2k03 and 2k05) simulations. Hydrogen bonding analysis for the 2k04_S simulations shows interactions that are similar to those seen in the fully sulfated (2k05) and in the partially sulfated (2k03) simulations (Table III). We next compared K–L divergences between the unsulfated 2k04 simulation and the 2k05 simulation (left), and the unsulfated 2k04 simulation and the sulfated 2k04 (2k04_S) simulation (right) [Fig. 2(b)]. Both panels show divergence in the same structural regions, indicating that the structural and dynamical changes caused by sulfation can be reproduced by sulfation of the originally unsulfated conformers.
Monomeric and dimeric CXCR4:SDF-1 complexes are structurally distinct
Studies have shown that the monomeric and dimeric forms of SDF-1 have distinct functional roles with respect to chemotaxis.4 We propose that these functional differences are a result of structural and dynamical differences that may be present in the different oligomeric states of the CXCR4:SDF-1 complex. Hydrogen bond analysis for simulation of monomeric complexes 2k04 and 2k05 (Tables IV and V) shows interactions for the sulfated monomeric complexes that are similar to those seen in the dimeric complexes (2k03 and 2k05) on each separate side of the dimer interface. The effects of interactions across the dimer interface, which can occur in the dimeric complexes only, can be seen in a comparison of K–L divergence between the simulations of the unsulfated and the sulfated dimers, and between simulations of the unsulfated and sulfated monomers. The left panel of Figure 4 shows the divergence between simulations of the unsulfated (2k04) and sulfated (2k05) dimers (half of the dimeric complex is shown for purpose of comparison with the monomer), and the right panel shows divergence between the simulations of the unsulfated (2k04) and sulfated monomers (2k05). The simulation of the sulfated monomeric complex shows minimal divergence from its unsulfated state as seen by the mostly white color in the figure. The components of the dimeric complex diverge more significantly from their unsulfated state, as sulfation sites on each side of the dimer perturb both sides of the dimer.
Table IV.
Hydrogen Bonds to Y7, Y12, and Y21 Side Chains for Simulation of Monomeric 2k04, Unsulfated
| Tyr | SDF-1 residue | Frames (%) |
|---|---|---|
| 7 | Asn30 | 7.3 ± 3.3 |
| 12 | Ile28 | 10.0 ± 4.6 |
| 21 | Glu15 | 11.2 ± 5.1 |
Table V.
Hydrogen Bonds to Y7, Y12, and Y21 Side Chains, for Simulation of Monomeric 2k05, Fully Sulfated
| sTyr | SDF-1 residue | Frames (%) |
|---|---|---|
| 7 | Lys1 | 2.4 ± 1.2 |
| 7 | Asn30 | 3.9 ± 1.8 |
| 7 | Asn67 | 2.8 ± 2.0 |
| 7 | Lys68 | 2.0 ± 1.7 |
| 12 | Arg8 | 8.4 ± 3.9 (8.3 ± 3.9) |
| 12 | Lys27 | 15.5 ± 4.2 |
| 12 | Arg41 | 6.3 ± 2.4 (5.2 ± 2.3) |
| 12 | Gln48 | 6.5 ± 2.4 |
| 21 | Asn44 | 3.3 ± 1.3 |
| 21 | Asn46 | 4.1 ± 1.9 |
| 21 | Arg47 | 10.4 ± 3.8 (9.3 ± 3.8) |
Values in parenthesis represent percentages of bidentate interactions.
Figure 4.
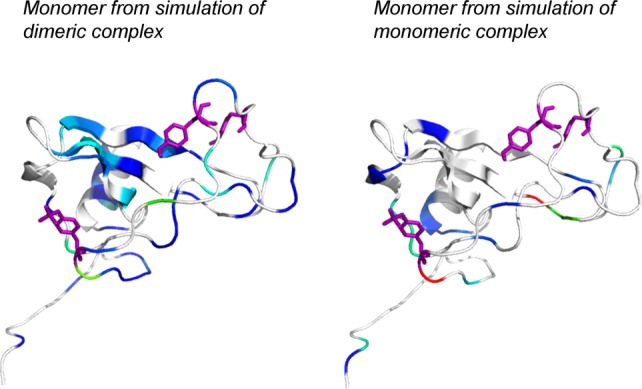
K–L Divergences between monomeric and dimeric CXCR4:SDF-1 complexes show minimal divergence for the monomeric complex. Divergence is shown between the unsulfated (2k04) and sulfated (2k05) conformers for simulation of the dimeric complex (left, half of the dimer is shown), and between the unsulfated (2k04) and sulfated (2k05) conformers for simulation of the monomeric complex (right). Y7, Y12, and Y21 are shown as purple sticks. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
We note that studies have shown that residues at the dimer interface form unique interactions with the CXCR4 N-terminus that are seen only in the monomeric, and not in the dimeric, complexes.21,22 The conformational changes which allow for these interactions occur on millisecond-microsecond time scales21,22 and thus would not be accounted for in the course of our simulation of CXCR4:SDF-1 monomeric complexes. However, our simulations of the monomeric complexes do reveal some hydrogen bonding interactions for Y7 and Y12 that are not observed in the simulation of the dimeric complexes (N67, K68, R8).
Stabilization of electrostatic binding energies
We hypothesized that the presence of sulfotyrosine interactions across the dimer interface would result in an enhancement in the electrostatic contribution to binding energies for the fully sulfated dimeric complexes, as compared to the unsulfated or partially sulfated systems. We report four binding energy terms (as described in Fig. 5) to address how sulfation contributes both to increased affinity of a single CXCR4 to a single SDF-1, and to stabilization of the dimeric CXCR4:SDF-1 complex. In Table VI, electrostatic energies are reported as averages with standard errors over six conformers, and for solvation energies over seven grid displacements for each conformer. Reported values do not reflect actual binding free energies due to factors (entropy, Lennard Jones energies etc.) that are not accounted for in the calculation; nonetheless, the relative energies show how partial or full sulfation contributes to stabilization of the SDF-1 dimer, the monomeric CXCR4:SDF-1 complex, and the dimeric CXCR4:SDF-1 complex.
Figure 5.
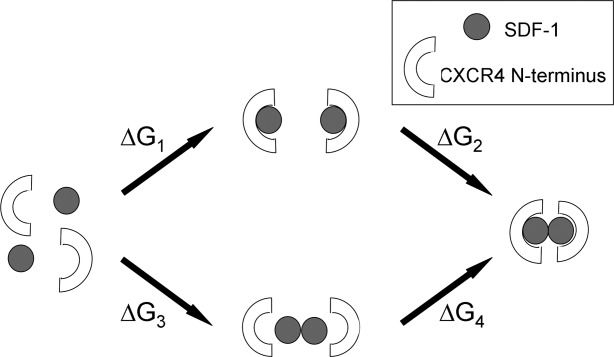
Schematic describing calculation of electrostatic binding energies for formation of monomeric CXCR4:SDF-1 complexes, SDF-1 dimer, and dimeric CXCR4:SDF-1 complex.
Table VI.
Solvation, Coulomb, and Total Electrostatic Binding Energies for Formation of Two CXCR4:SDF-1 Monomeric Complexes (ΔG1), Formation of the Dimeric Complex from Two Monomeric Complexes (ΔG2), Formation of the SDF-1 Dimer from Two Molecules of SDF-1 (ΔG3), and Formation of the Dimeric Complex from the SDF-1 Dimer and Two CXCR4 N-Terminal Chains (ΔG4)
| Binding energy (kcal/mol) | Y7, Y12, Y21 | Y21 | Unsulfated | |
|---|---|---|---|---|
| ΔG1 | Solvation | 2200 ± 20 | 1600 ± 20 | 1400 ± 18 |
| Coulomb | −2200 ± 20 | −1600 ± 20 | −1400 ± 18 | |
| Total | −13 ± 2.2 | −2.2 ± 2.1 | 2.5 ± 2.2 | |
| ΔG2 | Solvation | 280 ± 10 | 22 ± 9.6 | −23 ± 10 |
| Coulomb | −250 ± 11 | 24 ± 9.7 | 71 ± 26 | |
| Total | 38 ± 2.6 | 46 ± 2.6 | 48 ± 33 | |
| ΔG3 | Solvation | −870 ± 7.0 | −870 ± 7.0 | −870 ± 7.0 |
| Coulomb | 900 ± 6.5 | 900 ± 6.5 | 900 ± 6.5 | |
| Total | 28 ± 4.0 | 28 ± 4.0 | 28 ± 4.0 | |
| ΔG4 | Solvation | 3300 ± 23 | 2500 ± 22 | 2300 ± 21 |
| Coulomb | −3300 ± 24 | −2500 ± 24 | −2200 ± 29 | |
| Total | −3.5 ± 5.1 | 16 ± 4.4 | 22 ± 34 | |
Reported values are averages over six conformers, and for solvation energies over seven grid translations for each of six conformers.
Our results show that complete sulfation stabilizes binding of CXCR4 to SDF-1 by ∼16 kcal/mol for the monomeric CXCR4:SDF-1 complex (ΔG1), and ∼26 kcal/mol for binding of two CXCR4 N-terminal chains to a preformed SDF-1 dimer to make a dimeric CXCR4:SDF-1 complex (ΔG4). This enhancement in stabilization can be attributed to the interactions formed by Y7 and Y12 across the dimer interface that occur only in the dimeric complexes. Sulfation at Y21 alone stabilizes binding of CXCR4 to SDF-1 (ΔG1 and ΔG4) by 5–6 kcal for both the monomeric and dimeric complexes, which is consistent with interactions formed by Y21 on only one side of the dimer interface.
Binding energy terms for formation of the dimeric CXCR4:SDF-1 complexes from the monomeric complexes (ΔG2) show that with a greater degree of sulfation, solvation binding energies become less favorable, while coulombic binding energies become more favorable. This is to be expected due to the increased cost of desolvating the charged sulfate group during complex formation, and an increase in the number of favorable interactions that are formed within the complexes. Complete sulfation results in an enhancement of total electrostatic binding energies for dimer formation (ΔG2) of 10 kcal/mol, in contrast to sulfation at only Y21 which results in a much smaller (∼2 kcal/mol) enhancement. These results suggest sulfation at Y7 and Y12 might play a role in regulating dimer stability.
The increase in favorable interactions resulting from sulfation can be visualized in electrostatic potential maps (Figs. 6 and 7) which were generated using APBS25 and PyMOL,26 and are displayed at isovalue surfaces of ±1 kTe. Figure 6 shows the electrostatic complementarity (blue and red colors represent positive and negative potentials respectively) between a single SDF-1 molecule (chain A) and both CXCR4 N-terminal chains (chains B and D). This complementarity results from hydrogen bonding interactions between SDF-1 residues (displayed as sticks) and sulfotyrosine residues of both CXCR4 N-termini. Figure 7 shows how the negative electrostatic potential on the CXCR4 N-terminus increases with increasing sulfation, in particular from being sulfated at only Y21 to being fully sulfated.
Figure 6.
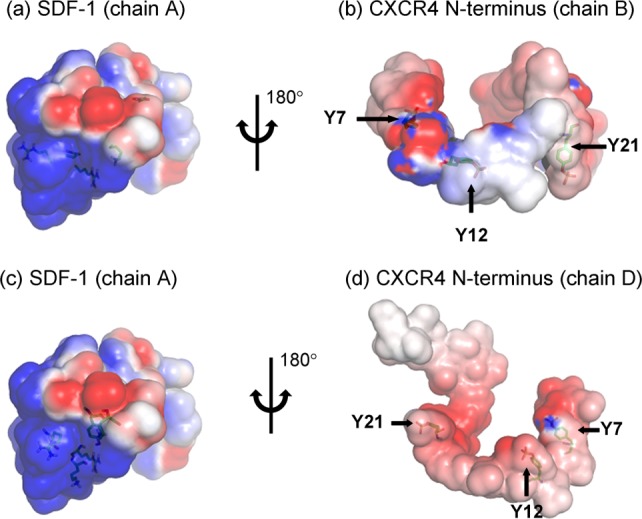
Electrostatic potential maps show complementarity between a single molecule of SDF-1 (chain A) and both CXCR4 N-termini (chains B and D). Sulfation sites on the CXCR4 N-termini, and SDF-1 residues which hydrogen bond to these sites, are shown as sticks. Figures are displayed at isovalue surfaces of ±1 kTe. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Figure 7.
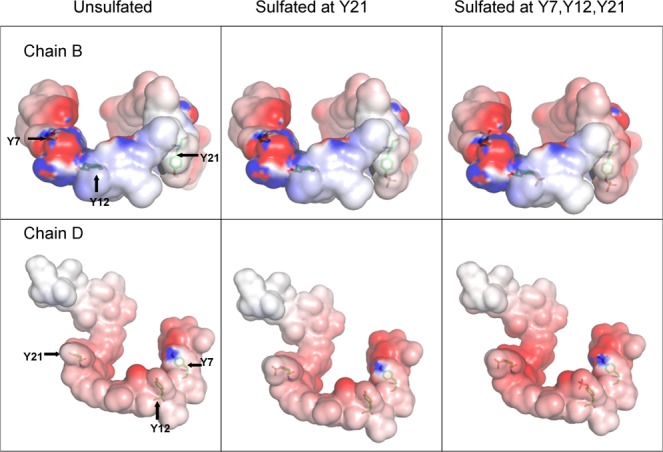
Electrostatic potential maps show increasing negative potential of the CXCR4 N-terminus with increasing sulfation. Sulfation sites on the CXCR4 N-termini are shown as sticks, and figures are displayed at isovalue surfaces of ±1 kTe. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Conclusion
Our results support a role for tyrosine sulfation in stabilization of the dimeric CXCR4:SDF-1 complex, and address the role of sulfation at sites Y7 and Y12, which are not conserved among chemokine receptors. Our findings can be tested experimentally by experimental determination of CXCR4:SDF-1 binding affinities in sulfated and unsulfated monomeric and dimeric complexes, or by functional assays comparing the biological effects of sulfated dimeric complexes with those in which dimerization is compromised by mutations in sulfation sites or key interacting residues. We would expect dimerization to be compromised by mutation of the sulfated tyrosine residues to phenylalanine, or by mutation of hydrogen bonding residues at the dimer interface (R20, K24, H25) to leucine or glutamic acid. Studies of this nature would provide important tests of the hypothesis suggested in this computational study.
Our findings are of particular importance as to date there have been limited studies characterizing the effects of tyrosine sulfation on protein–protein interactions. Though it is known that the monomeric and dimeric forms of SDF-1 have opposite effects on chemotaxis, it is not yet known whether dimerization of CXCR4 plays a role in modulating the oligomeric state of SDF-1 thereby regulating its activity. Studies of other GPCR complexes which exist in functionally different oligomeric states will be necessary to determine to what extent post-translational modifications, including but not limited to tyrosine sulfation, contribute to distinct physiological consequences through stabilization of a particular oligomeric state.
Methods
CXCR4:SDF-1 complexes
Starting structures for molecular dynamics simulation and ABPS calculations were taken from the first six conformers of PDB entries 2k03, 2k04, and 2k05.27 The entries contain dimers of the N-terminus of CXCR4 (chains B and D) in complex with SDF-1 (chains A and C), differing in the extent to which they are sulfated: 2k03 is sulfated at only Y21, 2k04 is unsulfated, and 2k05 is sulfated at Y7, Y12, and Y21. Monomers for molecular dynamics simulation were obtained from a single complex of CXCR4:SDF-1 (chains A and B only), for the first six conformers of entries 2k04 and 2k05.
As a control, the first six conformers of 2k04 were sulfated at Y7, Y12, and Y21, and then molecular dynamics simulations were performed (we refer to these as the 2k04_S simulations). This system was generated by replacing the residue name TYR with TYS in the structure files, and then adding the sulfotyrosine side chains using side chain prediction in PRIME.28,29
Molecular dynamics
The Desmond program30 was used for molecular dynamics using the OPLS-AA 2005 force field31,32 and SPC water model.32 Partial charges for sulfotyrosine were taken from the quantum mechanical calculations described in an earlier work.24 The system was set in an orthorhombic box with a 10 Å buffer region on each side and 0.15 M NaCl was added. Minimization was first performed with the solute positions restrained at 50 kcal/(mol Å), for 2000 steps or until forces were below 50 kcal/(mol Å2), with 10 steepest-descent steps, then with the solute positions unrestrained another 2000 steps or until forces were below 5 kcal/(mol Å2). Then solute heavy atoms were restrained at 50 kcal/mol/Å and NVT molecular dynamics at 10 K was performed for 12 ps using 1 fs time steps for the bonded and short-range nonbonded interactions and 3 fs time steps for the long-range interactions, with the Berendsen33 thermostat, a relaxation time of 0.1−1 ps, and a resampling period of 1 ps. Next, the timestep was increased to 2 fs for the bonded and short-range nonbonded interactions and 6 fs for long-ranged nonbonded interactions. NPT equilibration was performed at 10 K for 12 ps, with the Berendsen33 thermostat and barostat, using a thermostat relaxation rate of 0.1 ps−1, a barostat relaxation rate of 50 ps−1, and resampling period of 1 ps. The system was then simulated for another 12 ps at 300K using the same settings. Finally, heavy atom position restraints were removed and the system was simulated for an additional 24 ps at 300 K with a thermostat relaxation rate of 0.1 ps−1 and barostat relaxation rate of 2 ps−1.
After minimization and equilibration, production runs of 20 ns were performed on each system using the Martyna–Tobias–Klein integrator34 at 300 K (Nose–Hoover thermostat35) and 1 atm, with a barostat relaxation time of 2 ps and a thermostat relaxation time of 1 ps. Snapshots were output every 1 ps. All bonds involving hydrogen atoms were constrained, a 2 fs time step for the bonded and short-range nonbonded interactions was used, and long-range nonbonded interactions were updated every 6 fs using the RESPA multiple time step approach.36 Short-range columbic and van der Waals nonbonded interactions were cut off at 9.0 Å, and long-range electrostatics were computed using the smooth particle-mesh Ewald method. Pairlists were constructed using a distance of 10.5 Å and a migration interval of 12 ps.
Analysis of simulation trajectories
To validate our molecular dynamics trajectories against experimental data, we compared nuclear Overhauser enhancement (NOE) restraints used in the determination of the experimental structures (2k03, 2k04, 2k05) with proton–proton distances in our simulations, and report the extent to which restraints are violated. Violations in each range are reported for a single combined trajectory using every tenth frame from all six simulations. Violations are also reported as an average over simulations of the six conformers. Proton–proton distances were computed using AQUA70,37 and then ensemble averaged (using 1/r6 scaling) using a python script.
Hydrogen bond occupancies for side chains of Y7, Y12, and Y21, were calculated in VMD38 using a donor acceptor (D–A) cutoff distance of 3.0 Å and a D–H–A angle of less than 20°. Occupancies are reported for the converged part of each simulation, using block-averaging39 on the last seven nanoseconds for the simulations of the dimeric complexes, and the last 16 nanoseconds for the simulations of the monomeric simulations. We calculated the number of hydrogen bonds formed by residues Y7, Y12, and Y21 in the unsulfated (2k04) simulations, and then used plots of standard error vs. block size to determine an appropriate block length. Block lengths of 3500 and 4000 ps were used for the dimeric and monomeric simulations respectively, resulting in a total of 24 blocks for each system (for the dimers, two blocks of 3500 ps over six conformers and two halves of the dimer, and for the monomers, four blocks of 4000 ps over six conformers). We report percentage occupancies for any hydrogen bonding interaction appearing in more than 2% of frames in any one simulation (of all conformers of 2k03, 2k04, 2k05). We also report occupancies for bidentate hydrogen bonds with arginine; a bidentate hydrogen bond is an interaction in which two sulfate oxygen atoms on a single sulfated tyrosine residue interact with two nitrogen atoms on a single arginine residue.
Plots of root mean square deviation (RMSD) vs. time for all simulations were calculated in GROMACS40 for the protein backbone, using a least squares fit to the protein backbone. Protein tails that were shown by root mean square fluctuation values to be excessively floppy during the sulfated (2k05) simulation (residues 1–6 of SDF-1, and residues 1 and 33–38 of the CXCR4 N-termini) were excluded from the RMSD versus time calculation for all systems. The original conformers taken from the PDB are used as the reference structures for the 2k03, 2k04, 2k05 simulations, and the sulfated 2k04 conformers for the 2k04_S simulations.
Kullback−Leibler Divergence41 (K–L Divergence) was used to compare conformational ensembles generated by molecular dynamics simulations and to identify changes that result from sulfation. Analysis proceeded according to the previously published protocol. Briefly, we calculated the K–L Divergence between reference and target sets of simulations' dihedral bin populations, using 15-degree bins spaced uniformly from 0° to 360°, of residues' ϕ, θ, and ω torsion angles. This was followed by statistical filtering and corrections using the distribution of K–L Divergence under the null hypothesis of no difference between the ensembles, constructed by computing the K–L Divergence between all combinations of three out of the six runs from the reference dataset and their set complements. For simulations of the dimeric complex, divergence values are averaged for each residue over symmetric chains. Results are mapped onto the first conformer of PDB entry 2k03 using PyMOL's37 “b-factor putty” preset, using a color scheme in which white indicates a lack of statistically significant divergence and statistically significant divergence increases from blue to red. Structures appearing in the same figure are colored on the same scale.
APBS
The Adaptive Poisson–Boltzmann Solver (APBS) software program25 was used for the calculation of the electrostatic contribution to binding energies for the first six conformers of 2k05, and the same conformers sulfated at only Y21, or completely unsulfated. Partially sulfated and unsulfated conformers were generated from the 2k05 conformers by removing the relevant sulfate groups in the PDB structure file, and replacing the hydrogen atom of the hydroxyl group using the Maestro42 interface. All initial conformers were prepared in PRIME23,24 using the Protein Preparation Wizard. The prepared structural files were then used to create pqr files for APBS calculations using charges and radii from the OPLS-AA 2005 force field,26,27 and parameters for sulfated residues from quantum mechanical calculations described in an earlier work.24
Each complex or isolated subunit was mapped onto a three dimensional lattice of dimensions 129 × 161 × 129; course and fine grid lengths of 88.0, 99.9, 86.8, and 71.8, 78.8, 71.0, respectively; and the “mg_auto” option was used to focus the calculation and set grid size. The interior dielectric constant of the protein was set to 2 and the solvent dielectric constant was set to 78. Other parameters used included a smoothed molecular surface, a temperature of 298.15 K, and a solvent probe radius of zero. For each conformer, electrostatic binding energies were calculated for the (1) formation of two monomeric CXCR4:SDF-1 complexes from two CXCR4 N-terminal chains and two SDF-1 molecules, ΔG1, (2) formation of the dimeric CXCR4:SDF-1 complex from its component monomers, ΔG2, (3) formation of the SDF-1 dimer from two molecules of SDF-1, ΔG3, and (4) formation of the dimeric CXCR4:SDF-1 complex from the SDF-1 dimer and two CXCR4 N-terminal chains, ΔG4 (Fig. 5). Electrostatic binding energies are calculated as ΔΔGele = ΔΔGsolv + ΔΔGcol where ΔΔG represents a difference in free energy between a system and its component parts, for either the solvation, coulomb, or total electrostatic energies. Each calculation of solvation energy was performed seven times, once centering the calculation on the center of the complex or individual molecule, and then repeating the calculation using a center translated on the x-, y-, or z-axis of the grid by one third of the grid spacing in either the positive or negative direction. Solvation energy values are averaged over seven calculations of six conformers (42 calculations) and reported with standard errors. Values do not reflect actual binding free energies due to factors (entropy, Lennard Jones energies etc.) that are not accounted for in the calculation.
Acknowledgments
The authors thank Matt Jacobson for helpful discussions.
References
- 1.Wu BL, Chien EYT, Mol CD, Fenalti G, Liu W, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamatake M, Aoki T, Futahashi Y, Urano E, Yamamoto N, Komano J. Ligand independent higher order multimerization of CXCR4, a G protein-coupled chemokine receptor involved in targeted metastasis. Cancer Sci. 2009;100:95–102. doi: 10.1111/j.1349-7006.2008.00997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roussos ET, Condeelis JS, Patsialou A. Chemotaxis in cancer. Nat Rev Cancer. 2011;11:573–587. doi: 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drury LJ, Ziarek JJ, Gravel S, Veldkamp CT, Takekoshi T, Hwang ST, Heveker N, Volkman BF, Dwinell MB. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci USA. 2011;108:17655–17660. doi: 10.1073/pnas.1101133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mellado M, Vila-Coro AJ, Martinez C, Rodriguez-Frade JM. Receptor dimerization: a key step in chemokine signaling. Cell Mol Biol. 2001;47:575–582. [PubMed] [Google Scholar]
- 6.Wang JH, He LS, Combs CA, Roderiquez G, Norcross MA. Dimerization of CXCR4 in living malignant cells: control of cell migration by a synthetic peptide that reduces homologous CXCR4 interactions. Mol Cancer Ther. 2006;5:2474–2483. doi: 10.1158/1535-7163.MCT-05-0261. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez D, Gutierrez-de-Teran H. Characterization of the homodimerization interface and functional hotspots of the CXCR4 chemokine receptor. Proteins. 2012;80:1919–1928. doi: 10.1002/prot.24099. [DOI] [PubMed] [Google Scholar]
- 8.Babcock GJ, Farzan M, Sodroski J. Ligand-independent dimerization of CXCR4, a principal HIV-1 coreceptor. J Biol Chem. 2003;278:3378–3385. doi: 10.1074/jbc.M210140200. [DOI] [PubMed] [Google Scholar]
- 9.Toth PT, Ren D, Miller RJ. Regulation of CXCR4 receptor dimerization by the chemokine SDF-1α and the HIV-1 coat protein gp120: a fluorescence resonance energy transfer (FRET) study. J Pharmacol Exp Ther. 2004;310:8–17. doi: 10.1124/jpet.103.064956. [DOI] [PubMed] [Google Scholar]
- 10.Veldkamp CT, Peterson FC, Pelzek AJ, Volkman BF. The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci. 2005;14:1071–1081. doi: 10.1110/ps.041219505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC, Basnet H, Sakmar TP, Volkman BF. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008;1:ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farzan M, Babcock GJ, Vasilieva N, Wright PL, Kiprilov E, Mirzabekov T, Choe H. The role of post-translational modifications of the CXCR4 amino terminus in stromal-derived factor 1 alpha association and HIV-1 entry. J Biol Chem. 2002;277:29484–29489. doi: 10.1074/jbc.M203361200. [DOI] [PubMed] [Google Scholar]
- 13.Veldkamp CT, Seibert C, Peterson FC, Sakmar TP, Volkman BF. Recognition of a CXCR4 sulfotyrosine by the chemokine stromal cell-derived factor-1 alpha (SDF-1 alpha/CXCL12) J Mol Biol. 2006;359:1400–1409. doi: 10.1016/j.jmb.2006.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seibert C, Veldkamp CT, Peterson FC, Chait BT, Volkman BF, Sakmar TP. Sequential tyrosine sulfation of CXCR4 by tyrosylprotein sulfotransferases. Biochemistry. 2008;47:1251–1262. doi: 10.1021/bi800965m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choe H, Farzan M. Tyrosine sulfation of HIV-1 coreceptors and other chemokine receptors. Method Enzymol. 2009;461:147–170. doi: 10.1016/S0076-6879(09)05407-X. [DOI] [PubMed] [Google Scholar]
- 16.Sheinerman FB, Norel R, Honig B. Electrostatic aspects of protein–protein interactions. Curr Opin Struct Biol. 2000;10:153–159. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 17.Xu D, Lin SL, Nussinov R. Protein binding versus protein folding: The role of hydrophilic bridges in protein associations. J Mol Biol. 1997;265:68–84. doi: 10.1006/jmbi.1996.0712. [DOI] [PubMed] [Google Scholar]
- 18.Nohaile MJ, Hendsch ZS, Tidor B, Sauer RT. Altering dimerization specificity by changes in surface electrostatics. Proc Natl Acad Sci USA. 2001;98:3109–3114. doi: 10.1073/pnas.051624498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong CF, Hünenberger PH, Akamine P, Narayana N, Diller T, McCammon JA, Taylor S, Xuong NH. Computational Analysis of PKA−Balanol Interactions. J Med Chem. 2001;44:1530–1539. doi: 10.1021/jm000443d. [DOI] [PubMed] [Google Scholar]
- 20.Wang T, Tomic S, Gabdoulline RR, Wade RC. How Optimal Are the Binding Energetics of Barnase and Barstar? Biophys J. 2004;87:1618–1630. doi: 10.1529/biophysj.104.040964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baryshnikova OK, Sykes BD. Backbone dynamics of SDF-1alpha determined by NMR: interpretation in the presence of monomer-dimer equilibrium. Protein Sci. 2006;15:2568–2578. doi: 10.1110/ps.062255806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veldkamp CT, Ziarek JJ, Su J, Basnet H, Lennertz R, Weiner JJ, Peterson FC, Baker JE, Volkman BF. Monomeric structure of the cardioprotective chemokine SDF-1/CXCL12. Protein Sci. 2009;18:1359–1369. doi: 10.1002/pro.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altenburg JD, Jin Q, Alkhatib B, Alkhatib G. The Potent Anti-HIV Activity of CXCL12γ Correlates with Efficient CXCR4 Binding and Internalization. J Virol. 2010;84:2563–2572. doi: 10.1128/JVI.00342-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rapp C, Klerman H, Levine E, McClendon CL. 2013. Hydrogen bond strengths in phosphorylated and sulfated acid residues: a study of the pSer, pTyr and sTyr residues. 8:e57804 10.1371/journal.pone.0057804. [DOI] [PMC free article] [PubMed]
- 25.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.
- 27.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Databank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobson MP, Kaminski GA, Friesner RA, Rapp CS. Force field validation using protein side chain prediction. J Phys Chem B. 2002;106:11673–11680. [Google Scholar]
- 29.Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 30.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Brent A, Gregersen J, Klepeis L, Kolossváry I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. 2006. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In: Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa Florida.
- 31.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 32.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 33.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 34.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 35.Hoover WG. Canonical dynamics: equilibrium phase-space distributions. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 36.Tuckerman M, Berne BJ, Martyna GJ. Reversible multiple time scale molecular dynamics. J Chem Phys. 1992;97:1990–2001. [Google Scholar]
- 37.Laskowski RA, Rullmann JA, MacArthur MW, Kaptein R, Thornton JM. PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 38.Humphrey W, Dalke A, Schulten K. VMD - visual molecular dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 39.Grossfield A, Zuckerman DM. Quantifying uncertainty and sampling quality in biomolecular simulations. Annu Rep Comput Chem. 2009;5:23–48. doi: 10.1016/S1574-1400(09)00502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 41.McClendon CL, Hua L, Barreiro G, Jacobson MP. Comparing conformational ensembles using the Kullback-Leibler divergence expansion. J Chem Theory Comput. 2012;8:2115–2126. doi: 10.1021/ct300008d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maestro, version 9.2, Schrödinger, LLC, New York, NY, 2011.