

Abstract
Hyperthermia challenges the nervous system's ability to transmit action potentials faithfully. Neuromuscular diseases, particularly those involving demyelination have an impaired safety margin for action potential generation and propagation, and symptoms are commonly accentuated by increases in temperature. The aim of this study was to examine the mechanisms responsible for reduced excitability during hyperthermia. Additionally, we sought to determine if motor and sensory axons differ in their propensity for conduction block during hyperthermia. Recordings of axonal excitability were performed at normal temperatures and during focal hyperthermia for motor and sensory axons in six healthy subjects. There were clear changes in excitability during hyperthermia, with reduced superexcitability following an action potential, faster accommodation to long-lasting depolarization and reduced accommodation to hyperpolarization. A verified model of human motor and sensory axons was used to clarify the effects of hyperthermia. The hyperthermia-induced changes in excitability could be accounted for by increasing the modelled temperature by 6°C (and adjusting the maximum conductances and activation kinetics according to their Q10 values; producing a 2 mV hyperpolarization of resting membrane potential), further hyperpolarizing the voltage dependence of Ih (motor, 11 mV; sensory, 7 mV) and adding a small depolarizing current at the internode (motor, 20 pA; sensory, 30 pA). The modelling suggested that slow K+ channels play a significant role in reducing axonal excitability during hyperthermia. The further hyperpolarization of the activation of Ih would limit its ability to counter the hyperpolarization produced by activity, thereby allowing conduction block to occur during hyperthermia.
Key points
In six healthy subjects, the excitability of both motor and sensory axons was altered during hyperthermia, lowering their safety margin.
The results suggest that slow K+ channels play a significant role in these changes in axonal excitability during hyperthermia.
Inward rectification was reduced during hyperthermia, and the modelling suggests that the hyperpolarization-activated cation current, Ih, was reduced, thus hampering its ability to counter activity-dependent hyperpolarization.
Hyperthermia lowers the safety margin for action potential generation and propagation. Differences in their responses to hyperthermia suggest that motor axons undergo conduction block more readily than sensory axons during fever, particularly when the safety margin is already impaired.
Introduction
Signalling in axons of the peripheral nervous system has long been known to be sensitive to temperature, and hyperthermia can impact on the ability of axons to faithfully transmit impulses to or from the peripheral nervous system. Cooling increases the duration and amplitude of an action potential and slows conduction velocity, and it has been suggested that this is primarily due to the slowing of Na+ channel kinetics. Conversely, heating increases conduction velocity and decreases the response amplitude and duration of compound action potentials (Buchthal & Rosenfalck, 1966; Ludin & Beyeler, 1977; Rutkove et al]. 1997; Rutkove, 2001). In healthy individuals, the symptoms of fatigue and exhaustion can result from elevations in body temperature. In demyelinating disorders, increases in temperature can produce or exacerbate conduction block at sites of impairment of the safety margin for action potential generation. Experimentally demyelinated single nerve fibres may undergo reversible conduction block with elevations in nerve temperature as small as 0.5°C (Rasminsky, 1973). In a guinea-pig model of experimental allergic neuritis, Davis & colleagues (1975) found that the temperature for conduction block is a linear function of conduction velocity (which should be related to the degree of demyelination), confirming their earlier theoretical predictions (Schauf & Davis, 1974).
In the giant squid axon, Hodgkin & Katz (1949) found the resting membrane potential to be practically constant between 3 and 20°C, and then gradually depolarized by 10 to 15 mV up to 35°C. In contrast, they found the shape of an action potential to be much more affected by temperature. The temperature dependence of the falling phase of the action potential was much greater than of the rising phase, for which they postulated that different mechanisms were responsible. To account for the temperature dependence of ion channel gating, Hodgkin & Huxley (1952a,b) adopted a temperature coefficient (Q10) of 3 for all rate constants. However, subsequent studies in myelinated fibres found Na+ activation to be less temperature sensitive, with a Q10 of ∼2 (Frankenhaeuser & Moore, 1963, frog; Schwarz & Eikhof, 1987, rat). To a lesser extent, ion concentrations, Nernst potentials, ion channel permeability, axonal membrane capacity and axoplasmic resistance are also dependent on temperature (Hodgkin et al. 1952; Taylor & Chandler, 1962; Frankenhaeuser & Moore, 1963; Palti & Adelman, 1969).
In vivo threshold tracking techniques have been used to examine the underlying mechanisms of excitability in healthy and diseased human axons (Bostock et al. 1998; Kiernan et al. 2000; Krishnan et al. 2009). Cooling has a marked effect on the excitability of both motor axons and cutaneous afferents (Burke et al. 1999; Moldovan & Krarup, 2004) and even modest changes in temperature can have significant effects (Kiernan et al. 2001a). However, to date the effects of hyperthermia on axonal excitability in human subjects have not been studied, so that the responses of the nervous system during hyperthermia have to be inferred, primarily from studies of cooling.
In the present study, we examined the factors affecting the excitability of peripheral nerve axons when subjected to physiologically high temperatures. We find that slow potassium (Ks) channels contribute to a dampening of excitability at elevated temperatures, and that this is particularly evident during the recovery of excitability following an action potential. In addition, our modelling suggests that the voltage dependence of the hyperpolarization-activated current (Ih) is further hyperpolarized during hyperthermia. This would limit its ability to counter activity-dependent hyperpolarization and restore resting membrane potential (RMP). These changes would impact on the safety margin for action potential generation, and are likely to be factors in the conduction failure and fatigue of febrile illnesses, particularly in patients with diseases affecting axonal function, both central and peripheral.
Methods
The research followed the guidelines in the Declaration of Helsinki and ethics approval for the studies was obtained from the Human Research Ethics Committee of The University of Sydney, Australia. Experiments were performed on 27 separate occasions on six healthy subjects, all of whom provided written informed consent prior to the commencement of the study.
On each occasion we measured axonal excitability using the extended Trond protocol (Tomlinson et al. 2010; Howells et al. 2012) and a new protocol for studying polarized recovery cycles, first at a normal skin-surface temperature and then during focal hyperthermia. Sensory and motor axons of the median nerve of the six subjects were studied on separate occasions. To test the reproducibility of the results within subjects, measurements at normal temperature and during focal hyperthermia were performed on motor axons five times on separate days in two of the six subjects (subjects 1 and 2). The repeatability data for each subject were averaged before inclusion in the group data. The variability within an experiment was also tested five times in two subjects (subjects 2 and 3). Temperature was measured continuously near the site of stimulation using a skin-surface thermistor (YSI-409B; YSI Inc., Yellow Springs, OH, USA; see Fig. 1). Reusable ‘click-style’ sodium acetate heat pads (Maverick Co., Christchurch, New Zealand) were applied to the dorsal and palmar aspects of the distal forearm and hand to create focal hyperthermia at the site of stimulation, while the whole arm was wrapped in a blanket to slow temperature loss. The average skin surface temperatures recorded under control conditions were 32.5 ± 0.2°C (mean ± SEM) for motor axons and 32.6 ± 0.1°C for sensory axons. The temperatures recorded during heating ranged from 39.4 to 43.4°C. After application of the heat pads, measurements began within 5 min, when the skin-surface temperature had stabilized at its peak value (41.7 ± 0.1°C, mean ± SEM). Over the course of the experiment the mean temperature cooled slightly to 40.8 ± 0.1°C, with a maximal drop in any experiment of 2.5°C.
Figure 1. Recording arrangement.

Stimuli were delivered at the wrist (cathode) using Ag–AgCl electrodes with the anode 10 cm distal on radial edge of the forearm. CMAPs were recorded over the thenar eminence, with the reference electrode on the proximal phalanx of the thumb. CSAPs were recorded using disposable Ag–AgCl ring electrodes on digit 2. The same ground was used for both motor and sensory recordings. Reusable ‘click-style’ sodium acetate heat pads were applied to the dorsal and palmar aspects of the distal forearm and hand, at wrist level (as indicated by the dotted lines), and the thermistor recorded the temperature close to the site of stimulation.
Nerve excitability studies were performed using the QtracS software (© Hugh Bostock, Institute of Neurology, UCL, Queen Square, London, UK). Stimuli were applied to the median nerve at the wrist via non-polarizable Ag–AgCl adhesive electrodes (Cleartrace REF 1700–050, ConMed, Utica, NY, USA), with the anode placed 10 cm proximally on the radial edge of the forearm (see Fig. 1). Compound muscle action potentials (CMAPs) were recorded using the same Ag–AgCl electrodes with the active electrode over the thenar eminence, the reference electrode distally on the proximal phalanx of the thumb. Compound sensory action potentials (CSAPs) were recorded with disposable Ag–AgCl ring electrodes (RE-D; Electrode Store, EnumClaw, WA, USA) on digit 2 with the active electrode on the proximal phalanx, the reference approximately 4 cm distally. A ground electrode on the palm was used for both motor and sensory recordings. All electrodes were further secured to the skin with surgical tape (Transpore; 3M, St Paul, MN, USA) prior to commencement of the experiments.
A purpose-built isolated preamplifier with high common-mode rejection and low noise was used to amplify the compound action potentials (CMAP ×200; CSAP × 10,000) which then had mains-frequency noise removed with a HumBug Noise Eliminator (Quest Scientific, North Vancouver, BC, Canada) before being digitized by a data acquisition system (PCI-6221; National Instruments, Austin, TX, USA). QtracS and the data acquisition system provided the command signal for the delivery of stimuli by an isolated linear bipolar current stimulator (DS5; Digitimer, Welwyn Garden City, UK).
Excitability protocols
Excitability studies adjust the intensity of a test stimulus to track a constant fraction of the maximal compound action potential (target potential). The response to graded stimuli was measured first to determine stimulus–response (SR) properties, the size of the target potential and to optimize the tracking. For both motor and sensory studies, 1 ms-wide test stimuli were used, allowing simpler comparisons between motor and sensory axons and the use of strong hyperpolarization in sensory axons (Tomlinson et al. 2010; Howells et al. 2012). From the SR relationship, the target response was set to 50% of the maximal response (near the steepest part of the curve).
Routine axonal excitability assessment
The extended nerve excitability protocol (TrondNF) was used to study the excitability of axons at normal and hyperthermic temperatures. This protocol consists of five parts: stimulus–response relationship (SR; described above); strength–duration properties, which are plotted as charge versus duration (QT); threshold electrotonus (TE); current–threshold relationship (I-V; the threshold analogue of current-voltage) and the recovery cycle (RC).
The strength–duration properties of motor axons were studied by tracking the 50% target potential using stimuli of different widths: 1, 0.8, 0.6, 0.4 and 0.2 ms. There is a linear relationship between stimulus charge (stimulus strength × width) and stimulus width, with the slope and negative intercept on the x-axis representing rheobase and the strength–duration time constant (SDTC), respectively (Mogyoros et al. 1996). The strength–duration properties of sensory axons were made using narrower test widths (0.5, 0.4, 0.3, 0.2 and 0.1) to avoid dispersion of the CSAP that occurs with longer stimuli.
Threshold electrotonus was tested before, during and after long subthreshold conditioning currents. In addition to the standard conditioning currents of ±20 and ±40% of the unconditioned threshold (see Kiernan et al. 2000), two additional levels of hyperpolarization were included: a 200 ms-long hyperpolarizing current which was 70% of the control threshold, and a 300 ms long hyperpolarizing current which was 100% of the control threshold (Tomlinson et al. 2010; Howells et al. 2012).
The current–threshold relationship quantifies the rectifying conductances, both inward and outward, and the resting input conductance. The test threshold was measured at the end of a 200 ms conditioning current for 16 conditioning strengths from +50% (depolarizing) to −100% (hyperpolarizing) in 10% increments.
The recovery cycle measures the recovery of excitability following supramaximal stimulation at 18 conditioning–test intervals which change in a logarithmic fashion between 2 and 200 ms. Under normal conditions axons undergo a distinct pattern of excitability following an action potential (Adrian & Lucas, 1912). Initially the axons are inexcitable (absolute refractoriness), followed by reduced excitability (relative refractoriness), the end of which corresponds to the relative refractory period (RRP). After refractoriness axons undergo a period of superexcitability which is associated with the depolarizing afterpotential (Barrett & Barrett, 1982), and then a period of subexcitability ensues, attributed to the after-hyperpolarization.
Polarized recovery cycles
To determine whether changes in membrane potential contribute to the changes in the recovery cycle much as described in Bergmans (1970) and Kiernan & Bostock (2000), recovery cycles were measured in the same way as described above, at rest and during background polarization, at normal and hyperthermic temperatures. The recovery cycle was measured 200 ms after the onset of long +30% (depolarizing) and −30% (hyperpolarizing) currents which extended beyond the test stimulus.
Mathematical modelling
The Bostock model of the human motor axon (Bostock et al. 1991), as modified by Howells and colleagues (2012) for motor and sensory axons, was used to assist in the interpretation of the excitability changes during focal hyperthermia. This space-clamped model is well suited to the simulation of superficially located nerves stimulated with relatively large electrodes, and has been used to model the changes in axonal excitability in tetrodotoxin poisoning (Kiernan et al. 2005), porphyria (Lin et al. 2008), and following stroke and multiple sclerosis (Jankelowitz et al. 2007; Ng et al. 2008). The model consists of nodal and internodal compartments, which are linked by the Barrett–Barrett conductance, representing paranodal pathways through and under the myelin sheath (Barrett & Barrett, 1982; Mierzwa et al. 2010). To model the effects of focal hyperthermia on axonal excitability, the ion channel rate constants were increased according to their Q10 values (Hodgkin & Huxley, 1952a,b; Frankenhaeuser & Moore, 1963; Guttman, 1971; Hart, 1983; Schwarz & Eikhof, 1987): Na+ activation ‘m’, 2.2; Na+ inactivation ‘h’, 2.9; K+ (fast and slow) and HCN channels, 3. As the maximal conductances are affected by changes in passive diffusion, a Q10 of 1.4 was also applied to all voltage-gated ion channel (Hodgkin & Huxley, 1952a,b; Moore, 1958; Hart, 1983; Hille, 1992), leak and Barrett–Barrett conductances. The effect of temperature on capacitance is negligible (Hodgkin et al. 1952; Tasaki, 1955; Taylor & Chandler, 1962; Palti & Adelman, 1969), and capacitances in the model were therefore not adjusted for hyperthermia.
Statistical analysis
Group data are reported as mean (and SEM), except for measures that are better plotted on logarithmic scales to be normally distributed. Here geometric means were calculated and reported as mean [geometric SEM (as a factor)] (Kiernan et al. 2000). The mean values for the two groups (normal and hyperthermic) were compared with Student's t tests for paired data, and are reported in Table 1. The P values of related measures (as indicated by the parentheses in Table 1) were adjusted for multiple comparisons using the Holm-Bonferroni method, and the corrected values are reported in the text and Tables.
Table 1.
Axonal excitability measures at normal and hyperthermic temperatures
 |
For statistical analysis the extent of superexcitability was measured as the area below the x-axis following the relative refractory period. Similarly, the extent of subexcitability (which follows superexcitability) was measured as the area above the x-axis.
Results
CMAP recruitment
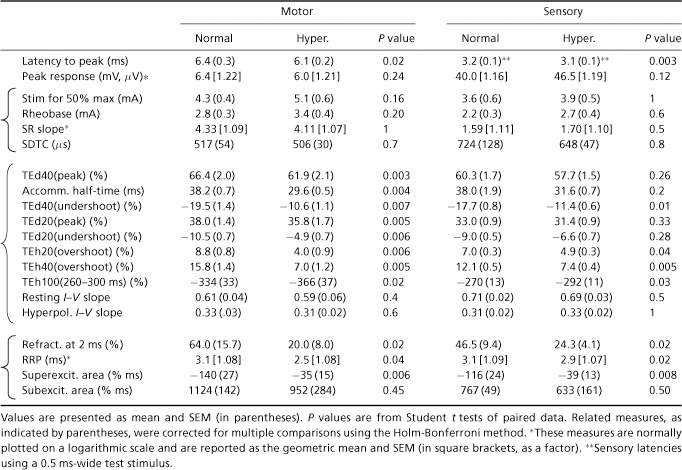
The latency from the onset of the test stimulus to the peak of the CMAP was 0.36 ms shorter when hyperthermic (P < 0.02), though the amplitude of the peak CMAP recorded at the normal and hyperthermic temperatures was not significantly different (P= 0.24; Fig. 2A and Table 1). The stimulus current required for a half-maximal response and rheobase were both higher during hyperthermia but not significantly so after adjustment for multiple comparisons (respectively, 5.1 ± 0.6 mA (hyperthermia) and 4.3 ± 0.4 mA (normal) and 3.4 ± 0.4 mA (hyperthermia) and 2.8 ± 0.3 mA (normal); see Fig. 2A (large circles), and B and Table 1). The stimulus–response slopes and strength–duration time constants were also not significantly different (see Fig. 2C and Table 1).
Figure 2. Stimulus–response relationship during hyperthermia.
Group data (n= 6). A, recruitment of CMAP (measured baseline to peak) by gradually increasing stimulus intensity (normal temperature, open circles; hyperthermia, filled circles). B, stimulus required to recruit a half-maximal response at normal and hyperthermic temperatures. The continuous and dashed horizontal lines indicate the mean and standard errors (the mean half-maximal stimulus is also indicated by the large circles around the symbols in A.). C, normalized stimulus–response relationship recorded at normal and hyperthermic temperatures (open and closed circles, respectively). The peak response is normalized to a percentage of maximum and the stimulus is normalized to the stimulus current required for a half-maximal potential, before being averaged.
Threshold electrotonus and current–threshold relationship in motor axons
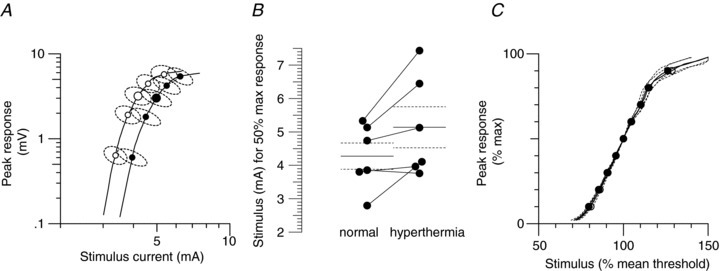
Accommodation to depolarizing currents was more rapid during hyperthermia than at normal temperatures (see Fig. 3A), such that the maximal threshold reduction was less (with +40%: 61.9 ± 2.1% and 66.4 ± 2%, respectively; with +20%: 35.8 ± 1.7% and 38 ± 1.4%, respectively; see Table 1) and accommodation half-time was shorter (the time to the midpoint between the peak threshold reduction and the average level at the end of the polarization; hyperthermia, 29.6 ms; normal, 38.2 ms; see Table 1). The undershoot after the 100 ms depolarizing current ended is due to the slow de-activation of accommodative conductances active at the end of the depolarization; the peak undershoot occurred earlier but was smaller for both levels of polarization during hyperthermia (P≤ 0.007; see Table 1).
Figure 3. Motor axon excitability during focal hyperthermia.
Group data (n= 6; mean ± SEM). Measurements of axonal excitability were made at normal resting temperature (open circles) and during focal hyperthermia (filled circles). A, threshold electrotonus for a depolarizing conditioning current of +40% of the unconditioned threshold. B, threshold electrotonus for hyperpolarizing conditioning currents of −40, −70 and −100%. C, current–threshold relationship for 200 ms-long conditioning currents from +50% (depolarizing) to −100% (hyperpolarizing) of the control threshold. D, the recovery of excitability following a supramaximal discharge. The dotted line represents the recovery of excitability during hyperthermia when normalized to the unconditioned threshold at ‘normal’ temperature (see Discussion). The corresponding data points in threshold electrotonus (B) and the current–threshold relationship (C) are highlighted by the shaded ellipses and rectangles for hyperpolarization by −70 and −100%, respectively.
In contrast, significant differences in the accommodation to hyperpolarization were only apparent for the strongest hyperpolarizing current and during the hyperpolarizing overshoots (see Fig. 3B and Table 1). Unexpectedly, the accommodation appeared slower during hyperthermia, and excitability was reduced at the end of the long −100% hyperpolarizing currents (i.e. at the intervals between 260 and 300 ms, ‘TEh100(260–300 ms)’; P= 0.02).
Figure 3C shows the current–threshold relationship (a threshold analogue of current/voltage, ‘I–V’, plots), measured at the end of a 200 ms-long conditioning current. Hyperthermia produced no significant differences in the resting and hyperpolarizing I–V slopes (P= 0.4 and 0.6, respectively; see Table 1).
The fact that the hyperpolarizing portion of the current–threshold relationship appears unaffected by hyperthermia is consistent with the equivalent data in threshold electrotonus (see ellipses and rectangles in Figs 3B and C and 6B and C), and probably reflects opposing effects on HCN channels which cancel each other out for all but the strongest and longest hyperpolarization (see Modelling and Discussion sections).
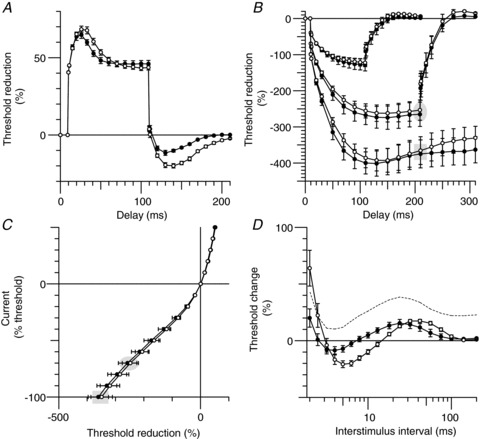
Figure 6. Sensory axon excitability during hyperthermia.
Group data (n= 6, mean ± SEM), open circles represent measures at normal temperatures and filled circles during hyperthermia. A, depolarizing threshold electrotonus with a conditioning current of +40% of the control stimulus. B, hyperpolarizing threshold electrotonus with currents −40, −70 and −100% of the control stimulus. C, current–threshold relationship tested at the end of 200 ms long conditioning currents (−100% to 50% of control threshold). D, recovery cycle after supramaximal stimulation. The corresponding data points in threshold electrotonus (B) and the current–threshold relationship (C) are highlighted by the shaded ellipses and rectangles for hyperpolarization by −70 and −100%, respectively.
The recovery cycle of motor axons
The hyperthermia-induced changes in excitability of motor axons were particularly obvious in the recovery after a conditioning discharge (Fig. 3D and Table 1). The recordings during focal hyperthermia were characterized by less refractoriness at 2 ms (20 ± 8% and 64 ± 16%), a shorter relative refractory period (2.5 ± 0.2 ms and 3.1 ± 0.2 ms), with earlier superexcitability of reduced area (−35 ± 15% ms and −140 ± 27% ms). Similarly, subexcitability occurred earlier during hyperthermia but its area was not significantly different (P= 0.45).
Reproducibility
In order to demonstrate the reproducibility within single subjects, all measurements for motor axons were performed five times on separate days in two of the six subjects (subjects 1 and 2; the mean data for each of these two subjects were used in the group data averages). The individual recordings and mean data for one subject (subject 1) are shown in Fig. 4. The results were quite reproducible and comparable to the group data. As is usually the case (Tomlinson et al. 2010; Howells et al. 2012), the greatest variability was seen for the strongest hyperpolarizing currents in threshold electrotonus (which is not surprising considering the number of possible modulators of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels). To explore the source of variability further, repeated measurements were made five times in the same experiment in each of two subjects (subjects 2 and 3). Fig. 5 compares the variability within an experiment, within a subject and between all subjects, and these data are summarized in Table 2. The data shows that the variability within an experiment is considerably lower than within the same subject on different days. We expected an increase in excitability during hyperthermia. Based on the within-experiment variability a power analysis using TEh100(260–300 ms) indicated that with six subjects we were sufficiently powered (80%) to detect a 7.9% increase in excitability at α= 0.05. The actual change during hyperthermia was much larger (32%), and in the opposite direction to that hypothesized. The variability within an experiment is the relevant measure for the present studies because the normal and hyperthermic studies were done within the same experiment.
Figure 4. Reproducibility of hyperthermia induced changes.
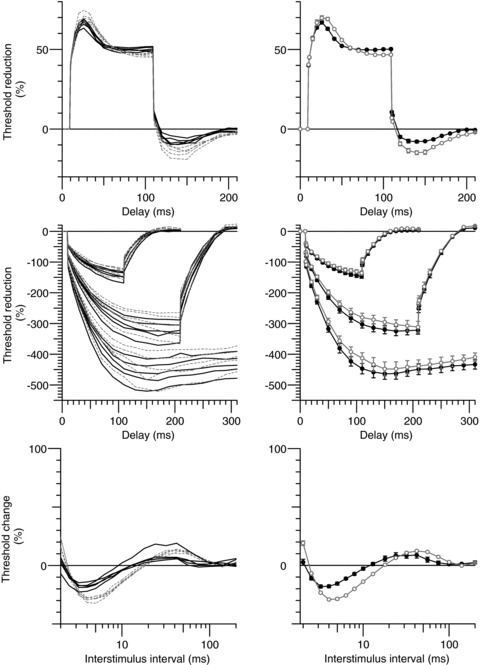
Left column, repeated measurements of axonal excitability in subject 1 on five separate occasions (normal temperature, dashed lines; hyperthermia, continuous lines). Right column, mean data (normal temperature, open circles; hyperthermia, filled circles; mean ± SEM). Top row, depolarizing threshold electrotonus (+40%; as in Fig. 2A). Middle row, hyperpolarizing threshold electrotonus (−40, −70 and −100%; as in Fig. 2B). Bottom row, recovery cycle.
Figure 5. Reproducibility of excitability measures within an experiment.
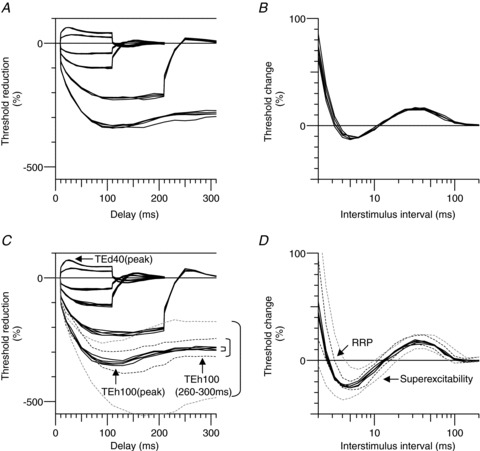
Five repeated measurements within the same experiment for subject 3 for threshold electrotonus (A) and the recovery cycle (B). Five repeated measurements (continuous lines) in subject 2 for threshold electrotonus (C) and the recovery cycle (D). The brackets in C highlight differences in the variability for the strongest hyperpolarization. The smallest bracket shows the within-experiment variability, the next largest bracket and the inner dashed lines indicate the within-subject variability (95% confidence limits) on different days, while the largest bracket and the outer dashed lines show the 95% confidence limits for all subjects. Note that the within-experiment variability is the measure relevant to the present studies.
Table 2.
Variability of excitability measures in motor axons
| Same subject, within experiment | Same subject, different days | Between subjects (n= 6) | |
|---|---|---|---|
| TEd40(peak) (%) | 0.8 (1.1%) | 1.4 (2.2%) | 4.8 (7.3%) |
| TEh100(peak) (%) | 8.2 (2.4%) | 23 (6.8%) | 79 (19.8%) |
| TEh100(260–300 ms) (%) | 5.5 (1.9%) | 18 (6.3%) | 81 (24.1%) |
| RRP (ms; geometric SD)* | 1.026 | 1.06 | 1.198 |
| Superexcitability (%) | 1.2 (5.2%) | 1.5 (6.9%) | 7.05 (35.8%) |
| Superexcit. area (% ms) | −12.5 (8.8%) | −16.4 (12.1%) | −65.4 (46.9%) |
Figures are for the standard deviation and the coefficient of variation is in parentheses. *RRP – geometric standard deviation (as a factor).
Excitability of sensory axons
The hyperthermia-induced changes in sensory excitability are shown in Fig. 6 and are summarized in Table 1. These changes were qualitatively similar to those described above for motor axons with, in the recovery cycle (Fig. 6D), reduced refractoriness (24 ± 4%vs. 46 ± 9%), a shorter relative refractory period (2.9 ± 0.2 ms vs. 3.2 ± 0.3 ms), reduced superexcitability (−8.3 ± 2.4%vs. −17.6 ± 2.8%) and, in threshold electrotonus, reduced undershoot (Fig. 6A; −11.4 ± 0.6%vs. −17.7 ± 0.8%) and overshoot (Fig. 6B; 7.4 ± 0.4%vs. 12.1 ± 0.5%). Hyperthermia had little effect on hyperpolarizing threshold electrotonus and the current–threshold relationship for sensory axons, except at the end of the strongest hyperpolarization of threshold electrotonus (Fig. 6B). The other measures showed trends in the same direction as with motor axons, but the changes were smaller and not significant.
Polarized recovery cycles
To examine the extent to which changes in membrane potential could contribute to the changes in the recovery cycle (the measure most distorted during hyperthermia), we made recordings while injecting depolarizing and hyperpolarizing currents to change membrane potential (Fig. 7). The effects of changes in temperature are generally explained by changes in Na+ currents, but it is probable that an enhanced Na+ current would produce a left-ward shift in the stimulus–response curve, rather than the trend for a shift to the right shown in Fig. 2A. The reduction in refractoriness could occur with hyperpolarization of RMP, but the reduced superexcitability and early accommodation in depolarizing threshold electrotonus favour depolarization. During depolarization (+30% threshold, approximately +1.2 mA for motor axons and 0.5 mA for sensory axons; triangles in Fig. 7) superexcitability was abolished in motor axons (Fig. 7A), and in five of the six subjects for sensory axons (Fig. 7B). The peak of subexcitability occurred earlier, as seen in hyperthermia (P= 0.002 for motor axons and P= 0.04 for sensory), but unlike hyperthermia, the extent of subexcitability was greater. These findings are similar to those of Kiernan & Bostock (2000). During hyperpolarization (–30% threshold, approximately −1.2 mA for motor axons and −0.5 mA for motor axons; squares in Fig. 7), the peak of superexcitability was larger and occurred at much the same conditioning-test intervals, while subexcitability was smaller and later. Hyperpolarization removes resting activation of Ks channels revealing recovery cycles that better represent the true extent of the decay of current through the Barrett–Barrett pathways (compare squares to circles in Fig. 7).
Figure 7. Polarized recovery cycles.
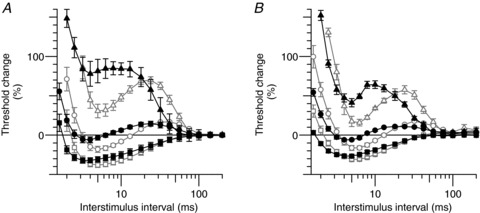
Group data (n= 6; mean ± SEM). Recovery from activation during polarization (depolarization, triangles; hyperpolarization, squares) and without polarization (circles) at normal temperature (open symbols) and during hyperthermia (filled symbols). A, motor axons. B, sensory axons. The recovery cycle was measured 200 ms after the onset of a long conditioning pulse of strength 30% (depolarization; triangles) and −30% (hyperpolarization; squares) of the unconditioned test stimulus, respectively.
Data from recordings at the normal temperature and during hyperthermia (open and filled symbols, respectively, in Fig. 7) demonstrate that changes in membrane potential could not restore the normal recovery cycle during hyperthermia.
Modelling
The Bostock model as modified by Howells and colleagues (2012) was used to interpret the hyperthermia-induced changes in motor and sensory axons. The average increase in skin surface temperature was 8.9°C, but the effects of hyperthermia were better modelled by an increase in nerve temperature of 6°C, the difference being an intuitively reasonable skin–nerve gradient during external heating. The rate constants of the gating processes in this model were increased according to the following Q10 values (Na+ activation, 2.2; Na+ inactivation, 2.9; fast K+, slow K+, Ih activation, 3) and a Q10 of 1.4 was applied to all of the maximal conductances (i.e. all voltage-gated ion channels, leak conductances and the Barrett–Barrett pathways through and under the myelin sheath).
Motor axons
Increasing the temperature by 6°C lowered the potassium equilibrium potential (EK; −94.8 mV from −93 mV), and this would have resulted in hyperpolarization of RMP by 1 mV. In combination with the increased conductances (particularly GKs), the resultant hyperpolarization of RMP was 1.7 mV.
The combined effect of hyperpolarization of EK and RMP, faster gating and larger conductances modelled well most of the hyperthermia-induced changes in the recovery cycle, accurately replicating the earlier relatively refractory period, earlier and reduced superexcitability and earlier (though reduced) late subexcitability. However the lesser inward rectification during both strong and long hyperpolarization in threshold electrotonus and the current-threshold relationship could not be reproduced by these changes alone. The most parsimonious explanation for this unexpected and seemingly paradoxical finding is a reduction in Ih. Accordingly all parameters associated with Ih were varied, and the best fit was obtained with hyperpolarization of the half-activation voltage of Ih (by −11 mV). This additional change in gating has precedent, and its extent compares favourably with the reported temperature sensitivity of the voltage activation of the cardiac isoform (If) in sheep Purkinje fibres (Hart, 1983; −8 mV between 27.5 and 36.4°C; see Fig. 8A in that paper).
Figure 8. Mathematical model of the hyperthermia-induced changes in excitability of motor axons.
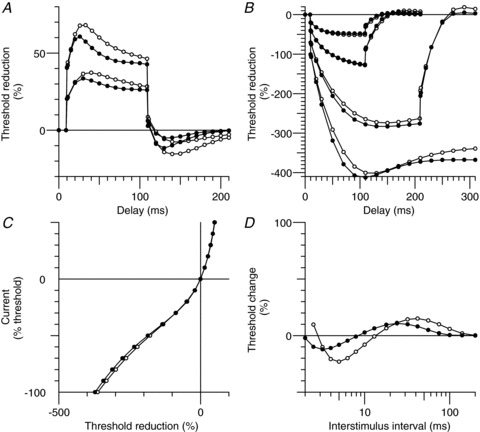
The open circles indicate the Bostock motor axon model at normal temperature, and the modelled changes during hyperthermia are shown as closed circles. Hyperthermia was modelled by an increase in temperature of 6°C (with the activation kinetics and maximum conductances increased according to their respective Q10 values; see the Methods section), a hyperpolarization of the half-activation voltage of Ih (11 mV) and a small additional depolarizing current at the internode of 20 pA. A, depolarizing threshold electrotonus for +40% and +20% of the control threshold. B, hyperpolarizing threshold electrotonus (−20%, −40%, −70% and −100%). C, current–threshold relationship. D, the recovery of excitability after a supramaximal stimulus.
In addition, a constant (DC) depolarizing current of 20 pA at the internode improved the fit to hyperpolarizing threshold electrotonus, the current–threshold relationship and the recovery cycle during hyperthermia in motor axons (see Fig. 8). This depolarizing influence could reflect greater activity of slow internodally located HCN isoforms (see Howells et al. 2012), and its extent may actually be greater than 20 pA because Na+–K+ pump activity probably increases, at least transiently, with elevated temperatures. It must be acknowledged however, that this constant depolarizing current is an approximation and, if indeed it does represent slow HCN isoforms, then it is also true that the current through a voltage-gated ion channel should diminish as the membrane potential approaches the equilibrium potential for HCN channels. The modelling suggests that the electrochemical driving force for HCN channels varies threefold (and as a consequence the current through slowly gated channels) during threshold electrotonus and is ∼−23 mV for the 40% depolarization and ∼−77 mV at the peak of the strongest hyperpolarization (100%). This probably explains why a greater separation of the modelled normal and hyperthermic excitability exists at the end of the depolarizing threshold electrotonus (90–100 ms) than for the observed recordings (compare Figs 8A and 9A with Figs 3A and 6A, respectively). This apparent discrepancy provides further support for the possibility of slower isoforms of HCN channels on motor axons.
Figure 9. Mathematical model of the hyperthermia-induced changes in excitability of sensory axons.

The open circles indicate the Bostock model as modified for sensory axons (Howells et al. 2012), and the modelled changes during hyperthermia are shown as closed circles. Hyperthermia was modelled by an increase in temperature of 6°C (with the activation kinetics and maximum conductances increased according to their Q10 values; see Methods section), a hyperpolarization of the half-activation voltage of Ih (7 mV) and a small additional depolarizing current of 30 pA. A, depolarizing threshold electrotonus for +40% and +20% of the control threshold. B, hyperpolarizing threshold electrotonus (−20%, −40%, −70% and −100%). C, current–threshold relationship. D, the recovery of excitability after a supramaximal stimulus.
Sensory axons
Similarly the hyperthermia-induced changes in excitability were modelled well by an increase in temperature of 6°C, increased gating kinetics and maximal conductances (according to the Q10 values), hyperpolarization of the voltage dependence of Ih (by −7 mV) and a constant (DC) current of 30 pA (see Fig. 9). It is noteworthy that, when normothermic, the voltage dependence of Ih in motor axons is likely to be some 13 mV more hyperpolarized than that of sensory axons (Howells et al. 2012), and this discrepancy would increase to ∼17 mV during hyperthermia.
The effect of hyperthermia on inward rectification in sensory axons was only apparent during strong and long hyperpolarization. The modelled hyperpolarization of the voltage dependence of Ih was therefore nearly perfectly balanced with any increase in Ih due to increased diffusion or faster kinetics.
Discussion
We have studied for the first time the effects of focal hyperthermia on the excitability of human peripheral myelinated nerve. There are studies of nerve conduction during hyperthermia in human subjects (Rutkove et al. 1997), but studies of the effect of temperature on the excitability of human axons have focused on either cooling (Burke et al. 1999) or small deviations around normal temperature (Kiernan et al. 2001a; Maurer et al. 2010), while, in the cat, there have been comparable studies of axonal excitability only during cooling (Moldovan & Krarup, 2004).
The action potential of mammalian myelinated axons can be modelled adequately incorporating only Na+ channels into the model (Schwarz et al. 1995), and the traditional view has been that the predominant effect of changes in temperature is likely to be due to changes in ion channel kinetics (Hodgkin & Huxley, 1952b; Schwarz & Eikhof, 1987; Burke et al. 1999; Kiernan et al. 2001a), and in particular those of Na+ channels. This may be so for small changes in axonal temperature. While changes in Na+ currents may well be the major factor shaping the action potential when temperature is close to normal, we suggest below that other currents exert a greater influence with marked increases in temperature. We suggest that these changes will reduce the safety margin for action potential generation and propagation, and will do so more for motor axons than sensory axons, impairing conduction and leading to fatigue in normal subjects and, particularly, in patients with diseases affecting central and peripheral axons.
Mechanisms underlying hyperthermia-induced changes in excitability
Resting membrane potential
Kiernan & Bostock (2000) found the sensitivity of the threshold for the half-maximal CMAP to polarization to be ∼8% per millivolt, which when applied to the small increase in threshold during hyperthermia would suggest a hyperpolarization of 2.2 mV. Applying the same logic to the sensitivity of rheobase to changes in RMP gives a hyperpolarization of 2.1 mV. There is a dearth of literature on Na+–K+ pump functions in axons during heating, but a temperature-dependent increase in Na+–K+ pump activity could account for some of this hyperpolarization. Nevertheless our modelling suggests that the effects of temperature on the equilibrium potentials (in particular EK) and on diffusion through channels open at rest are sufficient to account for 1.7 mV of hyperpolarization.
The hyperpolarization of RMP suggested by the modelling findings needs to be reconciled with the lack of a significant hyperpolarizing change in the threshold, rheobase and shift in the SR curve. These findings are not in conflict: the modelling suggested an offsetting tonic depolarization (which could be due to internodally located slow HCN isoforms) which would rein in the hyperpolarization of RMP to ∼0.6 mV. In any case, hyperpolarization by <2 mV is small, less than the range of holding potentials that result in inactivation of 30% Na+ channels (a surrogate for RMP) in experiments in vitro (e.g. −84 and −86 mV in studies on human sensory axons; Schwarz et al. 1995) and well within the likely difference in RMP for human sensory and motor axons (3–4 mV; Howells et al. 2012). A larger sample size may have yielded a statistically significant increase in the unconditioned threshold during hyperthermia, but the difference is likely to be small. The inability of polarization to restore normal recovery cycles during hyperthermia supports the view that a change in RMP was not the primary driver of the observed excitability changes during hyperthermia. The effects of temperature on channel function are more likely to have a greater impact on excitability and the safety margin (see below) than the small change in RMP.
The recovery cycle
The changes in axonal excitability during hyperthermia affected particularly the recovery cycle, possibly reflecting a disturbance to the finely tuned mechanisms underlying action potential generation and propagation.
Refractoriness
The relative refractory period was shorter and refractoriness was reduced during hyperthermia, findings consistent with studies during cooling (where the opposite changes occur; Burke et al. 1999; Moldovan & Krarup, 2004; Maurer et al. 2010) or around ‘normal’ temperatures (Kiernan et al. 2001a). Diffusion of Na+ ions would be greater and activation of Na+ channels would be faster with heating (Fig. 10). These changes would lead to an earlier onset of inactivation, accentuating any changes due to the kinetics of the inactivation gate. The changes in refractoriness are consistent with faster recovery from inactivation of Na+ channels. However temperature-induced changes in K+ currents would also affect refractoriness, much as they can superexcitability (see below).
Figure 10. Modelled action potential during hyperthermia.
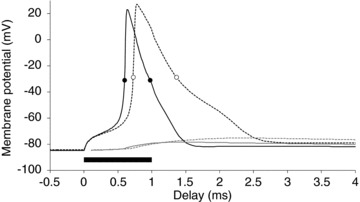
Nodal (black) and internodal (grey) membrane potentials during hyperthermia (continuous lines) and at normal temperature (dashed). Measurements of the half-maximal action potential width were made and are indicated by circles for hyperthermia (0.38 ms; filled) and normal temperature (0.64 ms; open). The filled bar depicts the stimulus duration.
Superexcitability
Superexcitability decreased during hyperthermia, a change unexpected from the earlier studies on human axons. Indeed, when the recovery cycle is normalized to the unconditioned threshold at ‘normal’ temperatures (dotted lines in Fig. 3D), there is only a period of ‘relative’ superexcitability with a peak that is actually an increase in threshold by +10.3 ± 8.9%. Increased diffusion through the paranodal seal during hyperthermia would serve to increase superexcitability. Decreased superexcitability could be due to a narrower action potential (Fig. 10), resulting in reduced charging of the capacitance of the internodal axolemma (Mitrovićet al. 1993; Honmou et al. 1994; Vogel & Schwarz, 1995; Kiernan et al. 2001b; McIntyre et al. 2002; Howells et al. 2012), but our modelling suggests that enhanced K+ currents are more likely to be responsible for the observed changes in superexcitability. By themselves increased diffusion through Na+ channels (transient and persistent) and their faster kinetics would result in a wider action potential, but increases in the potassium currents would provide a dampening effect on the depolarizing after-potential (David et al. 1995), counteracting any increases in the Na+ current. Fig. 10 shows the modelled membrane potential changes for a motor axon during hyperthermia in response to a 1 ms test stimulus. The action potential half-width (measured halfway between RMP and the peak of the action potential) decreased from 0.64 ms to 0.38 ms for normal and hyperthermic temperatures, respectively, corresponding to a Q10 of 2.4.
Late subexcitability
The peak of late subexcitability was earlier but its area was not significantly different at hyperthermic temperatures. Late subexcitability reflects after-hyperpolarization due to the decaying activation of Ks channels (Baker et al. 1987; Bowe et al. 1987; Stys & Waxman, 1994; David et al. 1995; Bostock et al. 1998). The earlier peak during hyperthermia is probably due to faster Ks kinetics. The lack of difference in the magnitude of late subexcitability may be due to opposing influences on Ks currents: hyperthermia will increase the Ks conductance, but this would hyperpolarize RMP, resulting in a smaller electrochemical gradient for K+ currents.
Accommodation to long-lasting depolarization
There was no significant difference in the excitability during the first 10 ms of depolarizing threshold electrotonus during hyperthermia and at normal temperatures (Fig. 3A), as would be expected if hyperthermia led to faster activation of Kf channels. However Baker and colleagues (1987) have shown that the fast outward rectification due to Kf currents would occur within the first millisecond at 30°C (see Fig. 5A in Baker et al. 1987) and presumably faster at warmer temperatures. Additionally, a similar increase in the maximal conductances of Na+ and Kf channels during hyperthermia could cancel any effect during early depolarizing threshold electrotonus. Increased diffusion through, and faster kinetics of, slow potassium (Ks) channels during hyperthermia can be seen in the faster accommodation to depolarizing currents, sufficient to limit the peak depolarization. The reduced peak depolarization would result in a smaller Ks current and correspondingly a smaller threshold undershoot when the polarizing current ends (Schwarz et al. 2006). The primary effect of hyperthermia is on the kinetics of Ks, and the current will be enhanced only if the overall depolarization is the same or greater.
The hyperpolarization-activated cation conductance, Ih
Unexpectedly, inward rectification was reduced during hyperthermia (Fig. 3B, Table 1). If the diffusion through Ih channels and their activation increase with temperature, one would expect greater inward rectification with heating. The reverse was seen. Our modelling suggests that this apparent paradox can be resolved by hyperpolarization of the voltage dependence of Ih channels and the addition of a small steady depolarizing current. The effects of temperature on the activation kinetics of voltage-gated ion channels have been well studied, but less is known about the effect of temperature on their voltage dependence. However, Hart (1983) reported a similar temperature dependence of the half-activation voltage of the cardiac isoform, If, in sheep Purkinje fibres. The voltage dependence of other channels could be similarly altered (Thomas et al. 2009), but the present study provides no compelling reasons for postulating such a change. The temperature dependence of HCN channels may involve the voltage sensor directly or indirectly via allosteric activation (Pian et al. 2006, 2007; Börjesson & Elinder, 2008). Some regulators of HCN channels can shift the voltage dependence of HCN channels by up to 50 mV (reviewed in Biel et al. 2009), and it is likely that the potential modulation of HCN channels is greater than occurs with other channels.
The modelling suggests that the reduced inward rectification seen in Figs 3 and 6 may be accompanied by greater activity of slow internodally located isoforms of Ih. This implies, firstly, that human axons are likely to express more than one HCN isoform, secondly, that current techniques document the properties of only the faster isoform(s) and, thirdly, that hyperthermia may have different effects on different isoforms. We are aware of no data to support the latter speculation.
The role of K+ channels in hyperthermia
Changes in Ks currents
Changes in Ks currents appear to be a major factor affecting excitability during hyperthermia, responsible for faster accommodation to depolarizing currents, the reduced super- and earlier late sub-excitability and possibly contributing to the shorter RRP, as discussed above. Sittl and colleagues (2010) tested axonal excitability in myelinated axons of rat sural nerve with the Kv7 (Ks) channel opener flupirtine, and reported increased threshold, reduced refractoriness and increased superexcitability, all consistent with hyperpolarization of RMP. In contrast the present study found reduced superexcitability, probably due to faster activation kinetics of both Kf and Ks channels.
Greater access to Kf channels at the paranode during hyperthermia
Increased diffusion through the paranodal seal to the periaxonal space could have an effect on excitability analogous to that of paranodal demyelination or a loosening of the myelin sheath attachment (Cappelen-Smith et al. 2001; Nodera et al. 2004; Stephanova & Daskalova, 2005). If so this could jeopardize action potential generation, thereby predisposing to conduction block, much as seen in those clinical studies.
Variability of hyperpolarizing threshold electrotonus measures
Tomlinson and colleagues (2010) examined the inter- and intra-subject variability of measures of hyperpolarizing threshold electrotonus and found that despite these measures appearing variable, that they are actually characteristic of an individual. The data in the present study suggests that the variability of hyperpolarizing threshold electrotonus measures is greater in experiments performed on different days than those in the same session. This is perhaps unsurprising given the number of modulators of HCN channel function, but it does have implications for studies which aim to compare hyperpolarizing threshold electrotonus on different occasions.
Clinical implications
The change in excitability in both sensory and motor axons during hyperthermia is the product of several mechanisms, all of which have implications for the security of conduction.
Safety margin and conduction block
Any reduction in the safety margin for action potential generation during hyperthermia may be of functional significance in healthy motor axons (see section on Fatigue below), but is likely to be even more important for already impaired axons. Our results suggest four mechanisms for a deterioration of the safety margin during hyperthermia. First, at rest, motor axons are probably hyperpolarized 3–4 mV relative to sensory axons (Howells et al. 2012), and even slight hyperpolarization during hyperthermia would induce conduction block more readily in motor axons than sensory. Second, the voltage dependence of HCN channels in motor axons is also more hyperpolarized than in sensory axons (Howells et al. 2012), and the further hyperpolarization during hyperthermia, would render them less able to counter activity-dependent hyperpolarization, making motor axons even more vulnerable to conduction block. Third, our modelling suggests that faster channel kinetics during hyperthermia will result in a narrower action potential (Fig. 10), such that the time integral of the driving current for the action potential would be reduced. Finally the narrow action potential would result in reduced charging of the internodal axolemma (Fig. 10), affecting superexcitability and the ability to transmit high firing frequencies (see below).
Fatigue
Both central and peripheral mechanisms contribute to the development of fatigue in healthy subjects (Gandevia, 2001) and fatigue can arise from defects at a number of levels in those affected by neuromuscular disease or febrile illness (Friman et al. 1977; Thomas & Zijdewind, 2006).
Todd and colleagues (2005) proposed that during hyperthermia achieving maximal force could impose a greater impulse load because a higher motor unit firing frequency would be required to achieve twitch fusion. This would require greater inward rectification to counter the greater activity-dependent hyperpolarization. However the ability to mobilize at least some HCN isoforms underlying Ih appears to be impaired by hyperthermia. As noted above, the reduction in superexcitability seen in the current study may also contribute to the failure of motor axons to achieve transient high firing rates of 50–200 Hz during hyperthermia. This could limit the ability of the motor axon to transmit action potentials at 50–200 Hz through branch points into nerve terminals (Adrian & Lucas, 1912; Swadlow et al. 1980; Zhou & Chiu, 2001, Debanne et al. 2011). Symptoms of fatigue in neuromuscular disease are often exacerbated by elevated temperatures. Exercise and heat are both recognized triggers of fatigue in sufferers of multiple sclerosis with already impaired safety margins (Uhthoff's sign; Guthrie & Nelson, 1995; Vucic et al. 2010), and exercise may accentuate weakness in chronic inflammatory demyelinating polyneuropathy and multifocal motor neuropathy due to the development of activity-dependent conduction block (Cappelen-Smith et al. 2000; Kaji et al. 2000). Even in amyotrophic lateral sclerosis with primarily lower motor neurone involvement, surviving motor axons may suffer from greater activity-dependent hyperpolarization because they are required to maintain an increased impulse load (Vucic et al. 2007).
Fever is a common feature of infectious diseases, such as influenza. Friman and colleagues (1977) reported increased ‘jitter’ in single-fibre EMG recordings in patients suffering from influenza. They attributed this to hyperthermia-induced blockade of neuromuscular transmission, but the current study provides support for the possibility of branch-point failure due to reduced excitability and a reduction in the safety margin for action potential propagation during hyperthermia.
Acknowledgments
The authors wish to thank Adelle Coster and Hans Coster for valuable discussions during the preparation of the manuscript.
Glossary
- CMAP
compound muscle action potential
- CSAP
compound sensory action potential
- EK
equilibrium potential for K+ ions
- HCN
hyperpolarization-activated cyclic nucleotide-gated channels
- Ih
hyperpolarization-activated cation current
- I-V
current-threshold relationship
- Kf
fast potassium
- Ks
slow potassium
- Q10
temperature coefficient over 10°C
- QT
charge–duration relationship
- RC
recovery cycle
- RMP
resting membrane potential
- RRP
relative refractory period
- SDTC
strength–duration time constant
- SR
stimulus–response relationship
- TE
threshold electrotonus
- TEd
depolarizing threshold electrotonus
- TEh
hyperpolarizing threshold electrotonus
- TEh100(260–300 ms)
average threshold reduction, measured at 260–300 ms after the onset of a 100% hyperpolarizing conditioning current
Additional information
Competing interests
None declared.
Author contributions
J.H., D.C. and D.B. contributed to the study design. J.H., D.C. and L.T. collected and analysed the data. J.H. and L.T. performed the mathematical modelling. All authors contributed to the interpretation of the data and the preparation of the manuscript and have approved the final version. The experiments were performed at The University of Sydney.
Funding
This research was supported by the National Health and Medical Research Council of Australia. D.C. was supported by a stipend from the Deutsche Gesellschaft für Klinische Neurophysiologie.
References
- Adrian ED, Lucas K. On the summation of propagated disturbances in nerve and muscle. J Physiol. 1912;44:68–124. doi: 10.1113/jphysiol.1912.sp001503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Bostock H, Grafe P, Martius P. Function and distribution of three types of rectifying channel in rat spinal root myelinated axons. J Physiol. 1987;383:45–67. doi: 10.1113/jphysiol.1987.sp016395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF, Barrett JN. Intracellular recording from vertebrate myelinated axons: mechanism of the depolarizing afterpotential. J Physiol. 1982;323:117–144. doi: 10.1113/jphysiol.1982.sp014064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmans J. The Physiology of Single Human Nerve Fibres. Belgium: University of Louvain, Vander; 1970. [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: From genes to function. Physiol Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- Börjesson SI, Elinder F. Structure, function, and modification of the voltage sensor in voltage-gated ion channels. Cell Biochem Biophys. 2008;52:149–174. doi: 10.1007/s12013-008-9032-5. [DOI] [PubMed] [Google Scholar]
- Bostock H, Baker M, Reid G. Changes in excitability of human motor axons underlying post-ischaemic fasciculations: evidence for two stable states. J Physiol. 1991;441:537–557. doi: 10.1113/jphysiol.1991.sp018766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of the human peripheral nerve. Muscle Nerve. 1998;21:137–158. doi: 10.1002/(sici)1097-4598(199802)21:2<137::aid-mus1>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Bowe CM, Kocsis JD, Waxman SG. The association of the supernormal period and the depolarizing afterpotential in myelinated frog and rat sciatic nerve. Neuroscience. 1987;21:585–593. doi: 10.1016/0306-4522(87)90144-8. [DOI] [PubMed] [Google Scholar]
- Buchthal F, Rosenfalck P. Evoked action potentials and conduction velocity in human sensory nerves. Brain Res. 1966;3:1–122. [Google Scholar]
- Burke D, Mogyoros I, Vagg R, Kiernan MC. Temperature dependence of excitability indices of human cutaneous afferents. Muscle Nerve. 1999;22:51–60. doi: 10.1002/(sici)1097-4598(199901)22:1<51::aid-mus9>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Cappelen-Smith C, Kuwabara S, Lin CS, Mogyoros I, Burke D. Activity-dependent hyperpolarization and conduction block in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. 2000;48:826–832. [PubMed] [Google Scholar]
- Cappelen-Smith C, Kuwabara S, Lin CS, Mogyoros I, Burke D. Membrane properties in chronic inflammatory demyelinating polyneuropathy. Brain. 2001;124:2439–2447. doi: 10.1093/brain/124.12.2439. [DOI] [PubMed] [Google Scholar]
- David G, Modney G, Scappaticci KA, Barrett JN, Barrett EF. Electrical and morphological factors influencing the depolarizing after-potential in rat and lizard myelinated axons. J Physiol. 1995;489:141–157. doi: 10.1113/jphysiol.1995.sp021037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FA, Schauf CL, Reed BJ, Kesler RL. Experimental studies of the effects of extrinsic factors on conduction in normal and demyelinated nerve. 1. Temperature. J Neuro Neurosurg Psychiatry. 1975;39:442–448. doi: 10.1136/jnnp.39.5.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Campanac E, Bialowas A, Carlier E, Alcaraz G. Axon physiology. Physiol Rev. 2011;91:555–602. doi: 10.1152/physrev.00048.2009. [DOI] [PubMed] [Google Scholar]
- Frankenhaeuser B, Moore LE. The effect of temperature on the sodium and potassium permeability changes in myelinated nerve fibres of Xenopus laevis. J Physiol. 1963;169:431–437. doi: 10.1113/jphysiol.1963.sp007269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friman G, Schiller HH, Schwartz MS. Distubed neuromuscular transmission in viral infections. Scand J Infect Dis. 1977;9:99–103. doi: 10.3109/inf.1977.9.issue-2.08. [DOI] [PubMed] [Google Scholar]
- Gandevia SC. Spinal and supraspinal factors in human muscle fatigue. Physiol Rev. 2001;81:1725–1789. doi: 10.1152/physrev.2001.81.4.1725. [DOI] [PubMed] [Google Scholar]
- Guthrie TC, Nelson DA. Influence of temperature on multiple sclerosis: critical review of mechanisms and research potential. J Neurol Sci. 1995;129:1–8. doi: 10.1016/0022-510x(94)00248-m. [DOI] [PubMed] [Google Scholar]
- Guttman R. The effect of temperature on the function of excitable membranes. In: Adelman WJ Jr, editor. Biophysics and Physiology of Excitable Membranes. New York: Van Nostrand Reinhold; 1971. pp. 320–336. [Google Scholar]
- Hart G. The kinetics and temperature dependence of the pace-maker current if in sheep Purkinje fibres. J Physiol. 1983;337:401–416. doi: 10.1113/jphysiol.1983.sp014631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 2nd edn. Sunderland, MA, USA: Sinauer Associates; 1992. [Google Scholar]
- Hodgkin AL, Katz B. The effect of temperature on the electrical activity of the giant axon of the squid. J Physiol. 1949;109:240–249. doi: 10.1113/jphysiol.1949.sp004388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol. 1952a;116:497–506. doi: 10.1113/jphysiol.1952.sp004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952b;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF, Katz B. Measurement of current–voltage relations in the membrane of the giant axon of Loligo. J Physiol. 1952;116:424–448. doi: 10.1113/jphysiol.1952.sp004716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honmou O, Utzschneider DA, Rizzo MA, Bowe CM, Waxman SG, Kocsis JD. Delayed depolarization and slow sodium currents in cutaneous afferents. J Neurophysiol. 1994;71:1627–1637. doi: 10.1152/jn.1994.71.5.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howells J, Trevillion L, Bostock H, Burke D. The voltage dependence of Ih in human myelinated axons. J Physiol. 2012;590:1625–1640. doi: 10.1113/jphysiol.2011.225573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankelowitz SK, Howells J, Burke D. Plasticity of inwardly rectifying conductances following a corticospinal lesion in human subjects. J Physiol. 2007;581:927–940. doi: 10.1113/jphysiol.2006.123661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji R, Bostock H, Kohara N, Murase N, Kimura J, Shibasaki H. Activity-dependent conduction block in multifocal motor neuropathy. Brain. 2000;123:1602–1611. doi: 10.1093/brain/123.8.1602. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Bostock H. Effects of membrane polarization and ischaemia on the excitability properties of human motor axons. Brain. 2000;123:2542–2551. doi: 10.1093/brain/123.12.2542. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Burke D, Andersen KJ, Bostock H. Multiple measures of axonal excitability: A new approach in clinical testing. Muscle Nerve. 2000;23:399–409. doi: 10.1002/(sici)1097-4598(200003)23:3<399::aid-mus12>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Cikurel K, Bostock H. Effects of temperature on the excitability properties of human motor axons. Brain. 2001a;124:816–825. doi: 10.1093/brain/124.4.816. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Isbister GK, Lin CS-Y, Burke D, Bostock H. Acute Tetrodotoxin-induced neurotoxicity after ingestion of puffer fish. Ann Neurol. 2005;57:339–348. doi: 10.1002/ana.20395. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Lin CS-Y, Andersen KV, Murray NMF, Bostock H. Clinical evaluation of excitability measures in sensory nerve. Muscle Nerve. 2001b;24:883–892. doi: 10.1002/mus.1085. [DOI] [PubMed] [Google Scholar]
- Krishnan AK, Lin CS-Y, Park SB, Kiernan MC. Axonal ion channels from bench to bedside: A translational neuroscience perspective. Prog Neurobiol. 2009;89:288–313. doi: 10.1016/j.pneurobio.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Lin CS-Y, Krishnan AV, Lee M-J, Zagami AS, You H-L, Yang C-C, Bostock H, Kiernan MC. Nerve function and dysfunction in acute intermittent porphyria. Brain. 2008;131:2510–2519. doi: 10.1093/brain/awn152. [DOI] [PubMed] [Google Scholar]
- Ludin HP, Beyeler F. Temperature dependence of normal sensory nerve action potentials. J Neurol. 1977;216:173–180. doi: 10.1007/BF00313618. [DOI] [PubMed] [Google Scholar]
- McIntyre CC, Richardson G, Grill WM. Modeling the excitability of mammalian nerve fibers: influence of afterpotentials on the recovery cycle. J Neurophysiol. 2002;87:995–1006. doi: 10.1152/jn.00353.2001. [DOI] [PubMed] [Google Scholar]
- Maurer K, Wacker J, Vastani N, Seifert B, Spahn DR. Changes in axonal excitability of primary sensory afferents with general anaesthesia in humans. Brit J Anaesth. 2010;105:648–656. doi: 10.1093/bja/aeq218. [DOI] [PubMed] [Google Scholar]
- Mierzwa A, Shroff S, Rosenbluth J. Permeability of the paranodal junction of myelinated nerve fibers. J Neurosci. 2010;30:15962–15968. doi: 10.1523/JNEUROSCI.4047-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrović N, Quasthoff S, Grafe P. Sodium channel inactivation kinetics of rat sensory and motor nerve fibres and their modulation by glutathione. Pflugers Arch. 1993;425:453–461. doi: 10.1007/BF00374872. [DOI] [PubMed] [Google Scholar]
- Mogyoros I, Kiernan MC, Burke D. Strength–duration properties of human peripheral nerve. Brain. 1996;119:439–447. doi: 10.1093/brain/119.2.439. [DOI] [PubMed] [Google Scholar]
- Moldovan M, Krarup C. Mechanisms of hyperpolarization in regenerated mature motor axons in cat. J Physiol. 2004;560:807–819. doi: 10.1113/jphysiol.2004.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JW. Temperature and drug effects on squid axon membrane ion conductances. Fed Proc. 1958;17:113. [Google Scholar]
- Ng K, Howells J, Pollard JD, Burke D. Up-regulation of slow K+ channels in peripheral motor axons: a transcriptional channelopathy in multiple sclerosis. Brain. 2008;131:3062–3071. doi: 10.1093/brain/awn180. [DOI] [PubMed] [Google Scholar]
- Nodera H, Bostock H, Kuwabara S, Sakamoto T, Asanuma K, Jia-Ying S, Ogawara K, Hattori N, Hirayama M, Sobue G, Kaji R. Nerve excitability properties in Charcot- Marie-Tooth disease type 1A. Brain. 2004;127:203–211. doi: 10.1093/brain/awh020. [DOI] [PubMed] [Google Scholar]
- Palti Y, Adelman WJ., Jr Measurement of axonal membrane conductances and capacity by means of a varying potential control voltage clamp. J Membrane Biol. 1969;1:431–458. doi: 10.1007/BF01869791. [DOI] [PubMed] [Google Scholar]
- Pian P, Bucchi A, DeCostanzo A, Robinson RB, Siegelbaum SA. Modulation of cyclic nucleotide-regulated HCN channels by PIP2 and receptors coupled to phospholipase C. Pflugers Arch. 2007;455:125–145. doi: 10.1007/s00424-007-0295-2. [DOI] [PubMed] [Google Scholar]
- Pian P, Bucchi A, Robinson RB, Siegelbaum SA. Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J Gen Physiol. 2006;128:593–604. doi: 10.1085/jgp.200609648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasminsky M. The effects of temperature on conduction in demyelinated single nerve fibers. Arch Neurol. 1973;28:287–292. doi: 10.1001/archneur.1973.00490230023001. [DOI] [PubMed] [Google Scholar]
- Rutkove SB. Effects of temperature on neuromuscular electrophysiology. Muscle Nerve. 2001;24:867–882. doi: 10.1002/mus.1084. [DOI] [PubMed] [Google Scholar]
- Rutkove SB, Kothari MJ, Shefner JM. Nerve, muscle, and neuromuscular junction electrophysiology at high temperature. Muscle Nerve. 1997;20:431–436. doi: 10.1002/(sici)1097-4598(199704)20:4<431::aid-mus5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Schauf CL, Davis FA. Impulse conduction in multiple sclerosis: a theoretical basis for modification by temperature and pharmacological agents. J Neurol Neurosurg Psychiatry. 1974;37:152–161. doi: 10.1136/jnnp.37.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JR, Eikhof G. Na currents and action potentials in rat myelinated nerve fibres at 20 and 37°C. Pflugers Arch. 1987;409:569–577. doi: 10.1007/BF00584655. [DOI] [PubMed] [Google Scholar]
- Schwarz JR, Glassmeier G, Cooper EC, Kao TC, Nodera H, Tabuena D, Kaji R, Bostock H. KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J Physiol. 2006;573:17–34. doi: 10.1113/jphysiol.2006.106815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JR, Reid G, Bostock H. Action potentials and membrane currents in the human node of Ranvier. Pflugers Arch. 1995;430:283–292. doi: 10.1007/BF00374660. [DOI] [PubMed] [Google Scholar]
- Sittl R, Carr RW, Schwarz JR, Grafe P. The Kv7 potassium channel activator flupirtine affects clinical excitability parameters of myelinated axons in isolated rat sural nerve. J Peripher Nerv Sys. 2010;15:63–72. doi: 10.1111/j.1529-8027.2010.00253.x. [DOI] [PubMed] [Google Scholar]
- Stephanova DI, Daskalova M. Differences in potentials and excitability properties in simulated cases of demyelinating neuropathies. Part II. Paranodal demyelination. Clin Neurophysiol. 2005;116:1159–1166. doi: 10.1016/j.clinph.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG. Activity-dependent modulation of excitability: implications for axonal physiology and pathophysiology. Muscle Nerve. 1994;17:969–974. doi: 10.1002/mus.880170902. [DOI] [PubMed] [Google Scholar]
- Swadlow HA, Kocsis JD, Waxman SG. Modulation of impulse conduction along the axonal tree. Ann Rev Biophys Bioeng. 1980;9:143–179. doi: 10.1146/annurev.bb.09.060180.001043. [DOI] [PubMed] [Google Scholar]
- Tasaki I. New measurements of the capacity and the resistance of the myelin sheath and the nodal membrane of the isolated frog nerve fibre. Am J Physiol. 1955;181:639–650. doi: 10.1152/ajplegacy.1955.181.3.639. [DOI] [PubMed] [Google Scholar]
- Taylor RE, Chandler WK. Effect of temperature on squid axon membrane capacity. Biophys Soc Abstr. 1962 TD1. [Google Scholar]
- Thomas CK, Zijdewind I. Fatigue of muscles weakened by death of motoneurons. Muscle Nerve. 2006;33:21–41. doi: 10.1002/mus.20400. [DOI] [PubMed] [Google Scholar]
- Thomas EA, Hawkins RJ, Richards KL, Xu R, Gazina EV, Petrou S. Heat opens axon initial segment sodium channels: A febrile seizure mechanism. Ann Neurol. 2009;66:219–226. doi: 10.1002/ana.21712. [DOI] [PubMed] [Google Scholar]
- Todd G, Butler JE, Taylor JL, Gandevia SC. Hyperthermia: a failure of the motor cortex and the muscle. J Physiol. 2005;563:621–631. doi: 10.1113/jphysiol.2004.077115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson S, Burke D, Hanna M, Koltzenburg M, Bostock H. In vivo assessment of HCN channel current (Ih) in human motor axons. Muscle Nerve. 2010;41:247–256. doi: 10.1002/mus.21482. [DOI] [PubMed] [Google Scholar]
- Vogel W, Schwarz JR. Voltage-clamp studies in axons: macroscopic and single-channel currents. In: Waxman SG, Kocsis JD, Stys PK, editors. The Axon. New York: Oxford University Press; 1995. pp. 257–280. [Google Scholar]
- Vucic S, Burke D, Kiernan MC. Fatigue in multiple sclerosis: mechanisms and management. Clin Neurophysiol. 2010;121:809–817. doi: 10.1016/j.clinph.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Vucic S, Krishnan AV, Kiernan MC. Fatigue and activity dependent changes in axonal excitability in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2007;78:1202–1208. doi: 10.1136/jnnp.2006.112078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Chiu SY. Computer model for action potential propagation through branch point in myelinated nerves. J Neurophysiol. 2001;85:197–210. doi: 10.1152/jn.2001.85.1.197. [DOI] [PubMed] [Google Scholar]