

Abstract
Inflammatory response has been strongly implicated in the pathogenesis of numerous diseases, including Alzheimer’s disease (AD). However, little is known about the molecular mechanisms initiating the generation of inflammatory molecules in the central nervous system, such as interleukine-1β (IL-1β). Previously we identified that palmitate (PA) can induce primary astrocytes to produce cytokines, causing AD-like changes in primary neurons. Here we investigated and identified that PA induced the activation of IPAF-ASC inflammasome in astrocytes leading to the maturation of IL-1β, thereby implicating not only pathogen-related factors can activate IPAF-ASC inflammasome. Moreover, downregulating IPAF (which was found to be regulated by CREB) in astrocytes through silencing to decrease IL-1β secretion from the astrocytes reduced the generation of amyloid β42 by primary neurons. Furthermore, the expression levels of IPAF and ASC were found significantly elevated in a subgroup of sporadic AD patients; suggesting an involvement of IPAF-ASC inflammasome in the inflammatory response associated with AD, and thus could be a potential therapeutic target for AD.
Keywords: Alzheimer’s disease, Fatty acid, inflammasome, IL-1β, IPAF
1. INTRODUCTION
Interleukin-1β (IL-1β) is a major proinflammatory cytokine that initiates and amplifies diverse cellular responses. Elevated IL-1β has been linked to diseases, such as type 2 diabetes, certain cancers, central nervous system (CNS) dysfunction and dementia (Mayeux, 2003; Vandanmagsar et al., 2011). Systemic inhibition of inflammation has been suggested to improve type 2 diabetes, enhance neuroprotection during brain injury, and delay the onset of Alzheimer’s disease (AD) (Hailer, 2008; Maedler et al., 2009; Weggen et al., 2001; Weiner and Frenkel, 2006). Blocking or neutralizing IL-1β reduced cognitive impairment and decreased AD-like pathological changes in AD mouse models (Gonzalez et al., 2009; Kitazawa et al., 2011). Similarly, knockout of IL-1β receptor antagonist in mice increased the neuronal damage induced by Aβ (Craft et al., 2005). These findings suggest that IL-1β could be involved in AD pathogenesis.
IL-1β is produced by various types of cells, including astrocytes (Liu et al., 2013; Tuppo and Arias, 2005; Wen et al., 2011). Astrocytes are resident brain cells that play crucial roles in synaptic formation and function, as well as neuronal cell survival and death. Upon activation of astrocytes by diverse factors, chemokines and cytokines are released, including IL-1β (Dowell et al., 2009; Parpura et al., 2012; Tuppo and Arias, 2005). As major contributors of IL-1β production in the CNS, activated astrocytes are involved in the pathogenesis of many neurodegenerative diseases, including AD (Tuppo and Arias, 2005). Elevated IL-1β levels have been detected in astrocytes of brains of AD patients and AD animal models (Halle et al., 2008; Hunter et al., 2012; Reilly et al., 2003). The inflammatory mediators secreted from activated astrocytes alter normal function of neurons and microglial cells, and trigger neurotoxicity, which further exacerbates the neurodegenerative pathology (Khandelwal et al., 2011).
IL-1β is derived from an inactive precursor, pro-IL-1β, by the multiprotein complex, inflammasome (Dinarello, 2009). Inflammasomes generally consist of nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) member and pro-caspase-1. Upon activation, caspase-1 cleaves pro-IL-1β resulting in the maturation of IL-1β. The composition of each inflammasome differs and is cell-type and stimulus-specific. The NLRP1 (NLR protein 1) and NLRP3 (NLR protein 3) inflammasome can be activated by pathogenic or danger-associated molecules (Masters et al., 2010). ASC (apoptosis-associated speck-like protein containing a CARD), the adaptor protein, is required for the activation of NLRP1 and NLRP3 inflammasome (Masters et al., 2010). The IPAF inflammasome (ice protease-activating factor, also known as NLRC4) has been demonstrated to play a role in host defense mechanism against pathogen-associated molecules (Jamilloux et al., 2013; Schroder and Tschopp, 2010). Although the ACS adaptor protein is not essential for the activation of the IPAF inflammasome, under certain conditions ASC is required for IPAF inflammasome activation (Martinon et al., 2009).
We previously reported that palmitate (PA) induced IL-1β maturation and secretion from primary rat astrocytes, which contributed to AD-like changes in primary rat neurons (Liu et al., 2013). PA is the most abundant saturated fatty acid in the diet. Studies have shown that high fat diet induces Aβ deposition and memory deficits in APP transgenic mice (Julien et al., 2010; Maesako et al., 2012a; Maesako et al., 2012b). PA and its metabolites, such as ceramides, regulate many gene expression and immunological pathways (Ajuwon and Spurlock, 2005). Elevated ceramide levels have been consistently reported in AD patients (Cutler et al., 2004; Geekiyanage and Chan, 2011; Han et al., 2002; He et al., 2010). Ceramide can be synthesized from serine and palmitoyl-CoA by serine palmitoyltransferase (SPT), the rate-limiting enzyme of de novo ceramide synthesis (Perry, 2000; Perry et al., 2000). We found that the effects of high fat diets, in particular PA, on AD-like pathology are mediated by ceramides through SPT (Geekiyanage and Chan, 2011; Geekiyanage et al., 2013; Liu et al., 2013; Patil et al., 2007). We reported that high fat diet containing 60% of the total fat as PA significantly increased ceramide and SPT levels in AD mouse model as compared with those fed a control chow diet and the elevated ceramide and SPT levels mediated the increase in Aβ levels (Geekiyanage and Chan, 2011; Geekiyanage et al., 2013). Inhibiting SPT in a high fat fed AD mouse model through subcutaneous administration of L-cylcoserine, an inhibitor of SPT, significantly down-regulated cortical Aβ42 and hyperphosphorlated tau levels (Geekiyanage and Chan, 2011; Geekiyanage et al., 2013).
To elucidate how PA, through ceramides, increased Aβ levels, we performed cell studies. We demonstrated that an upregulation of BACE1 and hyperphosphorylation of tau in primary neurons is mediated through conditioned media from astrocytes cultured with PA (Patil and Chan, 2005; Patil et al., 2006). Interestingly, culturing primary neurons directly with PA did not induce these AD-like pathologies (Patil and Chan, 2005; Patil et al., 2006). We determined that PA induced the release of cytokines, in particular IL-1β and TNFα, from the astrocytes into the media (i.e. conditioned media). In support saturated fatty acid PA also has been shown to induce pro-inflammatory cytokine release from microglia cells (Wang et al., 2012). Inhibiting SPT in the astrocytes to mitigate the elevated ceramide levels significantly reduced IL-1β release from the astrocytes cultured with PA. Congruently, neutralizing IL-1β and TNFα in the astrocyte conditioned media significantly reduced the upregulation of BACE1 in the primary neurons cultured in the neutralized astrocyte conditioned media (Liu et al., 2013). Taken together, the evidence suggest a potential connection exists between PA and AD pathogenesis mediate by ceramides and the generation of cytokines, thus we conducted experiments in this study to uncover the molecular mechanism by which PA triggers the production of IL-1β from primary astrocytes. We demonstrate that PA activates IPAF inflammasome in primary rat astrocytes to induce the maturation of IL-1β. This is the first report in which the IPAF inflammasome is induced by factors other than pathogen-associated molecules. Moreover, the expression of IPAF in the astrocytes upon exposure to PA is regulated by the transcription factor CREB. Downregulating IPAF in primary rat astrocytes significantly reduced the Aβ42 levels produced by the primary neurons cultured in conditioned media from PA-treated astrocytes. Finally, the levels of IPAF and ASC were found significantly upregulated in a subgroup of sporadic AD patient brains.
2. MATERIALS AND METHODS
2.1 Human samples
AD and control neocortical brain samples were obtained from the University of Kentucky Alzheimer’s disease center tissue bank (ADC). Diagnoses were confirmed by neurologists, neuropathologists, and neuropsychologists in the ADC clinic. Most samples have been obtained in less than 4hr postmortem interval, and the age for the patients ranged from 88-99 years. Table 1 lists the reference number, gender, Braak stage, Mini Mental State Examination scores, frontal neuritic plaque numbers, neurofibrillary tangle numbers, and ApoE genotype of the individuals. The information was provided by the ADC.
Table 1. Patient information.
| Group | Ref# | Age | Sex | PMI | Braak Stage |
Front NP count |
NFT counts |
ApoE alleles |
MMSE |
|---|---|---|---|---|---|---|---|---|---|
| Control | 1132 | 95 | F | 3.5 | 0 | 3.0 | 0 | 3/4 | 26 |
| Control | 1159 | 86 | F | 3.5 | 0 | 0.0 | 0 | 3/3 | 28 |
| Control | 1165 | 92 | M | 3.3 | 0 | 10.4 | 0 | 3/3 | 28 |
| Control | 1206 | 94 | F | 2.3 | 2 | 0.0 | 0.4 | 3/3 | 29 |
| Control | 1221 | 81 | M | 2.8 | 2 | 5.6 | 0 | 3/3 | 27 |
| AD | 1013 | 80 | M | 2.5 | 5 | 5.8 | 1 | ||
| AD | 1098 | 81 | F | 2.8 | 5 | 6.2 | 2 | 3/3 | 15 |
| AD | 1174 | 96 | M | 3.5 | 5 | 19.2 | 4 | 2/3 | 22 |
| AD | 1201 | 94 | M | 2.0 | 5 | 6.0 | 0.2 | 3/3 | 22 |
| AD | 1215 | 91 | F | 3.0 | 5 | 15.2 | 7.2 | 3/3 | 13 |
Patient information including reference number (Ref#), age, gender, Braak stage, postmortem interval (PMI), front neuritic plaque (NP) count, neurofibrillary tangle (NFT) count, ApoE genotype and Mini Mental State Examination (MMSE) score was provided by the UK ADC.
2.2 Isolation and culture of primary rat astrocytes and neurons, and astrocyte conditioned media (CM)
All procedures in the cell isolation were approved by the Institutional Animal Care and Use Committee at Michigan State University. Primary cortical astrocytes were isolated from postnatal day 0-2 newborn Sprague-Dawley rats as previously described (Liu et al., 2013; Patil et al., 2007). Approximately 4×104 cells/cm2 were seeded on poly-L-lysine (PLL, Cultrex, Gaithersburg, MD, USA) coated plates. Primary astrocytes were maintained in DMEM/F12 supplemented with 10% fetal bovine serum, 100μg/mL streptomycin and 100U/mL penicillin (Invitrogen, Carlsbad, CA, USA). When the astrocytes reach around 60-70% confluence, the astrocyte medium was removed and subsequently cultured in cortical media for 24hr (see below).The purity of the astrocytes monolayers were >90% as determined by GFAP immunoreactivity and flow cytometry (Supplementary figure 1).
Cortices from postnatal day 0 Sprague Dawley rats were used for neuronal culture. The primary neurons were plated on PLL coated plates at 2.5×105cells/cm2, and were cultured in neurobasalA medium with B27, 0.5mM glutamine, and PS for 3-4 days after isolation. Then the neurobasalA/B27 media was removed and the neurons were cultured in cortical media (see below) for 24hr prior to treatment (i.e. conditioned media from the astrocytes). The purity of the neurons was >90% as determined by βIII tubulin immunostaining and flow cytometry (Supplementary figure 1).
Cortical medium (DMEM 10313 supplemented with 10% horse serum, 10mM HEPE, 2mM glutamine, 100μg/mL streptomycin and 100U/mL penicillin, (Invitrogen)) was used to culture primary neurons and primary astrocytes for 24hr prior to treatment, i.e. conditioned media from astroyctes for treating primary neurons or PA for treating astrocytes. BSA (fatty acid-free bovine serum albumin) (Millipore, Billerica, MA, USA), or 0.4mM PA (Sigma, St. Louis, MO, USA) plus BSA as a carrier protein (molar ration is 3:1) was added to the cortical medium and used to incubate the astrocytes for 12hr. This astrocyte conditioned media (CM-B or CM-P) were used subsequently to treat primary neurons for 12hr.
Forskolin and isobutylmethylxanthine (IBMX) (Sigma) at concentrations of 10μM and 100μM, respectively, were used to increase the CREB level.
2.3 Total mRNA Extraction and Quantitative real time PCR
Total mRNA from cells was extracted using the RNeasy Plus kit (QiaGen, Valencia, CA, USA) according to the manufacturer’s instructions. Total mRNA was extracted from human brain neocortices using TRIzol and RNeasy Plus kit. The total mRNA was reverse transcribed into cDNA using the cDNA synthesis kit (BioRad, Hercules, CA, USA) as previously described (Wu et al., 2011; Zhang et al., 2011). The following primer sets (Operon, Huntsville, AL, USA) were used for PCR: rat actin (5′-ctcttccagccttccttcct-3′and 5′-aatgcctgggtacatggtg-3′), rat pro-IL-1β (5′-gcatccagcttcaaatctc-3′ and 5′-ggtgctgatgtaccagttg-3′), rat pro-caspase-1 (5′-gacaagatcctgagggcaaa-3′and 5′-ggtctcgtgccttttccata-3′), rat IPAF (5′-gcgaaacctgaagaagatgc-3′ and 5′-aacgctcagcttgaccaaat-3′), rat ASC (5′-gcaatgtgctgactgaagga-3′and 5′-tgttccaggtctgtcaccaa-3′), rat CREB (5′-tgttcaagctgcctctggt-3′and 5′-tctttcgtgctgcttcttca-3′), human actin (5′-tggacttcgagcaagagatg-3′ and 5′-aggaaggaaggctggaagag-3′), human IPAF (5′-agcttgctgaaggcttgttgct-3′ and 5′-tcacccatctggattgcaca-3′). Quantitative real-time PCR was performed using iQSYBR Green Supermix and Real-Time PCR Detection System (BioRad). The reaction program is: cycle1 (1×) 95°C, 15min; cycle 2 (40×) step1: 94°C, 15sec, step2: 57°C, 30sec, step3: 70°C, 30sec; cycle 3 (1×) 72°C, 7min; cycle 4 (80×) 55°C, 10min. The cycle threshold values were determined by the MyIQ software.
2.4 Western blot
Whole cell extracts from cells and from human brain neocoritices (homogenized) were measured for protein concentrations by Bradford assay. Protein samples 15-30μg were subjected to Western analysis as previously described (Bilgin et al., 2013; Wu et al., 2013) using pro-IL-1β antibody (Biovision, Milpitas, CA, USA), caspase-1 (Abcam, Cambridge, MA, USA), IPAF (Santa Cruz biotech, Dallas, Texas, USA), ASC (Novus Biologicals, Littleton, CO, USA), CREB and pCREB (Cell signaling, Danvers, MA, USA), beta-actin and TBP (TATA binding protein) (Sigma). Anti-mouse and anti-rabbit HRP-conjugated secondary antibodies were purchased from Thermo Scientific, and donkey anti-goat IgG-HRP was purchased from Santa Cruz biotech. The blots were visualized by SuperSignal West Femto maximum sensitivity substrate (Thermo Scientific, Asheville, NC, USA).
2.5 Enzyme-linked immunosorbent assay (ELISA) and Aβ42 assay
The levels of IL-1β in the astrocyte cultured supernatants from various treatments were analyzed by an ELISA kit (R&D system, Minneapolis, MN, USA) according to the manufacturer’s instructions. The sensitivity of the assay was 5pg/ml for IL-1β. Aβ42 levels were detected by Aβ42 ELISA kit (Invitrogen). Optical densities were measured by Spectra MAX Plus384 plate reader. Each sample concentration was calculated based on a standard curve of IL-1β or Aβ42 standards. All readings were normalized to the total protein levels determined by Bradford’s assay, and the data then was normalized to the control.
2.6 Endotoxin assay and Measurement of cytotoxicity
To detect for possible contamination of PA with lipopolysaccaride, the endotoxin assay was performed using ToxinSensor, an endotoxin detection system, following the manufacturer’s instructions (GenScript, Piscataway, NJ, USA). The endotoxin level in PA-BSA mixture was less than 0.022ng/ml. which is far below the concentration required to induce astrocytic activation (Bhat et al., 1998; Lieberman et al., 1989).
The cytotoxicity of the PA treatment was determined by intracellular and secreted lactate dehydrogenase (LDH assay) according to the manufacturer’s instructions (Roche, Indianapolis, IN, USA). LDH released into the medium and retained in the cells were denoted as LDHm and LDHc, respectively. Percentage of release was calculated with the following equation: LDH release %=LDHm/(LDHm+LDHc)*100
2.7 Caspase -1 fluorometric activity assay and Caspase-1 inhibition
Caspase-1 activity in astrocytes was determined using a Caspase-1 FLICA kit (Immunochemistry Technologies, Bloomington, MN, USA) according to the manufacturer’s instructions. Briefly, the astrocytes were incubated with BSA or 0.4mM PA for 12hr. FAM-FLICA Caspase-1 reagent, FAM-YVAD-FMK, was reconstituted with DMSO, and was further diluted with PBS to a concentration of 150μM. 150μM FAM-FLICA Caspase-1 reagent was added to the cultured media at a final concentration of 5μM. The cells were incubated with FAM-FLICA Caspase-1 reagent for 1hr at 37°C. The cells bearing active caspase-1 coupled to FLICA showed green. Fluorescence images were taken by confocal microscope Olympus FluoView 1000, and the green fluorescence signal was detected by Spectra MAX GEMINI EM plate reader. All readings were normalized to the protein levels obtained by Bradford assay. Caspase-1 activity in astrocytes was inhibited using either the general caspase inhibitor Z-VAD (R&D system) or the specific inhibitor Z-YVAD to caspase-1 (Biovision).
2.8 Transfection
SiTENOME SMARTpool siRNAs targeting ASC, SPT, IPAF, CREB and CASP1 were purchased from Dharmacon (Pittsburgh PA, USA), and scramble siRNA was purchased from Ambion. In brief, for one well in 6 well plate, 200-250pmol siRNA was diluted to 250μl Opti-MEM I reduced serum medium without serum (Invitrogen), 5μl lipofectamine RNAiMAX was added to 250μl Opti-MEM. Diluted siRNA and diluted RNAiMAX were combined, mixed gently and incubated for 10-20min at room temperature to form siRNA-RNAiMAX complex. When the confluence of the astrocytes reaches 70-80%, the astrocyte culture medium was changed to 2ml of astrocyte culture medium without antibiotics. The siRNA-RNAiMAX complexes 500μl were added to the 2ml of astrocyte culture medium without antibiotics. The final concentration of the siRNA in the medium was 80-100nM and the media containing siRNA-RNAiMAX complexes was used to treat the astrocytes for 18-24 hr. After silencing, either PA or BSA was used to culture the astrocytes for 12 hrs.
The empty vector pCMV and the constitutively active form CREB (VP16-CREB) were used for overexpression. Briefly, the cells were transfected with 0.8μg pCMV or VP16-CREB using Lipofectamine 2000 (Invitrogen) according to the manufacture’s instruction. Media were changed after 6hr and the cells were incubated in fresh astrocyte media for up to 24hr prior to treatment with PA.
2.9 Statistical analysis
All experiments were performed at least three times, and representative results are shown. Statistical analysis was performed by an unpaired, two tail student t-test (and ANOVA), and Mann-Whitney tests was used for human brain samples. * indicates p<0.05, ** indicates p<0.01 and *** indicates p<0.001.
3. RESULTS
3.1 Both pro-IL-1β and the mature form of IL-1β increase in astrocytes upon PA treatment
IL-1β can be produced by monocytes, macrophages, dendritic cells and astrocytes upon stimulation by diverse factors (Dong and Benveniste, 2001; Eder, 2009). PA can induce IL-1β production from several types of cells, including macrophages and microglia (Wang et al., 2012). Fatty acid concentration in normal human plasma in vivo generally ranges between 0.3-1.0 mM (Dole, 1956; Shultz, 1991; Tikanoja et al., 1989). We demonstrated that 0.2mM PA can induce IL-1β secretion from primary rat astrocytes (Liu et al., 2013). Here we confirm that IL-1β secreted from astrocytes increases in a dose-dependent manner with PA concentration as compared with the control (BSA) (Figure 1A). To determine whether PA promoted cell death and cytotoxicity, primary astrocytes were incubated with different concentrations of PA for 12hr and LDH release was measured. At concentrations of 0.2 and 0.4mM PA cell death was not induced while at 0.7mM concentrations of PA cell death increased significantly (Supplementary Figure 2), therefore 0.2 and 0.4mM PA concentrations were used in subsequent experiments. In addition to the mature form of IL-1β, the mRNA and protein levels of precursor IL-1β (pro-IL-1β) increased in the astrocytes upon culture with 0.4 mM PA and treatment time (6 and 12hr) (Figure 1B-D). Previously we reported that PA increased ceramide levels in primary astrocytes through SPT (Patil et al., 2007), the rate-limiting enzyme for de novo ceramide synthesis pathway (Perry, 2000; Perry et al., 2000). Inhibiting SPT to reduce ceramide levels by specific inhibitor L-cycloserine significantly reduced the upregulation in IL-1β (Liu et al., 2013), suggesting that SPT is involved in the generation of IL-1β in primary astrocytes treated with PA. Here we further demonstrated that transient silencing of SPT by siRNA significantly reduced secreted IL-1β from astrocytes cultured with PA (Figure 1E, F).
Fig. 1. Pro-IL-1β and IL-1β levels produced by astrocytes with PA treatment.
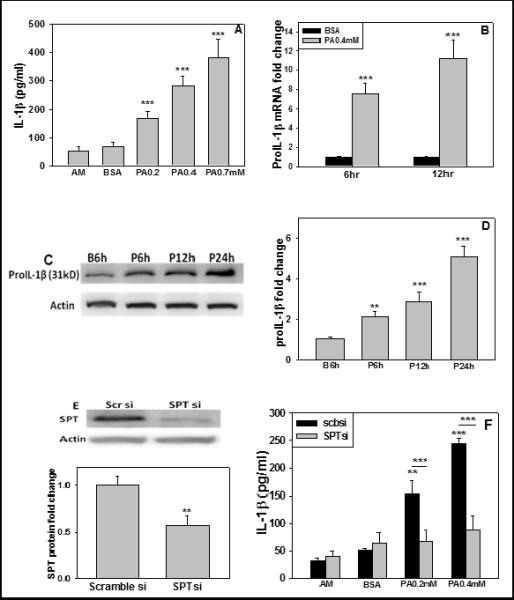
(A) Primary rat astrocytes were treated with regular astrocyte cultured medium (AM), or BSA (ctrl) or PA at the indicated concentration for 12hr. The mature form of IL-1β in cultured media was detected by ELISA assay (n≥3). (B, C, D) The mRNA and protein levels of pro-IL-1β in astrocytes treated with BSA (ctrl) or 0.4mM PA were detected by quantitative real-time PCR and western blot analysis, respectively (n≥3). (E and F) Silencing SPT decreased IL-1β level. (E) Representative western blot result and quantification of western blots of SPT levels in primary astrocytes treated with scramble siRNA (Scbsi) or SPT siRNA (SPTsi) for 24hr. (F) IL-1β protein expression level. Wild-type astrocytes or astrocytes silenced with scramble siRNA or SPT siRNA cultured with astrocyte medium (AM), BSA or PA for 12hr. IL-1β was detected by ELISA and normalized to astrocytes treated with BSA (ctrl) (n=3). **: p<0.01, ***: p<0.001. A line indicates comparison between the two bars connected by the line.
3.2 Upregulation in caspase-1 is involved in the release of IL-1β
The processing of pro-IL-1β usually requires cleavage by caspase-1, thus the activity of caspase-1 is critical to the inflammatory response. To determine if caspase-1 is involved in the maturation of IL-1β in astrocytes treated with PA, confocal microscopy and caspase-1 fluorometric activity assay were performed and established that the level of activated caspase-1 was elevated (Figure 2A, B). This is further supported by western blot analysis which showed increased caspase-1 as well as pro-caspase-1 mRNA and protein levels in astrocytes upon PA treatment (Figure. 2C-F). Primary astrocytes pre- and co-treated, for 30min and 12hr, respectively, with the general caspase-1 inhibitor Z-VAD or specific caspase-1 inhibitor Z-YVAD, along with PA, significantly reduced the IL-1β secreted (Figure 2G, H). To further confirm the inhibition results, pro-caspase-1 was transiently silenced in astrocytes by siRNA (Figure 2I-K), and the pro-caspase-1 silenced astrocytes were subsequently incubated with PA. Silencing caspase-1 reduced the IL-1β secreted by the PA-cultured astrocytes as compared with the PA-cultured astrocytes with scramble siRNA (Figure 2L). Taken together these results are consistent with caspase-1 involvement in the generation of IL-1β from astrocytes cultured with PA.
Fig. 2. The upregulated caspase-1 increased IL-1β level.
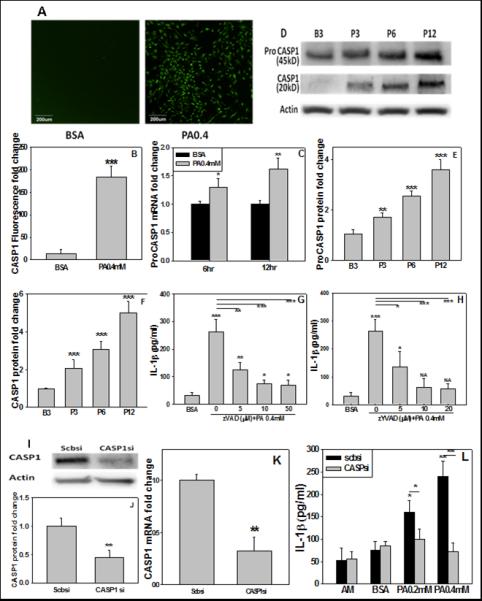
The primary rat astrocytes were treated with BSA or 0.4mM PA for 12hr and the caspase-1 was staining with FAM-YVAD-FMK (green fluorescence). (A) Images taken by confocal microscope. (B) Fuorescence fold change and the fluorescent signal detected by fluorescent plate reader. (C, D) Primary astrocytes treated with BSA or PA for the indicated time. The mRNA of pro-caspase-1 (ProCASP1) was monitored by real-time PCR and normalized to astrocytes treated with BSA (ctrl), and the protein expression was determined by immunoblot. Quantification of western blot results shown in (E, F). (G, H) Astrocytes pre- and co-treated with inhibitor ZVAD or ZYVAD and with PA for 12hr at the indicated concentration. The expression of IL-1β was monitored by ELISA and normalized to astrocytes treated with BSA (ctrl). (I, J) Astrocytes cultured with scramble siRNA (scbsi) or siRNA targeting CASP1 (CASPsi) for 24hr. Representative western blot results of CASP1 and the quantification of western blot results are shown (n=3). (K) The mRNA level of silenced CASP1 confirmed by real-time PCR (n=3). (L) The mature form of IL-1β in wild-type or silenced astrocytes. The astrocytes were silenced with scramble siRNA (scbsi) or CASP1 siRNA for 24hr, or wild-type astrocytes were treated with astrocyte medium (AM), BSA (ctrl), or PA for 12hr. ELISA was performed to detect IL-1β level and the results were normalized to astrocytes treated with BSA (ctrl) (n=3). NA: not available. *: p<0.05, **: p<0.01, ***: p<0.001. A line indicates comparison between the two bars connected by the line.
3.3 PA activates IPAF inflammasome to produce mature IL-1β
Inflammasomes are multiprotein complexes that recruit pro-caspase-1 and trigger the activation of caspase-1. Activation of cytoplasmic receptors of inflammasomes, which are initiators of inflammasome assembly, is cell-type and stimulus-dependent. In the CNS, NLRP1 (the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family protein 1) was reported to be expressed in neurons but was not detected in astrocytes (de Rivero Vaccari et al., 2008). A recent study uncovered IPAF inflammasome in microglial cells (Jamilloux et al., 2013). To date, the involvement of inflammasome receptors in astrocytes treated with PA has not been reported. Since PA can induce IL-1β secretion from astrocytes (Liu et al., 2013), we evaluated whether the other two receptors, IPAF and NLRP3, are involved in the processing of IL-1β in primary rat astrocytes. Notably, we found PA increased the mRNA level of IPAF significantly (Figure 3A). To further confirm the involvement of IPAF in the production of IL-1β by astrocytes, IPAF was transiently silenced with siRNA (Figure 3B, C, D) and the upregulation of IL-1β was abolished in the astrocytes cultured in PA (Figure 3E). In contrast, silencing NLRP3 in primary astrocytes did not reduce the upregulation of IL-1β upon cultured with PA (Figure 3F). These results suggest that IPAF, expressed in primary rat astrocytes, is activated by PA, and could recruit pro-caspase-1 and activate caspase-1 to lead to subsequent maturation and release of IL-1β.
Fig. 3. IPAF is involved in IL-1β maturation.

(A) The mRNA expression of IPAF in astrocytes treated with BSA or PA for 6 or 12hr. The mRNA levels in astrocytes treated with PA and were normalized to astrocytes treated with BSA (n=3). (B, C, D) mRNA and protein levels of silenced IPAF. Astrocytes were cultured with scramble siRNA (scbsi) or siRNA targeting IPAF (IPAF siRNA) for 20hr. The IPAF level was detected by immunoblot and real-time PCR (n=3). (E, F) IL-1β expression levels. Wild-type astrocytes or astrocytes silenced with scramble siRNA or IPAF siRNA or NLRP3 siRNA were cultured for 12hr with astrocyte medium (AM), BSA, or PA. ELISA was performed for IL-1β level and the expression levels of IL-1β were normalized to BSA treated wild-type or silenced astrocytes (n≥3). NA: not available. **: p<0.01. A line indicates comparison between the two bars connected by the line.
3.4 ASC is important for the activation of IPAF inflammasome
ASC (apoptosis-associated speck-like protein containing a CARD) is an adaptor protein required for the activation of the NLRP1 and NLRP3 inflammasomes, whereas the involvement of ASC in the activation of IPAF inflammasome is stimulus dependent (Sutterwala and Flavell, 2009). Upon exposure to bacteria, such as Salmonella typhimurium, Shigella flexneri or Pseudomonas aeruginosa, ASC is required for activation of the IPAF inflammasome (Schroder and Tschopp, 2010), but not for IPAF activation in response to Legionella pneumophila (Case et al., 2009). We found the levels of ASC protein increased dramatically in astrocytes cultured with PA for 6 and 12hr (Figure 4A, B). To determine if ASC is required for the activation of IPAF inflammasome in astrocytes cultured in PA, ASC was transiently silenced in astrocytes (Figure 4 C-E) prior to PA treatment for 12hr. This abolished the upregulation of IL-1β in the astrocytes cultured in PA (Figure 4F) implying that ASC is required for the activation of the IPAF inflammasome. In addition to ASC, Naip5 is sometimes needed for IPAF activation, depending on the stimulus (Lightfield et al., 2011; Sutterwala and Flavell, 2009). Upon transient silencing of Naip5 in primary astrocytes, as confirmed by a decrease in both its mRNA and protein levels, the ELISA analysis indicated, however, that the level of IL-1β was not reduced significantly in the astrocytes cultured in PA (Supplementary Figure 3). These results suggest that ASC, but not Naip 5 (NLR family, apoptosis inhibitory protein 5), is required for the activation of IPAF in primary astrocytes treated with PA. Currently the mechanisms by which ASC and Naip5 are involved in the activation of IPAF are unknown.
Fig. 4. ASC is involved in IL-1β production.
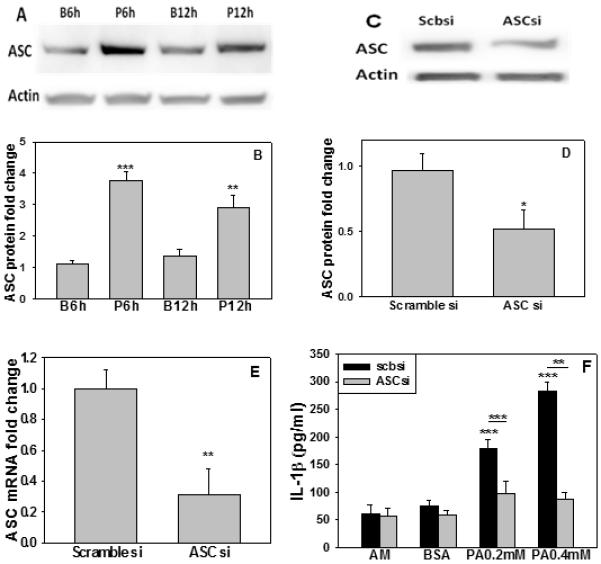
(A-E) ASC expression levels. (A, B) Representative western blot results of ASC protein levels in astrocytes treated with PA for the indicated time. Quantification of ASC western blots (n=3). (C, D) Representative western blot results of ASC protein levels in astrocytes cultured with scramble siRNA or siRNA targeting ASC for 20hr. Quantification of ASC immunoblots. (E) mRNA level of ASC detected by real-time PCR (n=3). (F) IL-1β expression levels. Wild-type astrocytes or astrocytes silenced with scramble siRNA (scbsi) or ASC siRNA (ASCsi) were cultured with astrocyte medium (AM), BSA or PA for 12hr. IL-1β was detected by ELISA and was normalized to astrocytes treated with BSA (ctrl) (n=3). *: p<0.05, **: p<0.01, ***: p<0.001. A line indicates comparison between the two bars connected by the line.
3.5 IPAF inflammasome regulates Aβ42 level in primary neurons
Many in vivo and in vitro studies suggest that inflammation is involved in the pathogenesis of AD. Specifically, IL-1β has been shown to induce tau hyperphosphorylation and enhance neurofibrillary tangles (Griffin et al., 2006; Salminen et al., 2008). We previously also reported that conditioned media from primary astrocytes cultured in PA contained elevated IL-1β, which induced an upregulation in the levels of tau hyperphosphorylation and Aβ42 in primary rat neurons (Patil et al., 2007). This study investigates whether the increase in the level of IPAF inflammasome in astrocytes cultured in PA affects the level of Aβ produced by primary rat cortical neurons treated with this astrocyte conditioned media. IPAF was silenced in primary rat astrocytes for 20hr, and the astrocytes were subsequently treated with either BSA or PA for 12hr, and then the astrocyte conditioned media was used to culture primary neurons for 12hr. A significant decrease in the level of extracellular Aβ42 was detected in primary neurons cultured in conditioned media from PA-treated astrocytes with IPAF silenced as compared with PA-treated astrocytes without IPAF silenced (Figure 5). This suggests that IL-1β generated from IPAF inflammasomes in the astrocytes is involved in enhancing the level of Aβ42 in the neurons.
Fig. 5. IPAF silencing in PA-treated astrocytes reduces neuronal Aβ42 levels.
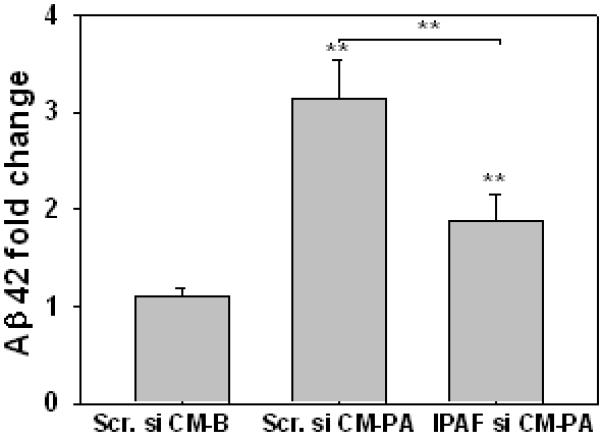
Primary astrocytes treated with scramble siRNA or IPAF siRNA for 20hr. After silencing, either BSA or 0.4 mM PA was used to culture the astrocytes for 12hr. Then the astrocyte conditioned medium: scramble siRNA-BSA (Scr.si CM-B), scramble siRNA-PA (Scr.si CM-PA), or IPAF siRNA-PA (IPAF si CM-PA) was transferred to treat primary rat neurons for 12hr. The supernatant was collected and the Aβ42 levels were monitored by ELISA (n=3). **: p<0.01. A line indicates comparison between the two bars connected by the line.
3.6 IPAF and ASC levels are elevated in sporadic AD patients
It was shown that the level of IL-1β in the brains of AD patients is significantly elevated (Cribbs et al., 2012; Griffin et al., 1989). Since the IPAF inflammasome in the astrocytes, induced by PA, is involved in upregulating Aβ42 levels in primary rat neurons through the astrocyte conditioned media, we measured the level of IPAF in the frontal brain cortices of a subgroup of sporadic AD patients and found both the mRNA and protein levels of IPAF were increased significantly as compared with the controls (Figure 6A, D and F, Table 1). Furthermore, the mRNA and protein levels of ASC were also significantly elevated (Figure 6B, D and F) while Naip5 did not changed significantly in this subgroup of sporadic AD brains as compared with normal controls (Supplementary Figure 4). These results raise the possibility that the IPAF inflammasome could be involved in the inflammatory response associated with AD. Another inflammasomes, NLRP3, was also evaluated and an increase in the mRNA and protein levels of NLRP3 was not detected in this subgroup of sporadic AD brains (Figure 6C, E and F). Similarly in a recent report, elevated NLRP3 level was not detected in AD patient brains, nevertheless Aβ was able to activate NLRP3 to release cytokines in a transgenic animal model (Cribbs et al., 2012; Heneka et al., 2013), suggesting that NLRP3 could still play a role in the inflammatory reactions involved in AD pathogenesis.
Fig. 6. IPAF and ASC are upregulated in sporadic AD brain.
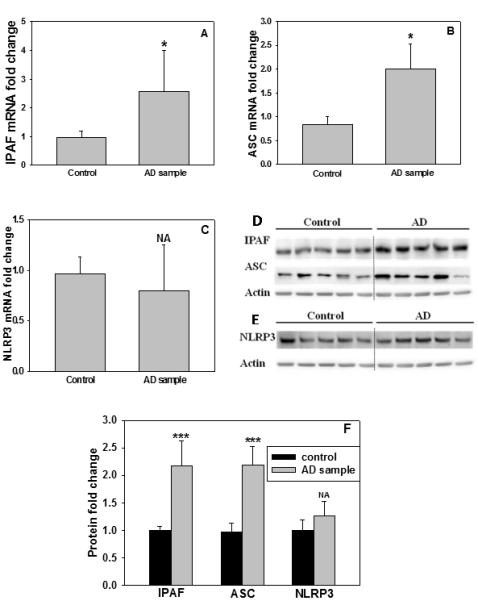
The neocortical brain samples were analyzed for IPAF, ASC and NLRP3. (A-C) The mRNA levels of IPAF, ASC and NLRP3 in neocotrical brain samples. The mRNA levels were measured by real-time PCR. (D, E) The protein levels of IPAF, ASC and NLRP3 in neocotrical brain samples. The protein levels were detected by western blot and (F) shows the quantification of the western blots (n=5). NA: not available. *: p<0.05, ***: p<0.001.
3.7 CREB regulates IPAF expression in astrocyte treated with PA
The mRNA level of IPAF is significantly increased, more than 5 times, in primary rat astrocytes treated with PA for 6hr (Figure 3A), and is also upregulated in a subgroup of sporadic AD brains (Figure 6A). Thus far, the regulation of IPAF has not been extensively studied. The transcription factor, AP1, has been reported to regulate IPAF in human leukemia cells treated with tumor necrosis factor α (Gutierrez et al., 2004). Furthermore several other transcription factors have been predicted, including CREB (cAMP response element-binding protein), Stat (Signal Transducer and Activator of Transcription), and E2F (Gutierrez et al., 2004), however, there has been no report on the regulation of IPAF in astrocytes. Therefore, we analyzed the promoter region of IPAF using JASPAR and TRANSFAC databases, and found that CREB could be a putative transcription factor of IPAF (Supplementary Figure 5). pCREB was detected by immunoblotting and the results showed an upregulation of pCREB in astrocytes treated with PA (Figure 7A, B). Forskolin and isobutylmethylxanthine (FI), well studied chemicals to increase CREB level, were used to treat primary astrocytes for 6hr. mRNA analysis indicates the level of IPAF increased 5-fold in the FI-treated over the untreated astrocytes (controls) (Figure 7C). To further confirm that CREB could be involved in the regulation of IPAF transcription, transient silencing of CREB and overexpression of a constitutively active form of CREB (VP16-CREB) were performed (Figure 7D-F). Overexpression of the constitutively active CREB significantly increased IPAF mRNA level while silencing CREB significantly reduced the IPAF transcript (Figure 7G). Similarly silencing CREB in the astrocytes prior to culturing with PA significantly reduced, while overexpressing CREB in the astrocytes enhanced the mRNA level of IPAF upon treatment with PA (Figure 7H), suggesting that CREB could regulate IPAF expression in primary rat astrocytes. To further monitor the effect of CREB on IL-1β expression in astrocytes cultured in PA, CREB was silenced or overexpressed in primary astrocytes, and the astrocytes were subsequently incubated in PA for 12hr. Silencing CREB significantly reduced the upregulation of IL-1β, while overexpressing CREB further enhanced the IL-1β secreted by astrocytes cultured with PA (Figure 7I, J), suggesting that CREB could regulate IL-1β expression in PA-treated astrocytes mediated by IPAF.
Fig. 7. CREB regulates IPAF expression in astrocytes.
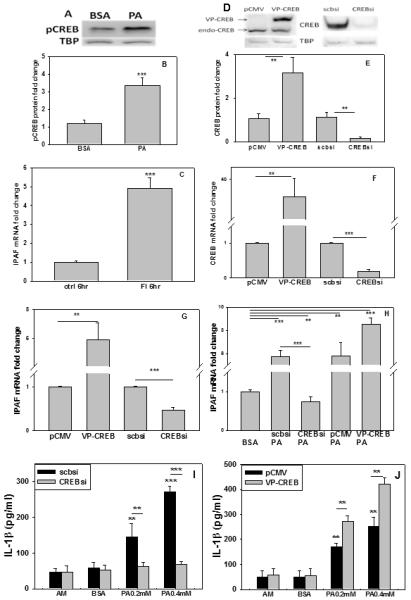
(A) pCREB protein levels in primary rat astrocytes upon treatment with PA for 12hr, and quantified in (B). TBP, TATA binding protein, was used as loading control. (C) Astrocytes incubated with forskolin and isobutylmethylxanthine (FI) for 6hr and mRNA of IPAF detected by real-time PCR. (D) Representative western blot results of silencing and overexpression of CREB in primary astrocytes. TBP was used as a loading control for the western blots. CREB overexpression was performed by transfection of the constitutively active VP16-CREB and silencing was achieved by CREB siRNA. pCMV and scramble siRNA were used as control. Endogenous CREB is denoted as endo-CREB. (E) Quantification of western blot. mRNA fold changes of (F) CREB and (G) IPAF levels in astrocytes overexpressing or silencing CREB. (H) Astrocytes treated with pCMV, VP16-CREB, scramble siRNA (scbsi), or CREB siRNA, followed by treatment with PA or BSA (ctrl) for 12hr. IPAF mRNA was detected by real-time PCR. (I) IL-1β expression level of astrocytes silenced with scramble siRNA or CREB siRNA. The cells were cultured for 12hr with astrocyte medium (AM), BSA or PA. ELISA was performed for IL-1β level and the expression levels were normalized to BSA treated wild-type or silenced astrocytes. (J) IL-1β expression level of astrocytes overexpressed with pCMV or VP16-CREB. The cells were cultured for 12hr with astrocyte medium (AM), BSA or PA. ELISA was performed for IL-1β level and expression levels were normalized to BSA treated wild-type or silenced astrocytes. (n=3). **: p<0.01, ***: p<0.001. A line indicates comparison between the two bars connected by the line.
4. DISCUSSION
We identify that the IPAF inflammasome is involved in mediating the effects of astrocytes in determining the outcome of primary neurons in response to environmental factors, i.e. fatty acids. IL-1β expression is regulated at both the transcriptional and posttranscriptional proteolytic processing levels, namely, it requires two signals, one to induce the expression of pro-IL-1β and the other to activate the inflammasome (Wen et al., 2011). We found that PA alone can activate both signals; increase the level of the precursor IL-1β protein and activate the IPAF inflammasome, leading to the maturation of IL-1β in primary rat astrocytes. The regulation of pro-IL-1β expression by transcription factor NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells) is well established (Basak et al., 2005), and NFκB is known to be activated by fatty acids, such as PA or high fat diet (Wang et al., 2012). In support, we observed that NFκB expression in the astrocytes is increased upon PA treatment (Supplementary Figure 6) and inhibiting NFκB significantly reduced the mRNA level of pro-IL-1β (Supplementary Figure 7). This indicates that NFκB could be involved in the regulation of pro-IL-1β expression in PA-treated astrocytes. Furthermore, treating primary astrocytes with PA elevates ASC expression in the primary astrocytes (Figure 4). The regulation of ASC expression is not as well studied, and an analysis of the promoter region of ASC identified that NFκB could be a potential regulator of ASC. Indeed inhibiting NFκB reduced the upregulation of ASC in astrocytes cultured in PA (Supplementary Figure 6), suggesting that NFκB could be involved in the regulation of both ASC and pro-IL-1β expression in PA-treated astrocytes. In addition to pro-IL-1β and ASC, the mRNA level of IPAF also increased in PA-treated astrocytes (Figure 3). Thus far, the regulation of IPAF has not been extensively studied. Several transcription factors, CREB, Stat and E2F have been predicted to potentially bind the human IPAF promoter region (Gutierrez et al., 2004). Our results suggest that CREB could transcriptionally regulate rat IPAF expression (Figure 7). Although the activation of IPAF inflammsome is not well understood, elevated levels of IPAF and ASC raise the possibility of enhanced assembly and activation of the IPAF inflammasome.
The activation of inflammasomes is critical for the maturation of IL-1β. NLRP1 and NLRP3 inflammasomes can be activated by diverse factors, involving both pathogen-associated molecular patterns, such as microbial and viral components, as well as danger-associated molecular factors, such as ATP, Aβ, and fibronectin (Masters et al., 2010). Prior studies of IPAF inflammasome showed that bacterial and viral pathogenic factors are involved (Jamilloux et al., 2013; Pereira et al., 2011), while our study demonstrate that non-pathogen associated factor, fatty acids, namely PA, also can activate IPAF inflammasome. To date, the signals and mechanisms leading to the activation of IPAF inflammasome in astroctyes are unclear. Aβ activates NLRP3 inflammasome in microglial cells through the phagocytosis-lysosome pathway (Schroder and Tschopp, 2010). A recent study in mice reported that the double-stranded RNA-dependent protein kinase (PKR) physically interacts with the inflammasome, including IPAF, to regulate inflammasome activity (Lu et al., 2012), and the phosphorylation of IPAF is critical for the activation of inflammasome (Qu et al., 2012). Thus diverse mechanisms may be involved in the activation of inflammasomes.
Many types of inflammasomes have been found in the CNS (Kummer et al., 2007). NRLP1 has been shown to be expressed in neurons, while NLRP3 and IPAF have been detected in microglial cells (Jamilloux et al., 2013; Masters et al., 2010). We found the IPAF inflammamsome is also expressed in primary astrocytes (Figure 3), suggesting that in the CNS the same type of inflammasome could be expressed in different cell types and a single cell type could express different inflammasomes. Astrocytes, acting as sentinels, and highly reactive to the microenvironment in their support of neurons and the blood-brain barrier, are important contributors to the release of IL-1β (Dinarello, 2009). However, the composition of the inflammasome expressed in PA-treated astrocytes had not been determined prior to this study. The compositions of NLRP1 and NLRP3 inflammasomes require ASC. For example, in spinal cord injury ASC was shown to be required in the activation of NLRP1 inflammasome in the neurons of rat spinal cord (de Rivero Vaccari et al., 2008; Silverman et al., 2009). However, the composition of IPAF is cell-type and stimulus-specific, i.e. under some but not all conditions ASC is required for its activation (Martinon et al., 2009). In addition to ASC, it was reported that activation of the IPAF inflammasome could require Naip5 (Schroder and Tschopp, 2010). In this study, we found that ASC, but not Naip 5, is required for the activation of the IPAF inflammasome in the astrocytes treated with PA. Currently the molecular mechanism and function of ASC and Naip5 in the activation of IPAF inflammasome remains unclear, although ASC has been suggested to stabilize or facilitate caspase-1 recruitment (Martinon et al., 2009).
Inflammation is an important characteristic of AD and has been implicated in the etiology of this neurodegenerative disease (Glass et al., 2010). Specifically, elevated IL-1β level has been reported in plasma, brains and cerebrospinal fluid of patients with AD and mild cognitive impairment. Blocking IL-1β rescued cognition, reduced Aβ and attenuated tau pathology in AD transgenic mice (Kitazawa et al., 2011). In addition, the IL-1 receptor antagonist knock-out mice studies exhibited increased mortality and neuroinflammation to Aβ-induced neuropathology (Craft et al., 2005). We previously showed PA induced AD-like pathological changes, such as BACE1, Aβ and hyperphosphorylated tau, in primary neurons mediated by the conditioned media from astrocytes cultured in PA, in particular we confirmed the involvement of IL-1β (Liu et al., 2013; Patil et al., 2007). Silencing IPAF in the astrocytes reduced IL-1β secretion, which significantly diminished Aβ42 level in the neurons cultured with the conditioned media from PA-treated astrocytes (Figure 5). We further confirmed the mRNA and protein levels of IPAF and ASC (but not Naip5) are upregulated in a subgroup of sporadic AD patient’s brains (Figure 6, Supplementary figure 4). In this study, IPAF inflammasome was detected in primary rat astrocytes cultured with PA, however recently, IPAF inflammasome, along with Naip 5, were reported in microglial cells to be involved in IL-1β production (Jamilloux et al., 2013). This suggests that multiple cell types in the CNS have the ability to express the same type of inflammasome. Thus, the upregulated IPAF level in human AD brains (Figure 6) could arise from astrocytes or microglial cells. Aβ was reported to trigger the activation of NLRP3 inflammasome in bone marrow derived dendritic and microglia cells (Masters et al., 2010). Although an elevated level NLRP3 was not detected in our subgroup of AD patient brains, nevertheless, NLRP3 could still play a role in the inflammatory reaction in AD pathogenesis triggered by other factors, such as Aβ (Heneka et al., 2013). Taken together, the results suggest that IPAF could be involved in the inflammatory response in AD.
Saturated fatty acids have been shown to induce cytokine release from several cell types (Wang et al., 2012). Diets rich in fat and brain trauma increase the levels of fatty acids (Jellinger, 2004), such as PA, in the brain. Since fatty acids can freely cross the blood-brain barrier, they are readily taken up by astrocytes, the major cell type in the brain that metabolizes fatty acids. Our results suggest that PA activates IPAF inflammasome in primary astrocytes to releases IL-1β, which in turn increases Aβ42 production in primary neurons. PA has been reported to induce IL-1β production in microglia (Wang et al., 2012), which raises the possibility that fatty acids could contribute to IL-1β production in the brain. IL-1β could further activate other cell types to release inflammatory molecules, e.g. chemotactic and neurotoxic molecules, to initiate and further propagate neuronal dysfunction and eventual neuronal death. In the CNS, there are many inflammasomes that could be activated in the different cell types. For examples, Aβ activates NLRP3-ASC inflammasome in microglia, brain trauma induces the activation of NLRP1-ASC inflammasome in neurons (de Rivero Vaccari et al., 2008), and we found that PA activates IPAF-ASC inflammasome in astrocytes and furthermore is upregulated in a subgroup of AD brains. Regardless of the complexity and which inflammasome is involved, ASC and caspase-1 are concomitantly implicated in the production of IL-1β in the CNS (de Rivero Vaccari et al., 2008; Mawhinney et al., 2011; Silverman et al., 2009). In support, neutralization of ASC spared the tissue and improved function in rat neurons exposed to inflammation induced by NLRP1-ASC inflammasome (de Rivero Vaccari et al., 2008). Taken together this suggests that IPAF, ASC and caspase-1 could be potential therapeutic targets to decrease inflammation in sporadic AD, as well as other CNS inflammatory diseases.
Supplementary Material
Acknowledgement
This study was supported in part by the National Institute of Health (R01GM079688 and R01GM089866), the National Science Foundation (CBET 0941055). We thank Dr. Peter Nelson and the UK ADC NIA P30-AG0-28383 for providing the human autopsy brain samples, Dr. David Ginty at Johns Hopkins University for providing the pCMV control vector and VP16-CREB plasmids, and Rebecca Martin and Garrett Kohler for assistant with the experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest and author contributions
The authors declare no actual or potential conflicts of interest. L.L. designed and performed all the experiments. C.C. guided the overall research design, contributed reagents and analytic tools. L.L. drafted and C.C edited the manuscript.
References
- Ajuwon KM, Spurlock ME. Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3T3-L1 adipocytes. J Nutr. 2005;135:1841–1846. doi: 10.1093/jn/135.8.1841. [DOI] [PubMed] [Google Scholar]
- Basak C, Pathak SK, Bhattacharyya A, Mandal D, Pathak S, Kundu M. NF-kappaB- and C/EBPbeta-driven interleukin-1beta gene expression and PAK1-mediated caspase-1 activation play essential roles in interleukin-1beta release from Helicobacter pylori lipopolysaccharide-stimulated macrophages. J Biol Chem. 2005;280:4279–4288. doi: 10.1074/jbc.M412820200. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilgin B, Liu L, Chan C, Walton SP. Quantitative, solution-phase profiling of multiple transcription factors in parallel. Anal Bioanal Chem. 2013;405:2461–2468. doi: 10.1007/s00216-013-6712-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case CL, Shin S, Roy CR. Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect Immun. 2009;77:1981–1991. doi: 10.1128/IAI.01382-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Hirsch E, Van Eldik LJ. Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid. J Neuroinflammation. 2005;2:15. doi: 10.1186/1742-2094-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, Cotman CW. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation. 2012;9:179. doi: 10.1186/1742-2094-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci. 2008;28:3404–3414. doi: 10.1523/JNEUROSCI.0157-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- Dole VP. A relation between non-esterified fatty acids in plasma and the metabolism of glucose. J Clin Invest. 1956;35:150–154. doi: 10.1172/JCI103259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Dowell JA, Johnson JA, Li L. Identification of astrocyte secreted proteins with a combination of shotgun proteomics and bioinformatics. J Proteome Res. 2009;8:4135–4143. doi: 10.1021/pr900248y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder C. Mechanisms of interleukin-1beta release. Immunobiology. 2009;214:543–553. doi: 10.1016/j.imbio.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Geekiyanage H, Chan C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid beta, novel targets in sporadic Alzheimer’s disease. J Neurosci. 2011;31:14820–14830. doi: 10.1523/JNEUROSCI.3883-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geekiyanage H, Upadhye A, Chan C. Inhibition of serine palmitoyltransferase reduces Abeta and tau hyperphosphorylation in a murine model: a safe therapeutic strategy for Alzheimer’s disease. Neurobiol Aging. 2013;34:2037–2051. doi: 10.1016/j.neurobiolaging.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez PV, Schioth HB, Lasaga M, Scimonelli TN. Memory impairment induced by IL-1beta is reversed by alpha-MSH through central melanocortin-4 receptors. Brain Behav Immun. 2009;23:817–822. doi: 10.1016/j.bbi.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Liu L, Li Y, Mrak RE, Barger SW. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation. 2006;3:5. doi: 10.1186/1742-2094-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, 3rd, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez O, Pipaon C, Fernandez-Luna JL. Ipaf is upregulated by tumor necrosis factor-alpha in human leukemia cells. FEBS Lett. 2004;568:79–82. doi: 10.1016/j.febslet.2004.04.095. [DOI] [PubMed] [Google Scholar]
- Hailer NP. Immunosuppression after traumatic or ischemic CNS damage: it is neuroprotective and illuminates the role of microglial cells. Prog Neurobiol. 2008;84:211–233. doi: 10.1016/j.pneurobio.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, D MH, McKeel DW, Jr., Kelley J, Morris JC. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J Neurochem. 2002;82:809–818. doi: 10.1046/j.1471-4159.2002.00997.x. [DOI] [PubMed] [Google Scholar]
- He X, Huang Y, Li B, Gong CX, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging. 2010;31:398–408. doi: 10.1016/j.neurobiolaging.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JM, Kwan J, Malek-Ahmadi M, Maarouf CL, Kokjohn TA, Belden C, Sabbagh MN, Beach TG, Roher AE. Morphological and pathological evolution of the brain microcirculation in aging and Alzheimer’s disease. PLoS One. 2012;7:e36893. doi: 10.1371/journal.pone.0036893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamilloux Y, Pierini R, Querenet M, Juruj C, Fauchais AL, Jauberteau MO, Jarraud S, Lina G, Etienne J, Roy CR, Henry T, Davoust N, Ader F. Inflammasome activation restricts Legionella pneumophila replication in primary microglial cells through flagellin detection. Glia. 2013;61:539–549. doi: 10.1002/glia.22454. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Head injury and dementia. Curr Opin Neurol. 2004;17:719–723. doi: 10.1097/00019052-200412000-00012. [DOI] [PubMed] [Google Scholar]
- Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, Calon F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010;31:1516–1531. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Khandelwal PJ, Herman AM, Moussa CE. Inflammation in the early stages of neurodegenerative pathology. J Neuroimmunol. 2011;238:1–11. doi: 10.1016/j.jneuroim.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol. 2011;187:6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, van Bruggen R, Tschopp J. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem. 2007;55:443–452. doi: 10.1369/jhc.6A7101.2006. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Pitha PM, Shin HS, Shin ML. Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad Sci U S A. 1989;86:6348–6352. doi: 10.1073/pnas.86.16.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfield KL, Persson J, Trinidad NJ, Brubaker SW, Kofoed EM, Sauer JD, Dunipace EA, Warren SE, Miao EA, Vance RE. Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect Immun. 2011;79:1606–1614. doi: 10.1128/IAI.01187-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Martin R, Chan C. Palmitate-activated astrocytes via serine palmitoyltransferase increase BACE1 in primary neurons by sphingomyelinases. Neurobiol Aging. 2013;34:540–550. doi: 10.1016/j.neurobiolaging.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, Ting JP, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Dharmadhikari G, Schumann DM, Storling J. Interleukin-1 beta targeted therapy for type 2 diabetes. Expert Opin Biol Ther. 2009;9:1177–1188. doi: 10.1517/14712590903136688. [DOI] [PubMed] [Google Scholar]
- Maesako M, Uemura K, Kubota M, Kuzuya A, Sasaki K, Asada M, Watanabe K, Hayashida N, Ihara M, Ito H, Shimohama S, Kihara T, Kinoshita A. Environmental enrichment ameliorated high-fat diet-induced Abeta deposition and memory deficit in APP transgenic mice. Neurobiol Aging. 2012a;33:1011, e1011–1023. doi: 10.1016/j.neurobiolaging.2011.10.028. [DOI] [PubMed] [Google Scholar]
- Maesako M, Uemura K, Kubota M, Kuzuya A, Sasaki K, Hayashida N, Asada-Utsugi M, Watanabe K, Uemura M, Kihara T, Takahashi R, Shimohama S, Kinoshita A. Exercise is more effective than diet control in preventing high fat diet-induced beta-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. J Biol Chem. 2012b;287:23024–23033. doi: 10.1074/jbc.M112.367011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, Mullooly N, Mielke LA, Harris J, Coll RC, Mills KH, Mok KH, Newsholme P, Nunez G, Yodoi J, Kahn SE, Lavelle EC, O’Neill LA. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawhinney LJ, de Vaccari Rivero JP, Dale GA, Keane RW, Bramlett HM. Heightened inflammasome activation is linked to age-related cognitive impairment in Fischer 344 rats. BMC Neurosci. 2011;12:123. doi: 10.1186/1471-2202-12-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeux R. Epidemiology of neurodegeneration. Annu Rev Neurosci. 2003;26:81–104. doi: 10.1146/annurev.neuro.26.043002.094919. [DOI] [PubMed] [Google Scholar]
- Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, Stout RF, Jr., Spray DC, Reichenbach A, Pannicke T, Pekny M, Pekna M, Zorec R, Verkhratsky A. Glial cells in (patho)physiology. J Neurochem. 2012;121:4–27. doi: 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S, Chan C. Palmitic and stearic fatty acids induce Alzheimer-like hyperphosphorylation of tau in primary rat cortical neurons. Neurosci Lett. 2005;384:288–293. doi: 10.1016/j.neulet.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Patil S, Melrose J, Chan C. Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur J Neurosci. 2007;26:2131–2141. doi: 10.1111/j.1460-9568.2007.05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S, Sheng L, Masserang A, Chan C. Palmitic acid-treated astrocytes induce BACE1 upregulation and accumulation of C-terminal fragment of APP in primary cortical neurons. Neurosci Lett. 2006;406:55–59. doi: 10.1016/j.neulet.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Pereira MS, Morgantetti GF, Massis LM, Horta CV, Hori JI, Zamboni DS. Activation of NLRC4 by flagellated bacteria triggers caspase-1-dependent and -independent responses to restrict Legionella pneumophila replication in macrophages and in vivo. J Immunol. 2011;187:6447–6455. doi: 10.4049/jimmunol.1003784. [DOI] [PubMed] [Google Scholar]
- Perry DK. The role of de novo ceramide synthesis in chemotherapy-induced apoptosis. Ann N Y Acad Sci. 2000;905:91–96. doi: 10.1111/j.1749-6632.2000.tb06541.x. [DOI] [PubMed] [Google Scholar]
- Perry DK, Carton J, Shah AK, Meredith F, Uhlinger DJ, Hannun YA. Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J Biol Chem. 2000;275:9078–9084. doi: 10.1074/jbc.275.12.9078. [DOI] [PubMed] [Google Scholar]
- Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, Louie S, Kayagaki N, Liu J, Komuves L, Cupp JE, Arnott D, Monack D, Dixit VM. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature. 2012;490:539–542. doi: 10.1038/nature11429. [DOI] [PubMed] [Google Scholar]
- Reilly JF, Games D, Rydel RE, Freedman S, Schenk D, Young WG, Morrison JH, Bloom FE. Amyloid deposition in the hippocampus and entorhinal cortex: quantitative analysis of a transgenic mouse model. Proc Natl Acad Sci U S A. 2003;100:4837–4842. doi: 10.1073/pnas.0330745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A. Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer’s pathology. J Cell Mol Med. 2008;12:2255–2262. doi: 10.1111/j.1582-4934.2008.00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Shultz TD. Physiological free fatty acid concentrations do not increase free estradiol in plasma. J Clin Endocrinol Metab. 1991;72:65–68. doi: 10.1210/jcem-72-1-65. [DOI] [PubMed] [Google Scholar]
- Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, Keane RW, Dahl G. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J Biol Chem. 2009;284:18143–18151. doi: 10.1074/jbc.M109.004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterwala FS, Flavell RA. NLRC4/IPAF: a CARD carrying member of the NLR family. Clin Immunol. 2009;130:2–6. doi: 10.1016/j.clim.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikanoja SH, Joutti A, Liewendahl BK. Association between increased concentrations of free thyroxine and unsaturated free fatty acids in non-thyroidal illnesses: role of albumin. Clin Chim Acta. 1989;179:33–43. doi: 10.1016/0009-8981(89)90020-x. [DOI] [PubMed] [Google Scholar]
- Tuppo EE, Arias HR. The role of inflammation in Alzheimer’s disease. Int J Biochem Cell Biol. 2005;37:289–305. doi: 10.1016/j.biocel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu D, Wang F, Liu S, Zhao S, Ling EA, Hao A. Saturated fatty acids activate microglia via Toll-like receptor 4/NF-kappaB signalling. Br J Nutr. 2012;107:229–241. doi: 10.1017/S0007114511002868. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Weiner HL, 2006 D. Frenkel. Immunology and immunotherapy of Alzheimer’s disease. Nat Rev Immunol. 6:404–416. doi: 10.1038/nri1843. [DOI] [PubMed] [Google Scholar]
- Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Liu L, Chan C. Identification of novel targets for breast cancer by exploring gene switches on a genome scale. BMC Genomics. 2011;12:547. doi: 10.1186/1471-2164-12-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Liu L, Hijazi H, Chan C. A multi-layer inference approach to reconstruct condition-specific genes and their regulation. Bioinformatics. 2013;29:1541–1552. doi: 10.1093/bioinformatics/btt186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Seitz LC, Abramczyk AM, Liu L, Chan C. cAMP initiates early phase neuron-like morphology changes and late phase neural differentiation in mesenchymal stem cells. Cell Mol Life Sci. 2011;68:863–876. doi: 10.1007/s00018-010-0497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.