

Abstract
Platelet attachment to von Willebrand factor (vWF) requires the interaction between the platelet GP1bα and exposed vWF-A1 domains. Structural insights into the mechanism of the A1-GP1bα interaction have been limited to an N-terminally truncated A1 domain that lacks residues Q1238 − E1260 that make up the linker between the D3 and A1 domains of vWF. We have demonstrated that removal of these residues destabilizes quaternary interactions in the A1A2A3 tridomain and contributes to platelet activation under high shear (Auton et al., J Biol Chem 2012;287:14579–14585). In this study, we demonstrate that removal of these residues from the single A1 domain enhances platelet pause times on immobilized A1 under rheological shear. A rigorous comparison between the truncated A1-1261 and full length A1-1238 domains demonstrates a kinetic stabilization of the A1 domain induced by these N-terminal residues as evident in the enthalpy of the unfolding transition. This stabilization occurs through site and sequence-specific binding of the N-terminal peptide to A1. Binding of free N-terminal peptide to A1-1261 has an affinity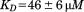 and this binding although free to dissociate is sufficient to suppress the platelet pause times to levels comparable to A1-1238 under shear stress. Our results support a dual-structure/function role for this linker region involving a conformational equilibria that maintains quaternary A domain associations in the inactive state of vWF at low shear and an intra-A1-domain conformation that regulates the strength of platelet GP1bα-vWF A1 domain associations in the active state of vWF at high shear.
and this binding although free to dissociate is sufficient to suppress the platelet pause times to levels comparable to A1-1238 under shear stress. Our results support a dual-structure/function role for this linker region involving a conformational equilibria that maintains quaternary A domain associations in the inactive state of vWF at low shear and an intra-A1-domain conformation that regulates the strength of platelet GP1bα-vWF A1 domain associations in the active state of vWF at high shear.
Keywords: von Willebrand factor, parallel plate flow chamber, catch bonds, pause time, thermodynamics, irreversible denaturation, peptide binding circular dichroism, fluorescence, differential scanning calorimetry
Introduction
The multimeric plasma glycoprotein, von Willebrand factor (vWF), is secreted from vascular endothelial cells into the blood and subendothelium as long filaments of covalently coupled monomeric units, each containing a conformationally regulated hook for platelet attachment. These monomeric units each contain a series of domains arranged in the order, D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-Cysteine Knot.1,2 Within this sequence, the A1 domain functions as the hook for capturing platelets through interaction with platelet glycoprotein 1b and contributes to the arrest of bleeding under the vascular shear stress of blood flow.3
Early studies of the domain structure of vWF isolated a tryptic fragment between residues V1212 and K1491 that retained functional binding to platelet GP1b.4,5 In a variety of studies designed to assess platelet adhesion, heparin and collagen binding, this sequence was engineered recombinantly by several investigators with various lengths of the N and C termini outside of the major disulfide loop that comprises the A1 domain.6–9 Eventually, optimal lengths of these N and C termini were obtained in a construct spanning residues Q1238–P1471 that eliminated disulfide linked aggregates and improved expression, purification, and solubility of the recombinant A1 domain as a monomeric species in solution.10 For the better part of the last two decades, this sequence has been demonstrated to retain the regulatory mechanism for platelet adhesion in response to the shear stress of blood flow. It has been demonstrated to form high strength bonds with platelet GP1b,11,12 shear-dependent platelet rolling on A1 domain coated surfaces,13,14 and force-dependent single molecule catch bonds with platelet GP1bα.14 Furthermore, it has provided internally consistent relationships between the interdependence of conformational thermodynamics with shear- and force-dependent binding of platelet GP1bα based on clinically identified mutations that cause opposite functional phenotypes in von Willebrand disease (vWD).15,16
Early efforts to obtain a crystal structure of this domain were unsuccessful until chymotrypsin was used to cleave off residues Q1238–Y1258 and diffractable crystals were obtained.17,18 Although this truncated domain spanning the structurally resolved residues D1261–P1471 can bind to GP1bα, platelet adhesion to it at high shear rates was not as efficient as the domain containing the N-terminal flanking region which showed similar adhesive activity as native vWF.19,20 Subsequent crystal structures of the truncated A1 domain and its cocrystal complex with GP1bα with and without Type 2 vWD mutations show that the structures are comparatively similar with the backbone RSMD less than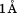 over much of the sequence within the disulphide loop (Fig. 1).18,21–23 Consequently, structural insights into the mechanisms of Type 2 vWD have been inadequate.
over much of the sequence within the disulphide loop (Fig. 1).18,21–23 Consequently, structural insights into the mechanisms of Type 2 vWD have been inadequate.
Figure 1.
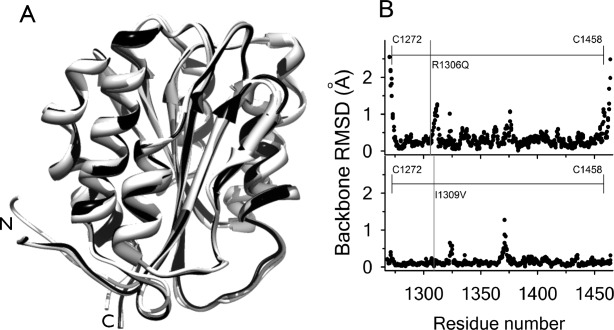
(A) Structural alignment of WT A1 (1auq, black), Type 2B I1309V A1 (1ijk, dark grey), and Type 2B R1306Q A1 extracted from the cocrystal complex with GP1bα (1m10, light grey). (B) Root mean squared deviation of all backbone atoms. Top: R1306Q—WT. Bottom: I1309V—WT. Diffraction resolutions are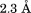 for WT,
for WT,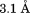 for R1306Q, and
for R1306Q, and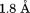 for I1309V. Horizontal line indicates residues within the disulfide bond.
for I1309V. Horizontal line indicates residues within the disulfide bond.
It has been known since the late 1980s that 15 residue peptides spanning the region between L1232–D1261 inhibit greater than 50% of the binding of vWF to platelet GP1bα. Furthermore, these peptides can completely inhibit the interaction between vWF and two monoclonal antibodies (NMC-4 and RG-46) known to block ristocetin and botrocetin-induced binding of vWF to GP1bα.24,25 Cocrystal structures of A1 in complex with NMC-4 moAb show that NMC-4 binds A1 at the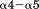 helix-loop-helix.26 Given the location of the structurally resolved N-terminal residues beginning at D1261, these data suggest that inhibition of the A1:NMC-4 interaction by the sequence Q1238–E1260 would involve a significant wrapping of the N-terminal sequence around the A1 domain. This type of interaction could result in a shielding effect that inhibits binding between A1 and GP1bα as has been demonstrated using vWF A domain fragments containing the N-terminal D'D3 domains.27 What is clear is that this N-terminal sequence, Q1238–E1260, is highly dynamic and its inhibition of crystal growth has precluded an accurate structure-based mechanism of GP1bα binding because of the inability to resolve its conformation in complex with the A1 domain.
helix-loop-helix.26 Given the location of the structurally resolved N-terminal residues beginning at D1261, these data suggest that inhibition of the A1:NMC-4 interaction by the sequence Q1238–E1260 would involve a significant wrapping of the N-terminal sequence around the A1 domain. This type of interaction could result in a shielding effect that inhibits binding between A1 and GP1bα as has been demonstrated using vWF A domain fragments containing the N-terminal D'D3 domains.27 What is clear is that this N-terminal sequence, Q1238–E1260, is highly dynamic and its inhibition of crystal growth has precluded an accurate structure-based mechanism of GP1bα binding because of the inability to resolve its conformation in complex with the A1 domain.
Our recent studies have shown that truncation of this N-terminal sequence destabilizes domain associations within the A1A2A3 tridomain resulting in a moderate increase in avidity for platelet GP1bα and fibrinogen-dependent platelet activation at high shear.28 Similarly, moAb 1C1E7 directed at the N-terminal sequence induces a vWF functional phenotype similar to Type 2B vWD with enhanced ristocetin-induced platelet agglutination (RIPA), shear-induced platelet agglutination (SIPA), and evidence of platelet activation via increased intracellular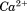 .29,30 Here, we show using flow chamber studies that this N-terminal flanking sequence suppresses the catch bond by decreasing the pause time of platelet interactions with surface immobilized A1 domain. Furthermore, we provide a thermodynamic comparison of the stability of the A1 domain with and without its N-terminal flanking region and demonstrate that A1 binds specifically to the N-terminal peptide in solution. The binding of the free N-terminal peptide by A1 lacking this sequence also suppresses the catch bond.
.29,30 Here, we show using flow chamber studies that this N-terminal flanking sequence suppresses the catch bond by decreasing the pause time of platelet interactions with surface immobilized A1 domain. Furthermore, we provide a thermodynamic comparison of the stability of the A1 domain with and without its N-terminal flanking region and demonstrate that A1 binds specifically to the N-terminal peptide in solution. The binding of the free N-terminal peptide by A1 lacking this sequence also suppresses the catch bond.
Results
Platelet adhesion to immobilized A1 under shear flow
To understand the effect of the N-terminal peptide on the function of the A1-domain of vWF, the protein was expressed with (A1-1238) and without the N-terminal peptide (A1-1261). We used a flow chamber to measure the dynamics of platelet interactions with A1-1238 and A1-1261. Each domain variant was immobilized on glass slides with a modified chelating chemistry that enabled surface capture via the N-terminally fused histidine tag. This method ensured reproducible capture of the proteins eliminating possible structural issues associated with nonspecific immobilization on plastic or glass. The slides were completely inert to platelets.
chelating chemistry that enabled surface capture via the N-terminally fused histidine tag. This method ensured reproducible capture of the proteins eliminating possible structural issues associated with nonspecific immobilization on plastic or glass. The slides were completely inert to platelets. of citrated whole blood followed by Tris-buffered saline was perfused at low shear to enable platelets to attach to the surface immobilized domains. After red cells were perfused away with buffer, the translocation of the remaining attached platelets over the surfaces was recorded at each shear rate and the velocities and pause times calculated from the coordinate data of each platelet on the surface as described in the methods. Figure 2 shows the pause time data as a function of shear rate along with the number of platelet tracks analyzed from each movie. A1-1261 has longer pause times than A1-1238 indicating that the presence of the covalently linked N-terminal sequence decreases the time for which platelets remain attached. In addition, the translocation velocities were increased for A1-1238 relative to A1-1261. To determine if this N-terminal sequence could inhibit the catch bond when free in solution, we incubated surface immobilized A1-1261 with excess free peptide and added the free peptide to
of citrated whole blood followed by Tris-buffered saline was perfused at low shear to enable platelets to attach to the surface immobilized domains. After red cells were perfused away with buffer, the translocation of the remaining attached platelets over the surfaces was recorded at each shear rate and the velocities and pause times calculated from the coordinate data of each platelet on the surface as described in the methods. Figure 2 shows the pause time data as a function of shear rate along with the number of platelet tracks analyzed from each movie. A1-1261 has longer pause times than A1-1238 indicating that the presence of the covalently linked N-terminal sequence decreases the time for which platelets remain attached. In addition, the translocation velocities were increased for A1-1238 relative to A1-1261. To determine if this N-terminal sequence could inhibit the catch bond when free in solution, we incubated surface immobilized A1-1261 with excess free peptide and added the free peptide to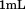 of whole blood and to our perfusion buffer and performed the assay. The result was a significant decrease in pause times to the levels of that obtained for A1-1238 and an increase in translocation velocities. Addition of free N-terminal peptide to A1-1238 or a peptide with the same amino acid composition, but a scrambled sequence, to either A1-1238 or A1-1261 did not significantly change the pause times or translocation velocities observed in the absence of peptide.
of whole blood and to our perfusion buffer and performed the assay. The result was a significant decrease in pause times to the levels of that obtained for A1-1238 and an increase in translocation velocities. Addition of free N-terminal peptide to A1-1238 or a peptide with the same amino acid composition, but a scrambled sequence, to either A1-1238 or A1-1261 did not significantly change the pause times or translocation velocities observed in the absence of peptide.
Figure 2.
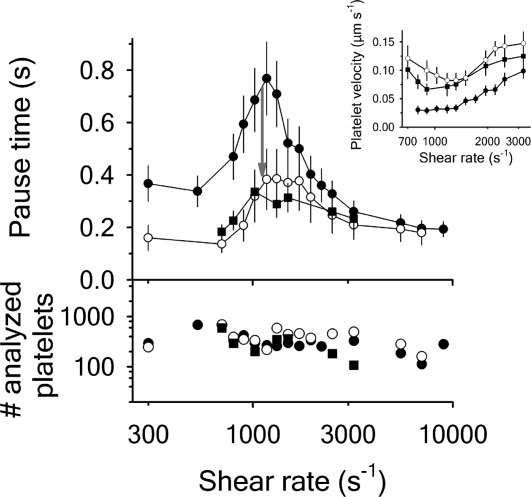
Platelet pause times and translocation velocities (Inset) for A1-1261 (black circles), A1-1238 (open circles) and A1-1261 in presence of 66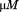 N-terminal peptide (black squares). The number of platelets analyzed at each shear rate is in the lower panel. Data are representative of three independent experiments from different donors. Both proteins were immobilized at a concentration of
N-terminal peptide (black squares). The number of platelets analyzed at each shear rate is in the lower panel. Data are representative of three independent experiments from different donors. Both proteins were immobilized at a concentration of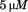 for
for and
and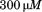 of N-terminal peptide were added to immobilized A1-1261 followed by
of N-terminal peptide were added to immobilized A1-1261 followed by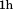 incubation. N-terminal peptide was added to
incubation. N-terminal peptide was added to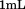 whole blood and buffer to a final concentration of
whole blood and buffer to a final concentration of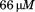 prior to flow.
prior to flow.
Structural characterization
Circular dichroism (CD) spectra obtained for A1-1261 and for A1-1238 are shown in Figure 3(A). Both proteins show a spectrum dominated by relatively high-α-helical content as indicated by the two minima at and
and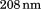 . Comparatively, the ellipticity of A1-1261 is slightly reduced relative to A1-1238, but a calculation of the secondary structure contributions for the two proteins (Supporting Information, Table SI) shows similar α-helical and parallel and antiparallel β-sheet structure.31 By contrast, the spectrum of the N-terminal peptide is dominated by the contribution of random coil-structure and spectrally equivalent to a peptide containing a scrambled sequence but identical amino acid composition.
. Comparatively, the ellipticity of A1-1261 is slightly reduced relative to A1-1238, but a calculation of the secondary structure contributions for the two proteins (Supporting Information, Table SI) shows similar α-helical and parallel and antiparallel β-sheet structure.31 By contrast, the spectrum of the N-terminal peptide is dominated by the contribution of random coil-structure and spectrally equivalent to a peptide containing a scrambled sequence but identical amino acid composition.
Figure 3.
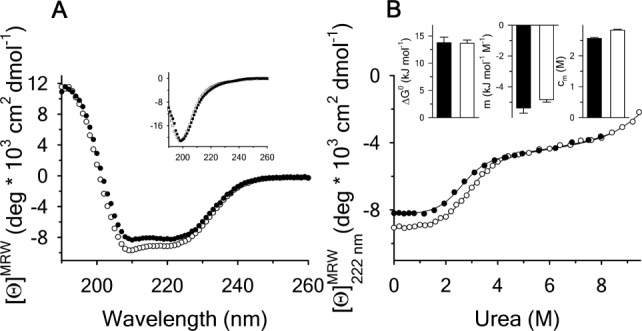
(A) Far-UV CD spectra at of A1-1261 (black circles), A1-1238 (open circles) and Inset: the N-terminal peptide (black squares) and of the scrambled peptide (open squares). (B) Urea denaturation at
of A1-1261 (black circles), A1-1238 (open circles) and Inset: the N-terminal peptide (black squares) and of the scrambled peptide (open squares). (B) Urea denaturation at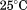 of A1-1261 (black circles) and of A1-1238 (open circles) via CD at
of A1-1261 (black circles) and of A1-1238 (open circles) via CD at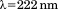 . Insets are the
. Insets are the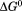 , m-value, and cm for the native to intermediate transition. A1-1261 (black bars) and A1-1238 (white bars).
, m-value, and cm for the native to intermediate transition. A1-1261 (black bars) and A1-1238 (white bars).
Urea-induced unfolding
A1-1261 and A1-1238 were chemically unfolded with urea to compare the obtained thermodynamic parameters. The unfolding was monitored at by CD at
by CD at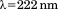 , Figure 3(B). For both proteins, unfolding through an intermediate state was observed, as has been previously reported.16 The thermodynamic parameters are illustrated in the insets of Figure 3(B) and listed in Supporting Information, Table SII. Relative to A1-1261, A1-1238 has a marginal loss in unfolding cooperativity (
, Figure 3(B). For both proteins, unfolding through an intermediate state was observed, as has been previously reported.16 The thermodynamic parameters are illustrated in the insets of Figure 3(B) and listed in Supporting Information, Table SII. Relative to A1-1261, A1-1238 has a marginal loss in unfolding cooperativity (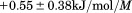 ) of the transition and the midpoint, cm, of A1-1238 is increased by
) of the transition and the midpoint, cm, of A1-1238 is increased by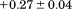 M. These two contributions to the
M. These two contributions to the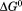 cancel each other so that the N-terminal peptide is neither stabilizing nor destabilizing the conformational transition.
cancel each other so that the N-terminal peptide is neither stabilizing nor destabilizing the conformational transition.
Temperature-induced unfolding
A1-1261 and A1-1238 were thermally unfolded using intrinsic protein tyrosine and tryptophan fluorescence with an excitation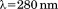 and emission
and emission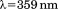 and CD monitored at
and CD monitored at . The observed thermal scans shown in Figure 4 demonstrate a strong dependency of the primary unfolding transition on the rate at which the temperature is changed indicating that the unfolding is kinetically controlled. As the scan rate increases, the midpoint of the transition (apparent TM) also increases. Closer inspection of the thermal transition in Figure 4 shows that while both spectroscopic methods capture the primary unfolding event between
. The observed thermal scans shown in Figure 4 demonstrate a strong dependency of the primary unfolding transition on the rate at which the temperature is changed indicating that the unfolding is kinetically controlled. As the scan rate increases, the midpoint of the transition (apparent TM) also increases. Closer inspection of the thermal transition in Figure 4 shows that while both spectroscopic methods capture the primary unfolding event between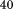 and
and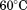 , fluorescence captures a second transition at high temperature in A1-1238 [Fig. 4(B)] which is also scan rate-dependent. This high temperature transition is absent in A1-1261 suggesting that the removal of the N-terminal sequence further exposes fluorophors in the protein [Fig. 4(A)]. Thermal scans of the peptide alone did not reveal any evidence of a conformational transition by either CD or fluorescence indicating that this high-temperature transition results from the exposure of the tyrosine in the N-terminal peptide when covalently linked to the A1 domain.
, fluorescence captures a second transition at high temperature in A1-1238 [Fig. 4(B)] which is also scan rate-dependent. This high temperature transition is absent in A1-1261 suggesting that the removal of the N-terminal sequence further exposes fluorophors in the protein [Fig. 4(A)]. Thermal scans of the peptide alone did not reveal any evidence of a conformational transition by either CD or fluorescence indicating that this high-temperature transition results from the exposure of the tyrosine in the N-terminal peptide when covalently linked to the A1 domain.
Figure 4.
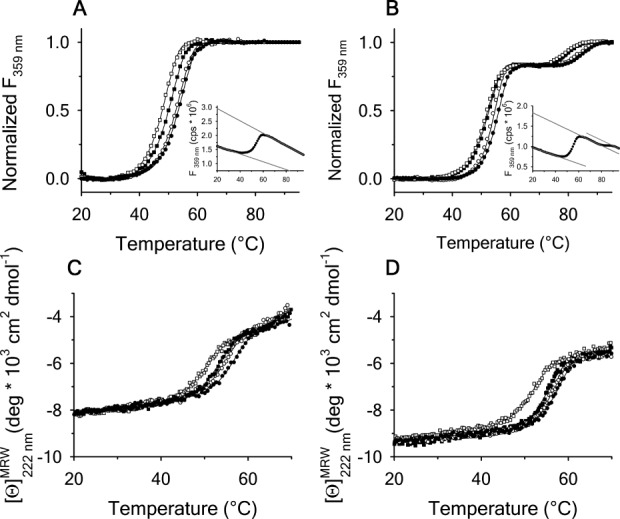
Scan rate dependency of normalized thermal unfolding transitions by fluorescence (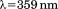 ) of A1-1261 (A) and A1-1238 (B) with excitation
) of A1-1261 (A) and A1-1238 (B) with excitation . Scan rates were
. Scan rates were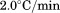 (black circles),
(black circles),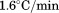 (open circles),
(open circles),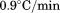 (black squares) and
(black squares) and for A1-1261 and
for A1-1261 and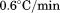 for A1-1238 (both with open squares). Insets show representative thermal scans and corresponding baselines before data normalization. Scan rate dependency of thermal unfolding transitions by CD at
for A1-1238 (both with open squares). Insets show representative thermal scans and corresponding baselines before data normalization. Scan rate dependency of thermal unfolding transitions by CD at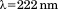 for A1-1261 (C) and A1-1238 (D). Scan rates were
for A1-1261 (C) and A1-1238 (D). Scan rates were (black circles),
(black circles),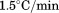 (open circles),
(open circles),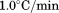 (black squares), and
(black squares), and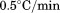 (open squares).
(open squares).
Thermal transitions were analyzed in terms of the observed midpoint of the transition, apparent TM, and also by a two-state irreversible model or a three-state irreversible model as described in Supporting Information, and the calculated parameters are summarized in Figure 5. Figure 5(A) demonstrates the increasing apparent TM as a function of scan rate for all methods used. If the protein unfolding was at equilibrium and reversible, this apparent TM would remain constant at all scan rates. Extrapolation of the TM to a scan rate of provides an indication of where reversible unfolding would occur under equilibrium conditions;
provides an indication of where reversible unfolding would occur under equilibrium conditions;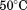 for A1-1238 and
for A1-1238 and for A1-1261 from both CD and fluorescence data. For the second transition observed by fluorescence that occurs with A1-1238, the TM extrapolates to
for A1-1261 from both CD and fluorescence data. For the second transition observed by fluorescence that occurs with A1-1238, the TM extrapolates to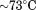 in the limit of zero scan rate. Analysis by the two-state and three-state irreversible models yields an
in the limit of zero scan rate. Analysis by the two-state and three-state irreversible models yields an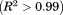 for both fluorescence and CD and a transition temperature,
for both fluorescence and CD and a transition temperature, , that is independent of the temperature scan rate. In addition, the enthalpies of the transitions are independent of scan rate within experimental error [Fig. 5(B)]. Figure 5(C,D) shows the resulting average
, that is independent of the temperature scan rate. In addition, the enthalpies of the transitions are independent of scan rate within experimental error [Fig. 5(B)]. Figure 5(C,D) shows the resulting average and
and ±the standard deviation obtained from the fitting of the primary thermal transitions in Figure 4 for CD and fluorescence and a total average for both methods. Comparing the transition temperatures for the primary unfolding transition shows that both A1-1261 and A1-1238 have an identical
±the standard deviation obtained from the fitting of the primary thermal transitions in Figure 4 for CD and fluorescence and a total average for both methods. Comparing the transition temperatures for the primary unfolding transition shows that both A1-1261 and A1-1238 have an identical within experimental error. A1-1261
within experimental error. A1-1261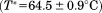 is not significantly different than A1-1238
is not significantly different than A1-1238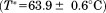 . However, the unfolding enthalpy was statistically larger for A1-1238
. However, the unfolding enthalpy was statistically larger for A1-1238 than for A1-1261
than for A1-1261 . The average
. The average and
and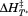 observed in the second fluorescence transition of A1-1238 is
observed in the second fluorescence transition of A1-1238 is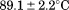 and
and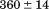 kJ/mol.
kJ/mol.
Figure 5.

The parameters obtained from fitting the thermal transitions in Figure 4 are reported for each applied method. (A) Scan rate dependence of the —values (filled symbols and black average and regression lines) and apparent TM—values (open symbols and gray average and regression lines). (B) Scan rate dependence of the enthalpy,
—values (filled symbols and black average and regression lines) and apparent TM—values (open symbols and gray average and regression lines). (B) Scan rate dependence of the enthalpy,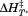 . Lines are solid for A1-1261 and dashed for A1-1238. Symbols are: fluorescence (FL), A1-1261 (circles); FL, A1-1238 (squares—dotted squares for the second transition); CD, A1-1261 (triangles); CD, A1-1238 (diamonds). Reported in (C) and (D) are the average
. Lines are solid for A1-1261 and dashed for A1-1238. Symbols are: fluorescence (FL), A1-1261 (circles); FL, A1-1238 (squares—dotted squares for the second transition); CD, A1-1261 (triangles); CD, A1-1238 (diamonds). Reported in (C) and (D) are the average and
and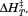 values ± standard deviation for each method. A1-1261 (black bars); A1-1238 (white bars).
values ± standard deviation for each method. A1-1261 (black bars); A1-1238 (white bars).
Thermal unfolding at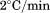 by differential scanning calorimetry (DSC) also shows that the
by differential scanning calorimetry (DSC) also shows that the is not significantly different between A1-1261 and A1-1238, but the enthalpy of unfolding is increased for A1-1238 (Fig. 6). The values obtained via DSC were not quantitatively comparable to those obtained from CD and fluorescence as a result of the aggregation of both proteins at the high temperatures and concentrations
is not significantly different between A1-1261 and A1-1238, but the enthalpy of unfolding is increased for A1-1238 (Fig. 6). The values obtained via DSC were not quantitatively comparable to those obtained from CD and fluorescence as a result of the aggregation of both proteins at the high temperatures and concentrations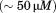 required to obtain reliable signal/noise in the calorimetric power compensation. It should also be noted that all thermal transitions remained irreversible in the presence of
required to obtain reliable signal/noise in the calorimetric power compensation. It should also be noted that all thermal transitions remained irreversible in the presence of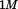 arginine, an excipient known to solubilize proteins, and a scan rate dependency was also observed in the presence of
arginine, an excipient known to solubilize proteins, and a scan rate dependency was also observed in the presence of urea confirming that thermal unfolding is intrinsically irreversible and not an artifact of any nonspecific aggregation.
urea confirming that thermal unfolding is intrinsically irreversible and not an artifact of any nonspecific aggregation.
Figure 6.
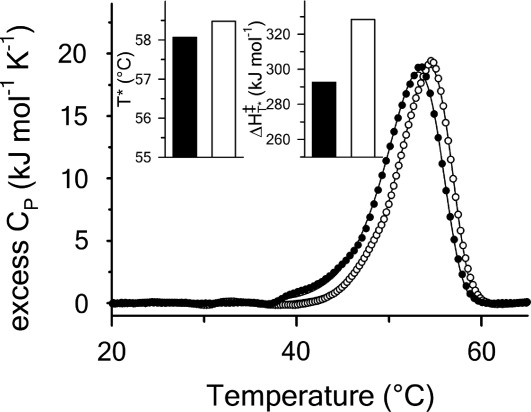
Excess molar heat capacity by DSC at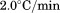 for A1-1261 (black circles) and A1-1238 (open circles). Insets are the transition temperature,
for A1-1261 (black circles) and A1-1238 (open circles). Insets are the transition temperature, , and enthalpy,
, and enthalpy, , of the native to intermediate transition. A1-1261 (black bars) and A1-1238 (white bars).
, of the native to intermediate transition. A1-1261 (black bars) and A1-1238 (white bars).
Interaction of A1-1261 with the N-terminal peptide
Because the thermal denaturation of A1 revealed that the covalently linked N-terminal peptide increases the enthalpy of the unfolding transition of the A1 domain, it was of interest whether this property could be used to detect binding of the peptide in solution to A1-1261. This would imply a specific interaction between this N-terminal sequence and the A1 domain when covalently linked in sequence. The only way to examine such a stabilization using spectroscopy was the measurement of thermal unfolding via fluorescence. Because the N-terminal peptide also contains a single tyrosine residue, the excitation wavelength was changed to to measure tryptophan fluorescence and the effect of the peptide on the unfolding of the A1-domain was observed indirectly. Thermal scans of
to measure tryptophan fluorescence and the effect of the peptide on the unfolding of the A1-domain was observed indirectly. Thermal scans of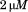 A1-1261 were performed as a function of increasing amounts of the N-terminal peptide at
A1-1261 were performed as a function of increasing amounts of the N-terminal peptide at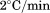 . Figure 7(A) shows that the primary transition shifts from an apparent
. Figure 7(A) shows that the primary transition shifts from an apparent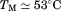 in the absence of peptide to
in the absence of peptide to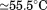 in the presence of saturating concentrations of peptide. The second transition becomes more prominent as the peptide concentration increases indicating that the peptide is binding to A1-1261. Thermal scans were also done in the presence of a peptide with the same amino acid composition, but a scrambled sequence. The inset of Figure 7(A) compares the raw fluorescence thermal scans in the presence of
in the presence of saturating concentrations of peptide. The second transition becomes more prominent as the peptide concentration increases indicating that the peptide is binding to A1-1261. Thermal scans were also done in the presence of a peptide with the same amino acid composition, but a scrambled sequence. The inset of Figure 7(A) compares the raw fluorescence thermal scans in the presence of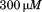 scrambled and N-terminal peptides. Unfolding in the presence of excess scrambled peptide was equivalent to unfolding in the absence of either peptide. Figure 7(B) shows the resulting
scrambled and N-terminal peptides. Unfolding in the presence of excess scrambled peptide was equivalent to unfolding in the absence of either peptide. Figure 7(B) shows the resulting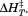 and
and obtained from these transitions as a function of the total concentration of N-terminal peptide. Although the
obtained from these transitions as a function of the total concentration of N-terminal peptide. Although the was constant within
was constant within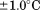 , the enthalpy of the transition changed sigmoidally on a log scale from
, the enthalpy of the transition changed sigmoidally on a log scale from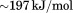 to
to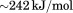 indicating that the unfolding enthalpy is sensitive to the binding of the N-terminal peptide. Fitting the enthalpy data to a simple single-site binding model yields a dissociation constant of
indicating that the unfolding enthalpy is sensitive to the binding of the N-terminal peptide. Fitting the enthalpy data to a simple single-site binding model yields a dissociation constant of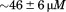 . Also the binding of the N-terminal peptide to A1-1261 was sequence specific as the scrambled peptide did not change the unfolding enthalpy at saturating concentrations. The N-terminal peptide did not change the unfolding enthalpy of A1-1238 suggesting that the binding is site specific and the covalently linked peptide occupies the binding site so that free N-terminal peptide cannot bind. In addition, the binding of the N-terminal peptide is fully reversible as excessive equilibrium dialysis of A1-1261 following incubation with
. Also the binding of the N-terminal peptide to A1-1261 was sequence specific as the scrambled peptide did not change the unfolding enthalpy at saturating concentrations. The N-terminal peptide did not change the unfolding enthalpy of A1-1238 suggesting that the binding is site specific and the covalently linked peptide occupies the binding site so that free N-terminal peptide cannot bind. In addition, the binding of the N-terminal peptide is fully reversible as excessive equilibrium dialysis of A1-1261 following incubation with peptide for 1 h resulted in a thermal scan with a
peptide for 1 h resulted in a thermal scan with a equivalent A1-1261 in the absence of peptide.
equivalent A1-1261 in the absence of peptide.
Figure 7.
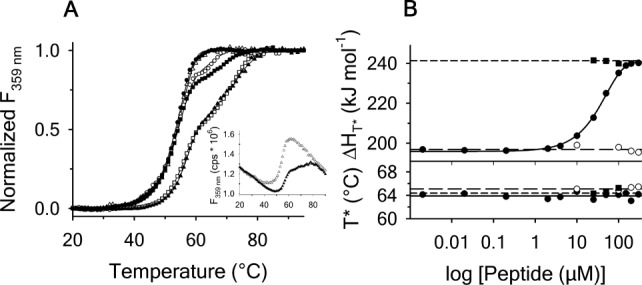
(A) Normalized thermal transitions of tryptophan fluorescence ( ) of
) of A1-1261 with excitation
A1-1261 with excitation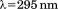 as a function of increasing concentrations of free N-terminal peptide. Concentrations are
as a function of increasing concentrations of free N-terminal peptide. Concentrations are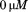 (black circles),
(black circles),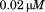 (open circles),
(open circles),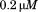 (black squares),
(black squares), (open squares),
(open squares),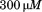 (black triangles) of the N-terminal peptide, and
(black triangles) of the N-terminal peptide, and of the scrambled peptide(open triangles). The inset shows transitions in presence of
of the scrambled peptide(open triangles). The inset shows transitions in presence of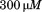 N-terminal peptide (closed triangles) and in presence of
N-terminal peptide (closed triangles) and in presence of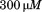 scrambled peptide (open triangles). (B) Determination of the binding affinity of the N-terminal peptide to A1-1261 from
scrambled peptide (open triangles). (B) Determination of the binding affinity of the N-terminal peptide to A1-1261 from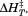 . Symbols represent A1-1261 + N-terminal peptide (black circles), A1-1261 + scrambled peptide (open circles; average—long dashed line) and A1 1238 + N-terminal peptide (black squares; average—short dashed line). The lower panel shows
. Symbols represent A1-1261 + N-terminal peptide (black circles), A1-1261 + scrambled peptide (open circles; average—long dashed line) and A1 1238 + N-terminal peptide (black squares; average—short dashed line). The lower panel shows values for all analyzed thermal scans. Symbols and lines are homolog to the upper panel.
values for all analyzed thermal scans. Symbols and lines are homolog to the upper panel.
The average and
and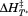 observed for the peptide-induced second fluorescence transition of A1-1261 are
observed for the peptide-induced second fluorescence transition of A1-1261 are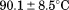 and
and kJ/mol. This second transition yields a comparable
kJ/mol. This second transition yields a comparable value to A1-1238
value to A1-1238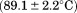 , although is less well-defined in cooperativity with a significant drop in
, although is less well-defined in cooperativity with a significant drop in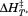 from
from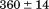 kJ/mol when covalently linked to
kJ/mol when covalently linked to kJ/mol when free to dissociate from the A1 domain over the full range of peptide concentrations studied.
kJ/mol when free to dissociate from the A1 domain over the full range of peptide concentrations studied.
Discussion
The results presented demonstrate the following important properties of the A1 domain.
The covalently linked N-terminal peptide suppresses the A1-GPIbα catch bond resulting in decreased pause times and faster translocation velocities. Specific binding of the N-terminal peptide free in solution to A1-1261 results in diminished pause times and enhanced velocities that are comparable to those obtained with A1-1238.
The N-terminal peptide, free in solution, is unstructured with
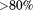 of the sequence having β and random coil content. In its native sequential context, this 23 residue peptide does not significantly alter the overall secondary structure content of the A1 domain. Therefore, this peptide is likely unstructured in the natural sequential context relative to the A1 domain.
of the sequence having β and random coil content. In its native sequential context, this 23 residue peptide does not significantly alter the overall secondary structure content of the A1 domain. Therefore, this peptide is likely unstructured in the natural sequential context relative to the A1 domain.Reversible urea denaturation illustrates that both A1-1261 and A1-1238 have an equivalent thermodynamic stability at
 despite small changes in the cm and the m-value. Irreversible thermal denaturation illustrates that both A1-1261 and A1-1238 have equivalent transition temperatures
despite small changes in the cm and the m-value. Irreversible thermal denaturation illustrates that both A1-1261 and A1-1238 have equivalent transition temperatures that define where the rate of unfolding is unity. Interestingly, the enthalpy of unfolding is larger for A1-1238 than A1-1261 which indicates that the rate of unfolding of 1261 to the intermediate state at temperatures lower than the
that define where the rate of unfolding is unity. Interestingly, the enthalpy of unfolding is larger for A1-1238 than A1-1261 which indicates that the rate of unfolding of 1261 to the intermediate state at temperatures lower than the will be faster than A1-1238.
will be faster than A1-1238.Although urea denaturation of A1 is reversible, thermal denaturation is irreversible and kinetically controlled. Kinetic irreversibility is evident by the significant scan rate dependence of the apparent melting temperature. Analysis of CD and fluorescence transitions using an irreversible two-state model defined by a first-order rate constant yield transition temperatures that are scan rate independent. Furthermore, the observed thermal transitions represent the native to intermediate conformational change observed in urea denaturation. This is evident by the significant amount of CD ellipticity remaining in the thermally denatured state and the presence of a second high temperature transition from the intermediate state to denatured observed by fluorescence. The fact that this high temperature transition is not observed with A1-1261 indicates that the N-terminal peptide is involved in this
 transition. In addition, extrapolation of the first apparent Tm of A1-1238 to zero scan rate (equilibrium) gives
transition. In addition, extrapolation of the first apparent Tm of A1-1238 to zero scan rate (equilibrium) gives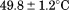 . This is in agreement with the
. This is in agreement with the obtained from the reversible
obtained from the reversible urea-temperature phase diagram determined previously.16 The apparent Tm of the second transition of A1-1238 is also comparable to that obtained from the
urea-temperature phase diagram determined previously.16 The apparent Tm of the second transition of A1-1238 is also comparable to that obtained from the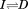 urea-temperature phase diagram although the error associated with that transition midpoint is larger.16
urea-temperature phase diagram although the error associated with that transition midpoint is larger.16In solution, the N-terminal peptide binds specifically to the truncated A1-1261. This binding is observed in the unfolding transition enthalpy which changes sigmoidally with respect to the natural log of the peptide concentrations from the enthalpy associated with A1-1261 unfolding to an enthalpy characteristic of A1-1238 unfolding. Scrambling the peptide sequence did not have any change in the unfolding properties of A1-1261 and the N-terminal peptide of correct sequence did not bind to A1-1238. These results indicate site and sequence specific binding of the peptide to A1-1261 at a site that is normally occupied by the covalently linked N-terminal peptide in A1-1238.
In vWD, the effect of inherited Type 2B and 2M mutations on the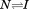 conformational equilibrium is now established for several mutations.16 These subtypes of mutations in the A1 domain cause clinically opposite phenotypes resulting in bleeding. From the perspective of reversible urea denaturation at variable temperature, it has been shown that Type 2M mutations shift the equilibrium in favor of the native state and decrease the affinity of A1 for platelet GP1bα. Conversely, Type 2B mutations shift the equilibrium in the opposite direction toward the intermediate state and increase the affinity of A1 for platelet GP1bα. Here, we have studied the irreversible thermal stability of A1 in the absence of urea and have demonstrated that truncating the N-terminal linker peptide from the A1 domain decreases the enthalpy of the
conformational equilibrium is now established for several mutations.16 These subtypes of mutations in the A1 domain cause clinically opposite phenotypes resulting in bleeding. From the perspective of reversible urea denaturation at variable temperature, it has been shown that Type 2M mutations shift the equilibrium in favor of the native state and decrease the affinity of A1 for platelet GP1bα. Conversely, Type 2B mutations shift the equilibrium in the opposite direction toward the intermediate state and increase the affinity of A1 for platelet GP1bα. Here, we have studied the irreversible thermal stability of A1 in the absence of urea and have demonstrated that truncating the N-terminal linker peptide from the A1 domain decreases the enthalpy of the transition without altering the transition temperature. This will necessarily increase the rate of unfolding from the native to intermediate state at temperatures below the transition temperature. However, the thermal stability of A1-1261 in solution can be restored to that of A1-1238 through specific binding of the free N-terminal peptide. The rate-dependent effect of this intramolecular interaction between the A1 domain and its N-terminal linker establishes the possibility of an on/off conformational switch that is affected by vWD mutations and by rheological shear stress on vWF multimers. In the crystal structure, the most frequently identified Type 2B mutations cluster in the loops and turns near the interface between the lower surface of the domain and the N-terminus. In addition to their destabilization of the globular domain structure, part of their effect on vWF conformation could be to favor dissociation of this N-terminal linker resulting in the uncoupling of A and possibly D domains within vWF which would enhance interactions between A1 and platelet GP1bα. We have observed that a monospecific antibody (A108), directed against the sequence D1444–E1452 within the
transition without altering the transition temperature. This will necessarily increase the rate of unfolding from the native to intermediate state at temperatures below the transition temperature. However, the thermal stability of A1-1261 in solution can be restored to that of A1-1238 through specific binding of the free N-terminal peptide. The rate-dependent effect of this intramolecular interaction between the A1 domain and its N-terminal linker establishes the possibility of an on/off conformational switch that is affected by vWD mutations and by rheological shear stress on vWF multimers. In the crystal structure, the most frequently identified Type 2B mutations cluster in the loops and turns near the interface between the lower surface of the domain and the N-terminus. In addition to their destabilization of the globular domain structure, part of their effect on vWF conformation could be to favor dissociation of this N-terminal linker resulting in the uncoupling of A and possibly D domains within vWF which would enhance interactions between A1 and platelet GP1bα. We have observed that a monospecific antibody (A108), directed against the sequence D1444–E1452 within the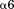 helix of the A1 domain, has a greater reactivity to the truncated 1261-A1A2A3 tridomain relative to the longer 1238-A1A2A3 tridomain. It also has a greater reactivity to purified plasma vWF in the presence of 0.5 mg/mL ristocetin.28 Therefore, it is plausible that this structural region of the A1 domain plays a role for the binding of the N-terminal linker.
helix of the A1 domain, has a greater reactivity to the truncated 1261-A1A2A3 tridomain relative to the longer 1238-A1A2A3 tridomain. It also has a greater reactivity to purified plasma vWF in the presence of 0.5 mg/mL ristocetin.28 Therefore, it is plausible that this structural region of the A1 domain plays a role for the binding of the N-terminal linker.
A corollary to the effect of vWD mutations on A1 domain conformational equilibria is their effect on the shear stress-dependent platelet rolling velocities on immobilized A1 and force-dependent single bond lifetimes between vWF A1 and platelet GP1bα. These properties are proportional to the strength of the interaction. We previously demonstrated on a single molecule scale that this interaction becomes stronger and increases the lifetime of the protein complex up to a critical threshold force above which the bond weakens and the lifetime decreases.14 This force sensing binding process referred to as “catch-bonding” regulates platelet detachment, rolling velocities, and overall adhesiveness of vWF as a function of shear stress. Type 2B mutations shift this threshold to lower forces due to the low stability of the native state. Consequently, at lower shear, attached platelets persist due to the higher bond strength and increase the risk for microthrombi. The stabilization of the native state caused by Type 2M mutations shifts this threshold to higher forces and weakens the bond strength at physiological shear stress thereby decreasing the adhesive capacity of vWF for platelet GP1bα.15
Here, we show that the platelet pause time to immobilized A1 has a similar behavior to the single molecule bond lifetime measurements with a sudden increase in platelet pause time at a critical shear threshold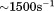 .14 Truncation of the N-terminal linker from the A1 domain enhances the pause times at low shear and shear rates that define the catch bond regime. However, addition of saturating amounts of free N-terminal peptide to the surface immobilized A1-1261 restores the pause times to those observed for A1-1238. Considering both of these observations, the fact that the binding of the free peptide in solution to A1 restores both the kinetic thermal stability and the catch-bond properties of the truncated domain to that of the full-length A1 domain demonstrates that the binding of this N-terminal linker is specific and critical for modulating the rheological dependence platelet adhesion to vWF.
.14 Truncation of the N-terminal linker from the A1 domain enhances the pause times at low shear and shear rates that define the catch bond regime. However, addition of saturating amounts of free N-terminal peptide to the surface immobilized A1-1261 restores the pause times to those observed for A1-1238. Considering both of these observations, the fact that the binding of the free peptide in solution to A1 restores both the kinetic thermal stability and the catch-bond properties of the truncated domain to that of the full-length A1 domain demonstrates that the binding of this N-terminal linker is specific and critical for modulating the rheological dependence platelet adhesion to vWF.
Our previous studies on the A1A2A3 tridomain fragment of vWF have demonstrated that similar to gain of function mutations,32 truncation of this sequence also destabilizes the quaternary association of these domains resulting in effective inhibition of RIPA and fibrinogen-dependent platelet activation under high shear.28 These studies confirmed a structural role for this N-terminal sequence to maintain interdomain associations and keep the tridomain in a thermodynamically stable binding incompetent conformation. The observations reported here highlight another role of the N-terminal linker as a force sensor for the regulation of platelet adhesion to the A1 domain in the open active state of vWF.
In conclusion, the rotational and elongational forces on plasma vWF and the tensile forces on vascular wall tethered Unusually Large vWF multimers that occur in the presence of rheological shear likely activate vWF in a two step process that first dissociates the A domains and subsequently modulates the kinetic stability of the A1 domain and its affinity for platelet GP1bα. These processes appear to be regulated by an on/off conformational equilibria between the A1 domain and its N-terminal flanking sequence although this may be a relatively small contribution to the overall process of conformational activation of full-length vWF in vivo.
Materials and Methods
Proteins
Recombinant human vWF A1-1238 (amino acids Q1238–P1471) and its N-terminal truncated variant A1-1261 (amino acids D1261–P1471) were expressed in Escherichia coli M-15 cells as fusion proteins containing a N-terminal 6×His-Tag using BamHI and HindIII restriction sites in the Qiagen pQE-9 plasmid vector.33,34 Proteins were purified from inclusion bodies by solubilization in 6M GuHCl followed by refolding by excessive dilution into Tris-buffered saline with 0.5% Tween 20 (TBS-T) and isolated by affinity chromatography using
Tris-buffered saline with 0.5% Tween 20 (TBS-T) and isolated by affinity chromatography using -chelated Sepharose followed by a second purification via Heparin-Sepharose. The purity of the recombinant proteins was confirmed via reducing Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and also by size exclusion chromatography using a Phenomenex SEC S3000 column on a BioCAD Sprint perfusion chromatography system. Proteins were stored at
-chelated Sepharose followed by a second purification via Heparin-Sepharose. The purity of the recombinant proteins was confirmed via reducing Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and also by size exclusion chromatography using a Phenomenex SEC S3000 column on a BioCAD Sprint perfusion chromatography system. Proteins were stored at in
in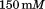 NaCl,
NaCl,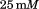 Tris HCl,
Tris HCl,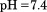 , and dialyzed overnight against a temperature stable buffer-mixture of
, and dialyzed overnight against a temperature stable buffer-mixture of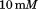 sodium acetate,
sodium acetate,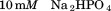 ,
,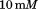 Glycine,
Glycine,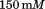 NaCl,
NaCl, EDTA, pH = 8 before all measurements.
EDTA, pH = 8 before all measurements.
The N-terminal peptide (QEPGGLVVPPTDAPVSPTTLYVE) corresponding to residues Q1238–E1260 and the scrambled peptide (QLPTGVLGEPSDAVPTVYEVTPPG) were synthesized at the Mayo Clinic Proteomics Core Lab with a free N-terminal amine and C-terminal acid, checked for purity via reversed-phase High Pressure Liquid Chromatography and their correct masses of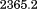 and
and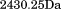 were verified via electrospray ionization mass spectrometry.
were verified via electrospray ionization mass spectrometry.
Protein and peptide concentrations were quantified using a Shimadzu UV2101PC spectrophotometer with the Pace method35 from absorption at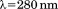 minus twice the absorption at
minus twice the absorption at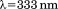 for correction of light scattering. Extinction coefficients for A1-1238 (
for correction of light scattering. Extinction coefficients for A1-1238 (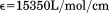 ) and A1-1261 (
) and A1-1261 (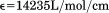 ) and the N-terminal peptide (
) and the N-terminal peptide (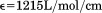 ) were calculated from the number of tyrosines (A1-1238 = 8, A1-1261 = 7, and N-terminal peptide = 1) and a single tryptophan.
) were calculated from the number of tyrosines (A1-1238 = 8, A1-1261 = 7, and N-terminal peptide = 1) and a single tryptophan.
Determination of platelet pause times
A Glycotech† rectangular parallel-plate flow chamber was mounted to slides with a chelated 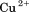 surface obtained from Microsurfaces Inc.# to which the A1-1261 and A1-1238 domains in TBS were immobilized by the 6×His-Tag. Citrated whole blood (
surface obtained from Microsurfaces Inc.# to which the A1-1261 and A1-1238 domains in TBS were immobilized by the 6×His-Tag. Citrated whole blood (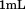 ) obtained from informed consent of healthy donors with approval from the Baylor College of Medicine institutional review board followed by buffer was perfused through the chamber at
) obtained from informed consent of healthy donors with approval from the Baylor College of Medicine institutional review board followed by buffer was perfused through the chamber at  for A1-1261 and
for A1-1261 and 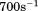 for A1-1238 shear rate to attach platelets to the surface immobilized proteins using a syringe-driven variable flow rate pump. The shear was increased incrementally and
for A1-1238 shear rate to attach platelets to the surface immobilized proteins using a syringe-driven variable flow rate pump. The shear was increased incrementally and  movies were recorded at 25 frames/s in phase contrast with 2 × 2 pixel binning on a Zeiss Axiocam MRm camera attached to a Zeiss Axio Observer D1 inverted microscope. Tracking analysis was performed using Mediacybernetics ImagePro Premier§. The resulting X-Y coordinate data for each platelet track was differentiated into instantaneous velocities using a Savitzky–Golay algorithm36 with a five data point window size and a second-order polynomial implemented into a Mathematica§ notebook written in our lab. These platelet tracks were retained for statistical analysis if the platelet was present for at least
movies were recorded at 25 frames/s in phase contrast with 2 × 2 pixel binning on a Zeiss Axiocam MRm camera attached to a Zeiss Axio Observer D1 inverted microscope. Tracking analysis was performed using Mediacybernetics ImagePro Premier§. The resulting X-Y coordinate data for each platelet track was differentiated into instantaneous velocities using a Savitzky–Golay algorithm36 with a five data point window size and a second-order polynomial implemented into a Mathematica§ notebook written in our lab. These platelet tracks were retained for statistical analysis if the platelet was present for at least  and travelled a total distance greater than
and travelled a total distance greater than 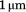 . Distance travelled in pixels was converted to
. Distance travelled in pixels was converted to 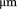 given the camera pixel size
given the camera pixel size 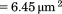 , pixel binning, and microscope magnification
, pixel binning, and microscope magnification 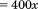 . Pause times were determined by the amount of time
. Pause times were determined by the amount of time  a platelets velocity was
a platelets velocity was 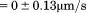 , within the noise. Pause times for all platelet tracks are reported as the mean of the medians. Examples of this analysis for individual platelets and for multiple platelets are provided in the Supporting Information.
, within the noise. Pause times for all platelet tracks are reported as the mean of the medians. Examples of this analysis for individual platelets and for multiple platelets are provided in the Supporting Information.
Protein denaturation
Urea and thermal unfolding of A1-1238 and A1-1261 was monitored by CD and fluorescence spectroscopy and DSC using an Aviv Biomedical Model 420C CD spectrometer, a Horiba Jobin-Yvon Fluorolog 3 spectrofluorimeter equipped with a Wavelength Electronics Model LF1–3751 temperature controller and a TA Instruments NanoDSC.
Isothermal urea-induced unfolding of A1-1261 and A1-1238 at was monitored via CD at
was monitored via CD at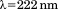 using a quartz cuvette with a path length of
using a quartz cuvette with a path length of and a protein concentration of
and a protein concentration of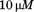 after an overnight equilibration. CD-signal was averaged for
after an overnight equilibration. CD-signal was averaged for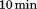 , corrected for the corresponding CD-signal of the buffer, and converted into mean molar ellipticities per amino acid residue (
, corrected for the corresponding CD-signal of the buffer, and converted into mean molar ellipticities per amino acid residue (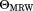 ).
).
Prior to all spectroscopic thermal scans, protein samples were equilibrated at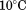 for
for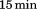 to obtain a stable baseline. CD thermal scans between 10 and
to obtain a stable baseline. CD thermal scans between 10 and were recorded at
were recorded at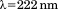 using a protein concentration of 1 or
using a protein concentration of 1 or in a
in a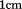 quartz cell under moderate stirring. Scan rates were 0.5, 1.0, 1.5, and
quartz cell under moderate stirring. Scan rates were 0.5, 1.0, 1.5, and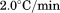 . Integration time for each data point was
. Integration time for each data point was with a
with a bandwidth. Fluorescence thermal scans between 10 and
bandwidth. Fluorescence thermal scans between 10 and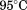 were recorded at an emission
were recorded at an emission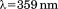 after excitation
after excitation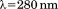 or
or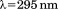 using
using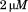 protein in a
protein in a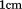 quartz cell with moderate stirring. Scan rates were 0.4 or 0.6, 0.9, 1.6 and
quartz cell with moderate stirring. Scan rates were 0.4 or 0.6, 0.9, 1.6 and . At each temperature, relative fluorescence intensity was collected for
. At each temperature, relative fluorescence intensity was collected for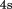 and averaged. Thermal scans in the presence of N-terminal peptide at
and averaged. Thermal scans in the presence of N-terminal peptide at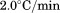 were preceded by
were preceded by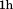 incubations at
incubations at with
with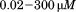 peptide.
peptide.
DSC was performed at pressure. Protein samples (40 to
pressure. Protein samples (40 to ), and buffers were degassed under moderate stirring prior to use. The calorimeter was equilibrated overnight with buffer in the sample and reference cell to obtain a stable baseline, and the protein sample was loaded during an equilibration step between scans. One measurement per second was recorded at
), and buffers were degassed under moderate stirring prior to use. The calorimeter was equilibrated overnight with buffer in the sample and reference cell to obtain a stable baseline, and the protein sample was loaded during an equilibration step between scans. One measurement per second was recorded at . DSC-traces were background corrected with the following irreversible scan that was used as a baseline. The molar heat capacity was calculated from the calorimetric power compensation
. DSC-traces were background corrected with the following irreversible scan that was used as a baseline. The molar heat capacity was calculated from the calorimetric power compensation using the relation,
using the relation, . The excess molar heat capacity,
. The excess molar heat capacity,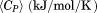 was obtained by subtracting a polynomial baseline fit to the pre- and post-transition regions of the heat capacity traces using a Mathematica** notebook written in our laboratory.
was obtained by subtracting a polynomial baseline fit to the pre- and post-transition regions of the heat capacity traces using a Mathematica** notebook written in our laboratory.
Isothermal urea unfolding was analyzed using a three state reversible model as previously described.16,37 All thermal unfolding transitions were analyzed according to the irreversible models provided in the Supporting Information.38–41
as previously described.16,37 All thermal unfolding transitions were analyzed according to the irreversible models provided in the Supporting Information.38–41
Glossary
- CD
circular dichroism
- DSC
differential scanning calorimetry
- FL
fluorescence
- RIPA
ristocetin-induced platelet agglutination
- SIPA
shear-induced platelet agglutination
- vWD
von Willebrand disease
- vWF
von Willebrand factor.
Footnotes
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Bonthron DT, Handin RI, Kaufman RJ, Wasley LC, Orr EC, Mitsock LM, Ewenstein B, Loscalzo J, Ginsburg D, Orkin SH. Structure of pre-pro-von Willebrand factor and its expression in heterologous cells. Nature. 1986;324:270–273. doi: 10.1038/324270a0. [DOI] [PubMed] [Google Scholar]
- 2.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 3.Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289–297. doi: 10.1016/s0092-8674(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 4.Fujimura Y, Titani K, Holland LZ, Russell SR, Roberts JR, Elder JH, Ruggeri ZM, Zimmerman TS. von Willebrand factor. A reduced and alkylated 52/48-kDa fragment beginning at amino acid residue 449 contains the domain interacting with platelet glycoprotein Ib. J Biol Chem. 1986;261:381–385. [PubMed] [Google Scholar]
- 5.Fujimura Y, Titani K, Holland LZ, Roberts JR, Kostel P, Ruggeri ZM, Zimmerman TS. A heparin-binding domain of human von Willebrand factor. Characterization and localization to a tryptic fragment extending from amino acid residue Val-449 to Lys-728. J Biol Chem. 1987;262:1734–1739. [PubMed] [Google Scholar]
- 6.Pietu G, Meulien P, Cherel G, Diaz J, Baruch D, Courtney M, Meyer D. Production in Escherichia coli of a biologically active subfragment of von Willebrand factor corresponding to the platelet glycoprotein Ib, collagen and heparin binding domains. Biochem. Biophys Res Commun. 1989;164:1339–1347. doi: 10.1016/0006-291x(89)91816-0. [DOI] [PubMed] [Google Scholar]
- 7.Sugimoto M, Ricca G, Hrinda ME, Schreiber AB, Searfoss GH, Bottini E, Ruggeri ZM. Functional modulation of the isolated glycoprotein Ib binding domain of von Willebrand factor expressed in Escherichia coli. Biochemistry. 1991;30:5202–5209. doi: 10.1021/bi00235a013. [DOI] [PubMed] [Google Scholar]
- 8.Gralnick HR, Williams S, McKeown L, Kramer W, Krutzsch H, Gorecki M, Pinet A, Garfinkel LI. A monomeric von Willebrand factor fragment, Leu-504–Lys-728, inhibits von Willebrand factor interaction with glycoprotein Ib-IX. Proc Natl Acad Sci USA. 1992;89:7880–7884. doi: 10.1073/pnas.89.17.7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azuma H, Dent JA, Sugimoto M, Ruggeri ZM, Ware J. Independent assembly and secretion of a dimeric adhesive domain of von Willebrand factor containing the glycoprotein Ib-binding site. J Biol Chem. 1991;266:12342–12347. [PubMed] [Google Scholar]
- 10.Cruz MA, Handin RI, Wise RJ. The interaction of the von Willebrand factor-A1 domain with platelet glycoprotein Ib/IX. The role of glycosylation and disulfide bonding in a monomeric recombinant A1 domain protein. J Biol Chem. 1993;268:21238–21245. [PubMed] [Google Scholar]
- 11.Arya M, Anvari B, Romo GM, Cruz MA, Dong JF, McIntire LV, Moake JL, Lopez JA. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood. 2002;99:3971–3977. doi: 10.1182/blood-2001-11-0060. [DOI] [PubMed] [Google Scholar]
- 12.Arya M, Kolomeisky AB, Romo GM, Cruz MA, Lopez JA, Anvari B. Dynamic force spectroscopy of glycoprotein Ib-IX and von Willebrand Factor. Biophys J. 2005;88:4391–4401. doi: 10.1529/biophysj.104.046318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coburn LA, Damaraju VS, Dozic S, Eskin SG, Cruz MA, McIntire LV. GPIb-vWF rolling under shear stress shows differences between type 2B and 2M von Willebrand disease. Biophys J. 2011;100:304–312. doi: 10.1016/j.bpj.2010.11.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yago T, Lou J, Wu T, Yang J, Miner JJ, Coburn L, López JA, Cruz MA, Dong JF, McIntire LV, McEver RP, Zhu C. Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J Clin Invest. 2008;118:3195–3207. doi: 10.1172/JCI35754. 1,9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auton M, Zhu C, Cruz MA. The mechanism of VWF-mediated platelet GPIbalpha binding. Biophys J. 2010;99:1192–1201. doi: 10.1016/j.bpj.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Auton M, Sedlak E, Marek J, Wu T, Zhu C, Cruz MA. Changes in thermodynamic stability of von willebrand factor differentially affect the force-dependent binding to platelet GPIbalpha. Biophys J. 2009;97:618–627. doi: 10.1016/j.bpj.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emsley J, Cruz M, Handin R, Liddington R. Crystal structure of the von Willebrand Factor A1 domain and implications for the binding of platelet glycoprotein Ib. J Biol Chem. 1998;273:10396–10401. doi: 10.1074/jbc.273.17.10396. [DOI] [PubMed] [Google Scholar]
- 18.Fukuda K, Doggett TA, Bankston LA, Cruz MA, Diacovo TG, Liddington RC. Structural basis of von Willebrand factor activation by the snake toxin botrocetin. Structure. 2002;10:943–950. doi: 10.1016/s0969-2126(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 19.Kim J, Zhang CZ, Zhang X, Springer TA. A mechanically stabilized receptor-ligand flex-bond important in the vasculature. Nature. 2010;466:992995. doi: 10.1038/nature09295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyata S, Ruggeri ZM. Distinct structural attributes regulating von Willebrand factor A1 domain interaction with platelet glycoprotein Ibalpha under flow. J Biol Chem. 1999;274:6586–6593. doi: 10.1074/jbc.274.10.6586. [DOI] [PubMed] [Google Scholar]
- 21.Celikel R, Ruggeri ZM, Varughese KI. von Willebrand factor conformation and adhesive function is modulated by an internalized water molecule. Nat Struct Biol. 2000;7:881–884. doi: 10.1038/79639. [DOI] [PubMed] [Google Scholar]
- 22.Huizinga EG, Tsuji S, Romijn RA, Schiphorst ME, de Groot PG, Sixma JJ, Gros P. Structures of glycoprotein Ibalpha and its complex with von Willebrand factor A1 domain. Science. 2002;297:1176–1179. doi: 10.1126/science.107355. [DOI] [PubMed] [Google Scholar]
- 23.Dumas JJ, Kumar R, McDonagh T, Sullivan F, Stahl ML, Somers WS, Mosyak L. Crystal structure of the wild-type von Willebrand factor A1-glycoprotein Ibalpha complex reveals conformation differences with a complex bearing von Willebrand disease mutations. J Biol Chem. 2004;279:23327–23334. doi: 10.1074/jbc.M401659200. [DOI] [PubMed] [Google Scholar]
- 24.Mohri H, Fujimura Y, Shima M, Yoshioka A, Houghten RA, Ruggeri ZM, Zimmerman TS. Structure of the von Willebrand factor domain interacting with glycoprotein Ib. J Biol Chem. 1988;263:17901–17904. [PubMed] [Google Scholar]
- 25.Fujimura Y, Usami Y, Titani K, Niinomi K, Nishio K, Takase T, Yoshioka A, Fukui H. Studies on anti-von Willebrand factor (vWF) monoclonal antibody NMC-4, which inhibits both ristocetin- and botrocetin-induced vWF binding to platelet glycoprotein Ib. Blood. 1991;77:113–120. [PubMed] [Google Scholar]
- 26.Celikel R, Varughese KI, Madhusudan, Yoshioka A, Ware J, Ruggeri ZM. Crystal structure of the von Willebrand factor A1 domain in complex with the function blocking NMC-4 Fab. Nat Struct Biol. 1998;5:189–194. doi: 10.1038/nsb0398-189. [DOI] [PubMed] [Google Scholar]
- 27.Ulrichts H, Udvardy M, Lenting PJ, Pareyn I, Vandeputte N, Vanhoorelbeke K, Deckmyn H. Shielding of the A1 domain by the D'D3 domains of von Willebrand factor modulates its interaction with platelet glycoprotein Ib-IX-V. J Biol Chem. 2006;281:4699–4707. doi: 10.1074/jbc.M513314200. [DOI] [PubMed] [Google Scholar]
- 28.Auton M, Sowa KE, Behymer M, Cruz MA. The N-terminal flanking region of A1 domain in Von Willebrand Factor stabilizes the structure of the A1A2A3 complex and modulates platelet activation under shear stress. J Biol Chem. 2012;287:14579–14585. doi: 10.1074/jbc.M112.348573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tornai I, Arnout J, Deckmyn H, Peerlinck K, Vermylen J. A monoclonal antibody recognizes a von Willebrand factor domain within the amino-terminal portion of the subunit that modulates the function of the glycoprotein IB- and IIB/IIIA-binding domains. J Clin Invest. 1993;91:273–282. doi: 10.1172/JCI116181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ulrichts H, Harsfalvi J, Bene L, Matko J, Vermylen J, Ajzenberg N, Baruch D, Deckmyn H, Tornai I. A monoclonal antibody directed against human von Willebrand factor induces type 2Blike alterations. J Thromb Haemost. 2004;2:1622–1628. doi: 10.1111/j.1538-7836.2004.00865.x. [DOI] [PubMed] [Google Scholar]
- 31.Böhm G, Muhr R, Jaenicke R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 1992;5:191–195. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
- 32.Auton M, Sowa KE, Smith SM, Sedlak E, Vijayan KV, Cruz MA. Destabilization of the A1 domain in von Willebrand factor dissociates the A1A2A3 tri-domain and provokes spontaneous binding to glycoprotein Ibalpha and platelet activation under shear stress. J Biol Chem. 2010;285:22831–22839. doi: 10.1074/jbc.M110.103358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morales LD, Martin C, Cruz MA. The interaction of von Willebrand factor-A1 domain with collagen: mutation G1324S (type 2M von Willebrand disease) impairs the conformational change in A1 domain induced by collagen. J Thromb Haemost. 2006;4:417–425. doi: 10.1111/j.1538-7836.2006.01742.x. [DOI] [PubMed] [Google Scholar]
- 34.Cruz MA, Diacovo TG, Emsley J, Liddington R, Handin RI. Mapping the glycoprotein Ib-binding site in the von willebrand factor A1 domain. J Biol Chem. 2000;275:19098–19105. doi: 10.1074/jbc.M002292200. [DOI] [PubMed] [Google Scholar]
- 35.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savitzky A, Golay MJE. Smoothing and differentiation of data by simplifies least squares procedures. Anal Chem. 1964;34:1627–1639. [Google Scholar]
- 37.Auton M, Cruz MA, Moake J. Conformational stability and domain unfolding of the von willebrand factor A domains. J Mol Biol. 2007;366:986–1000. doi: 10.1016/j.jmb.2006.10.067. [DOI] [PubMed] [Google Scholar]
- 38.Atkins PW. Chapter 25: the rates of chemical reactions. In: Atkins PW, editor. Physical chemistry. 5 ed. New York: W. H. Freeman and Company; 1994. pp. 861–897. [Google Scholar]
- 39.Lyubarev AE, Kurganov BI. Modeling of irreversible thermal protein denaturation at varying temperature. I. The model involving two consecutive irreversible steps. Biochemistry (Mosc) 1998;63:434–440. [PubMed] [Google Scholar]
- 40.Kurganov BI, Lyubarev AE, Sanchez-Ruiz JM, Shnyrov VL. Analysis of differential scanning calorimetry data for proteins. Criteria of validity of one-step mechanism of irreversible protein denaturation. Biophys Chem. 1997;69:125–135. doi: 10.1016/s0301-4622(97)80552-2. [DOI] [PubMed] [Google Scholar]
- 41.Rainville ED, Bedient PE. Chapter 2: Equations of Order One. In: (1989) Elementary differential equations. 7 ed. New York: Macmillan Publishing Company; p. 1746. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.