

Abstract
We show that DNA catalysts (deoxyribozymes, DNA enzymes) can phosphorylate tyrosine residues of peptides. Using in vitro selection, we identified deoxyribozymes that transfer the γ-phosphoryl group from a 5′-triphosphorylated donor (a pppRNA oligonucleotide or GTP) to the tyrosine hydroxyl acceptor of a tethered hexapeptide. Tyrosine kinase deoxyribozymes that use pppRNA were identified from each of N30, N40, and N50 random-sequence pools. Each deoxyribozyme requires Zn2+, and most additionally require Mn2+. The deoxyribozymes have little or no selectivity for the amino acid identities near the tyrosine, but they are highly selective for phosphorylating tyrosine rather than serine. Analogous GTP-dependent DNA catalysts were identified and found to have apparent Km(GTP) as low as ca. 20 μM. These findings establish that DNA has the fundamental catalytic ability to phosphorylate the tyrosine side chain of a peptide substrate.
Achieving site-specific covalent modification of large protein substrates is a substantial chemical challenge. Important approaches such as native chemical ligation and its variants1 enable joining of a small peptide segment (which may be prepared directly via chemical synthesis, incorporating chemical modifications2) to the remainder of a large protein. Nature takes a considerably different approach, using enzymes such as kinases,3 glycotransferases,4 and many others to perform post-translational modifications at particular residues of otherwise fully intact proteins. Site-specific modifications of intact proteins are difficult to achieve with small-molecule reagents or catalysts,5 and tailoring natural protein enzymes for different functions (e.g., by directed evolution6) is not always feasible. Deoxyribozymes are specific DNA sequences with catalytic activity that have been used to catalyze a variety of reactions,7 including self-5′-phosphorylation.8 We have initiated a research program to use deoxyribozymes to perform covalent modification of amino acid side chains in peptide and protein substrates.9 In the present study, we report that deoxyribozymes can be identified for phosphorylation of tyrosine residues in peptide substrates, using as the phosphoryl donor either a 5′-triphosphorylated RNA oligonucleotide (pppRNA) or guanosine 5′-triphosphate (GTP). These findings are a critical first step in the application of deoxyribozymes for practical phosphorylation of larger protein substrates.
We previously reported in vitro selection to identify numerous deoxyribozymes that use pppRNA as a substrate for RNA ligation.10 The RNA reacts at the α-phosphorus of its 5′-triphosphate, with departure of pyrophosphate and attachment of the α-phosphorus (along with the remainder of the RNA oligonucleotide donor) to the nucleophilic 3′-hydroxyl or 2′-hydroxyl group of the acceptor substrate strand (Figure 1A, top, X = RNA). Analogously, our efforts on modification of amino acid side chains have used pppRNA to form nucleopeptide linkages with peptide (tyrosine or serine hydroxyl) substrates, again with reaction of the pppRNA at its α-phosphorus (Figure 1A, bottom, X = RNA).9 In all of these cases, the selection method—which relied upon polyacrylamide gel electrophoresis (PAGE) shift to separate catalytically active DNA sequences—was incapable of identifying deoxyribozymes that catalyze reaction at the γ-phosphorus of the 5′-triphosphate. This alternative reaction involves departure of the oligonucleotide 5′-diphosphate as the leaving group and transfer of the γ-phosphoryl group to the nucleophilic amino acid residue (Figure 1B, X = RNA). Presumably, distinct deoxyribozymes that catalyze oligonucleotide phosphorylation were present in our previous selection experiments that identified RNA ligase activity, but the PAGE-shift selection method did not allow such oligonucleotide kinase DNA enzymes to emerge.
Figure 1.
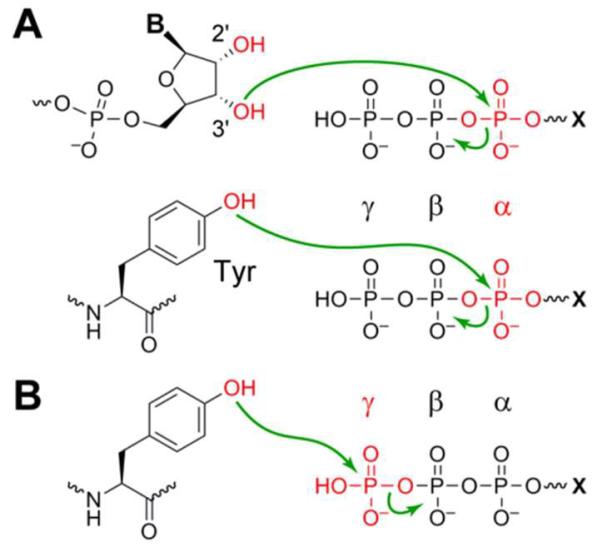
Reactions of phosphoryl donor substrates (5′-triphosphorylated RNA or GTP, where X = RNA or G). (A) Reaction of a nucleophile at the α-phosphorus, (leaving group = pyrophosphate). In RNA ligation (top), the nucleophile is an RNA hydroxyl group (could also be an internal 2′-OH group). In nucleopeptide formation as illustrated with tyrosine (bottom), the nucleophile is an amino acid hydroxyl group. (B) Reaction of a tyrosine nucleophile at the γ-phosphorus, phosphorylating the tyrosine (leaving group = oligonucleotide 5′-diphosphate or GDP).
Here we have identified deoxyribozymes that achieve phosphorylation of tyrosine (Tyr, Y) within peptide substrates via the reaction shown in Figure 1B, where X = RNA or G for a phosphoryl donor that is, respectively, pppRNA or GTP. For this purpose, we developed a two-stage selection strategy (Figure 2).11 As in our previous work, we initially used 5′-triphosphorylated RNA as the phosphoryl donor because the RNA oligonucleotide can readily be bound via Watson-Crick base pairs by one of the deoxyribozyme pool's binding arms (Figure 2A). The hexapeptide substrate, CAAYAA, that includes the Tyr acceptor was covalently attached via a disulfide linkage and a hexa(ethylene glycol) [HEG] tether to a DNA anchor oligonucleotide also bound by Watson-Crick interactions to the other pool binding arm. We wished to retain PAGE shift as the physical basis of these selection experiments, because in our hands PAGE shift has been a reliable approach to identify DNA catalysts.9–10 However, similar to the situation with oligonucleotide phosphorylation as described above, transfer of a phosphoryl group to the Tyr hydroxyl of the DNA-anchored peptide substrate does not induce a sufficiently large PAGE shift to enable selection. Therefore, we exploited a separate deoxyribozyme that is capable of attaching an RNA strand specifically to phosphorylated tyrosine, TyrP, within a peptide substrate (Figure 2B) while discriminating strongly against the unphosphorylated analogue, TyrOH. The 8VP1 deoxyribozyme12 was suitable for this purpose with ~40% capture yield, which is easily sufficient to enable selection. For the present study, we refer to 8VP1 as a “capture deoxyribozyme”, because during the selection process 8VP1 allows us to capture (by PAGE shift) specific DNA catalyst sequences that phosphorylate Tyr.
Figure 2.

Identifying DNA catalysts with Tyr kinase activity. (A) Key in vitro selection step in which DNA catalyst sequences transfer the γ-phosphoryl group from pppRNA to the tyrosine hydroxyl group of a tethered peptide. Analogously (not depicted), free GTP can be used as the phosphoryl donor, in which case the remainder of the RNA oligonucleotide is absent. See Figure S1 for structure and synthesis of the peptide substrate and Figure S2 for nucleotide details. (B) Use of 8VP1 as a “capture deoxyribozyme” that selectively attaches an RNA strand to TyrP (phosphorylated) but not TyrOH (unphosphorylated), thereby enabling PAGE-shift separation of DNA catalyst sequences that phosphorylate Tyr.
We recently showed that for DNA-catalyzed nucleopeptide linkage formation, the length of the initially random region is a critical experimental variable.13 Therefore, here the initial pool of DNA sequences included either 30, 40, or 50 random nucleotides (N30, N40, or N50) embedded between two fixed-sequence binding arms that enable PCR amplification during each selection round (Figure S2). The selection process, after extensive optimization of the experimental details (see Supporting Information), led to numerous DNA catalysts of all three random-region lengths that transfer the γ-phosphoryl group of the pppRNA donor to the Tyr acceptor within the DNA-anchored hexapeptide substrate. The key selection step of Figure 2A was performed using incubation conditions of 70 mM HEPES, pH 7.5, 1 mM ZnCl2, 20 mM MnCl2, 40 mM MgCl2, and 150 mM NaCl at 37 °C for 14 h. Using these conditions, each of the N30, N40, and N50 selection experiments led to robust catalytic activity (22%, 20%, and 27% yield at rounds 6, 7, and 8, respectively; Figure S3) In contrast, analogous selections with 50 mM CHES, pH 9.0, 40 mM MgCl2, and 150 mM NaCl at 37 °C for 14 h (omitting Zn2+ and Mn2+ because at pH 9.0 these ions precipitate or oxidize, respectively) led to no activity and were discontinued.
Each of the three successful selection experiments was cloned, and individual deoxyribozymes were characterized. Five, three, and nine unique sequences were identified from the N30, N40, and N50 selections, respectively. Tyr phosphorylation activity was verified by mass spectrometry and 8VP1 reactivity (Table S1). Sequence alignment (Figure S4A) revealed a conserved 11 nt segment at the 5′-end of the initially random region, which is near the 5′-triphosphate of the pppRNA substrate (see Figure 2A). This conservation was observed despite the three different lengths of initially random region; the gaps within the shorter N30 and N40 sequences were located almost exclusively at the 3′-side of the conserved region. For all but one sequence, kobs values were 0.1–0.5 h−1; the lone exception was the 6CF134 deoxyribozyme from the N30 selection, with higher kobs of 1.54 ± 0.22 h−1 (n = 7; Figure 3A) The metal ion dependences of the new deoxyribozymes were examined (Figure 3B). Both Zn2+ and Mn2+ were required whereas Mg2+ was generally dispensable for activity; in a few cases Mg2+ could replace Mn2+, albeit with substantially reduced yield. Again 6CF134 provided the sole exception, requiring only Zn2+ for its catalysis. The deoxyribozymes had slightly different Zn2+ concentration dependence profiles; 0.5 mM Zn2+ was highly effective in all tested cases (Figure S7A) and therefore used for all assays. When all new deoxyribozymes except 6CF134 were tested at pH 7.0 (HEPES) or pH 6.5 (MOPS or MES), activity was not detected (<0.5%; data not shown). In contrast, 6CF134 maintained activity at the lower pH values, with a linear relationship between log(kobs) and pH (slope near 1; Figure S8).
Figure 3.

Characterization of individual tyrosine kinase deoxyribozymes that use pppRNA as the phosphoryl donor. All assays were performed in trans, i.e., without the dispensable loop of Figure 2. (A) Kinetic analysis. The PAGE image shows three representative time points (t = 30 s, 5 h, and 24 h) for each of six new DNA catalysts. YOH = substrate; YP = product. The kinetic plots are for the same six deoxyribozymes. Incubation conditions: 70 mM HEPES, pH 7.5, 0.5 mM ZnCl2, 20 mM MnCl2, 40 mM MgCl2, and 150 mM NaCl at 37 °C. Kinetic plots for all 17 new deoxyribozymes are in Figure S5. (B) Metal dependence. Each deoxyribozyme was assayed in the presence of the indicated combinations of 0.5 mM Zn2+, 20 mM Mn2+, and 40 mM Mg2+. Shown are time points at t = 24 h; full kinetic plots along with data for the other eleven deoxyribozymes are in Figure S6. The combination Mn/Mg had no activity in all cases (<0.5%; data not shown). Reproducibility of such measurements is conservatively estimated as ±10% yield.
The six deoxyribozymes of Figure 3 were assayed with regard to their dependence on peptide sequence, peptide length, and length of the tether connecting the peptide to the DNA anchor oligonucleotide. Each deoxyribozyme was inactive with serine in place of tyrosine at the peptide phosphorylation site (<0.5%; data not shown). On the other hand, the deoxyribozymes were generally very tolerant of different amino acids at the positions flanking the tyrosine (Figure 4A and Figure S9A). Regarding peptide length, the deoxyribozymes were assayed with the parent HEG-tethered CAAYAA hexapeptide as well as the shorter CYA tripeptide and CY dipeptide (Figure 4B and Figure S9B). All were substantially active with the shorter peptides; in addition, 6CF134 alone was slightly active (albeit with ca. 100-fold lower kobs) with a nonpeptidic DNA 3′-hydroxyl group. Finally, different tether lengths were evaluated. All of the deoxyribozymes performed similarly with a shorter C3 tether in place of the longer HEG tether (<2-fold increase in kobs and <20% increase in yield; Figure S10). However, a longer poly(ethylene glycol) [PEG] tether comprising ten HEG units did not support detectable catalysis. Consistent with this observation, no activity was observed with an entirely free peptide substrate, i.e., when the hexapeptide substrate was not covalently attached to the DNA anchor oligonucleotide (HPLC assay; data not shown).
Figure 4.
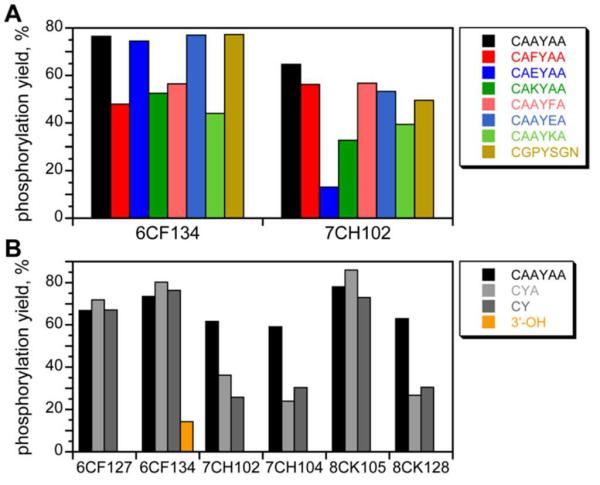
Dependence of the tyrosine kinase deoxyribozymes on peptide sequence and length. All data were acquired using the incubation conditions of Figure 3A (A) Peptide sequence dependence. The substrate included the indicated peptide on a HEG tether (t = 24 h). Two behaviors were observed, represented by 6CF134 and 7CH102; data for the other deoxyribozymes is in Figure S9A. 6CF134 was tolerant of replacing either of the flanking Ala (A) with any of Phe (F), Glu (E), or Lys (K). 7CH102 was tolerant of all of these substitutions except for Glu to the N-terminal side of the Tyr. (B) Peptide length dependence. The substrate included the indicated peptide on a HEG tether, or the substrate provided only a DNA 3′-hydroxyl group for phosphorylation (t = 12 h). Kinetic plots are in Figure S9B.
Each of the new deoxyribozymes was evaluated for Tyr kinase activity using HEG-tethered CAAYAA and free 1 mM GTP as the phosphoryl donor. In these experiments, GTP was provided in place of the pppRNA used during selection, which has G at its 5′-terminus; the remainder of the RNA substrate was included as a separate oligonucleotide. In all cases, no kinase activity was observed (data not shown). Similarly, the 5′-pppGA… of the RNA could not be replaced with any of 5′-pppGGA…, 5′-pppAA…, or 5′-pppA… (data not shown), indicating a particular structural requirement for presentation of the 5′-triphosphate group. We speculate that the conserved 5′-end of the initially random region interacts with the pppRNA donor during catalysis.
In parallel with all of the above efforts, separate new N30, N40, and N50 selections were performed in which 1 mM GTP was directly provided in place of pppRNA as the phosphoryl donor for the Tyr hexapeptide acceptor. In these efforts, successful catalysis requires that the initially random DNA region both bind GTP and catalyze phosphoryl transfer, whereas with the pppRNA donor, Watson-Crick binding interactions with the RNA oligonucleotide are preprogrammed. Despite this increased challenge, DNA catalysts emerged from each of the pools, with 5%, 6%, and 8% yield at rounds 8, 21, and 16 for N30, N40, and N50, respectively (Figure S3). Cloning revealed a single sequence from each selection, unrelated to each other and to the pppRNA-dependent DNA catalysts (Figure S4B). The N30 and N50 deoxyribozymes, 8EA101 and 16EC103, each had substantial catalytic activity, with kobs = 0.2 h−1 and yield as high as 45% (Figure 5). The N40 deoxyribozyme, 21EB121, had more modest 5% yield and was not examined further (data not shown). Kinase activity was verified for all three by mass spectrometry (Table S1). At pH 7.5 and 37 °C in the presence of 0.5 mM Zn2+, 20 mM Mn2+, 40 mM Mg2+, and 150 mM Na+, each of 8EA101 and 16EC103 had a substantial GTP concentration dependence, with apparent Km(GTP) values of approximately 20 μM and 42 μM, respectively, as well as substantial inhibition above 100–200 μM GTP (Figure S11). These apparent Km values are similar to (or better than) those for many natural protein kinases with ATP.14 8EA101 and 16EC103 had optimal Zn2+ concentrations of 0.3–0.5 mM in the presence of Mn2+, Mg2+, and 100 μM GTP (Figure S7B) and were inhibited when [GTP] > 100 μM. The identification of 8EA101 and 16EC103 establishes that DNA-catalyzed Tyr phosphorylation activity is not restricted to a pppRNA phosphoryl donor that is bound by predetermined Watson-Crick interactions. Instead, N30–N50 DNA sequences can simultaneously support both high-affinity GTP binding and phosphoryl transfer catalytic function.
Figure 5.
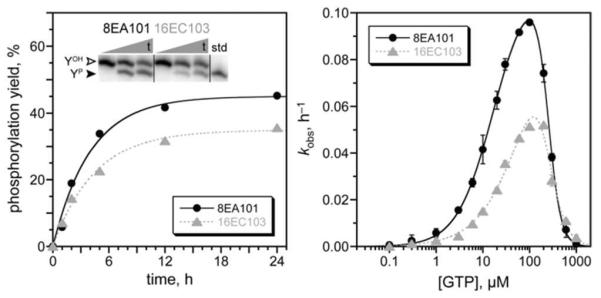
Activities of 8EA101 and 16EC103, the N30 and N50 deoxyribozymes that use free GTP as the phosphoryl donor for Tyr phosphorylation. Kinetic analysis as in Figure 3 (+ 100 μM GTP). kobs values (n = 3): 8EA101 0.23 ± 0.01 h−1; 16EC103 0.23 ± 0.04 h−1. See Figure S11 for apparent Km analysis, which used initial-rate kinetics (0 to 2 h).
In summary, we have shown that DNA can have tyrosine kinase activity, using either pppRNA or GTP as the phosphoryl donor. Along with our recent report on DNA-catalyzed dephosphorylation,15 these observations expand DNA catalysis to include important regulatory post-translational modifications. Many challenges remain in these efforts. Mechanistic studies of all of these deoxyribozymes are desirable, although without high-resolution structural information yet available for any deoxyribozyme of any kind,16 such studies are a longer-term objective. Our continuing efforts pursue tyrosine kinase deoxyribozymes with peptide sequence selectivity, improved reactivity of small-molecule phosphoryl donors (including via integration of an intact NTP aptamer domain near the random region), and DNA-catalyzed phosphorylation of free peptide and protein substrates.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by grants to S.K.S. from the National Institutes of Health (R01GM065966), the Defense Threat Reduction Agency (HDTRA1-09-1-0011), and the National Science Foundation (CHE0842534).
Footnotes
Supporting Information
Experimental details and additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Kent SBH. Chem. Soc. Rev. 2009;38:338–351. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- (2).Szewczuk LM, Tarrant MK, Cole PA. Methods Enzymol. 2009;462:1–24. doi: 10.1016/S0076-6879(09)62001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]; (b) Tarrant MK, Cole PA. Annu. Rev. Biochem. 2009;78:797–825. doi: 10.1146/annurev.biochem.78.070907.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Graves LM, Duncan JS, Whittle MC, Johnson GL. Biochem. J. 2013;450:1–8. doi: 10.1042/BJ20121456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Thibodeaux CJ, Melancon CE, Liu HW. Nature. 2007;446:1008–1016. doi: 10.1038/nature05814. [DOI] [PubMed] [Google Scholar]
- (5).Stephanopoulos N, Francis MB. Nat. Chem. Biol. 2011;7:876–884. doi: 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]
- (6).(a) Goldsmith M, Tawfik DS. Curr. Opin. Struct. Biol. 2012;22:406–412. doi: 10.1016/j.sbi.2012.03.010. [DOI] [PubMed] [Google Scholar]; (b) Cobb RE, Si T, Zhao H. Curr. Opin. Chem. Biol. 2012;16:285–291. doi: 10.1016/j.cbpa.2012.05.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Silverman SK. Chem. Commun. 2008:3467–3485. doi: 10.1039/b807292m. [DOI] [PubMed] [Google Scholar]; (b) Schlosser K, Li Y. Chem. Biol. 2009;16:311–322. doi: 10.1016/j.chembiol.2009.01.008. [DOI] [PubMed] [Google Scholar]; (c) Silverman SK. Angew. Chem. Int. Ed. 2010;49:7180–7201. doi: 10.1002/anie.200906345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Li Y, Breaker RR. Proc. Natl. Acad. Sci. USA. 1999;96:2746–2751. doi: 10.1073/pnas.96.6.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang W, Billen LP, Li Y. Chem. Biol. 2002;9:507–517. doi: 10.1016/s1074-5521(02)00127-8. [DOI] [PubMed] [Google Scholar]; (c) Achenbach JC, Jeffries GA, McManus SA, Billen LP, Li Y. Biochemistry. 2005;44:3765–3774. doi: 10.1021/bi0483054. [DOI] [PubMed] [Google Scholar]; (d) McManus SA, Li Y. Biochemistry. 2007;46:2198–2204. doi: 10.1021/bi061613c. [DOI] [PubMed] [Google Scholar]; (e) McManus SA, Li Y. J. Mol. Biol. 2008;375:960–968. doi: 10.1016/j.jmb.2007.10.080. [DOI] [PubMed] [Google Scholar]; (f) McManus SA, Li Y. J. Am. Chem. Soc. 2013;135:7181–7186. doi: 10.1021/ja311850u. [DOI] [PubMed] [Google Scholar]
- (9).(a) Pradeepkumar PI, Höbartner C, Baum DA, Silverman SK. Angew. Chem. Int. Ed. 2008;47:1753–1757. doi: 10.1002/anie.200703676. [DOI] [PubMed] [Google Scholar]; (b) Sachdeva A, Silverman SK. Chem. Commun. 2010;46:2215–2217. doi: 10.1039/b927317d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wong OY, Pradeepkumar PI, Silverman SK. Biochemistry. 2011;50:4741–4749. doi: 10.1021/bi200585n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Silverman SK. Acc. Chem. Res. 2009;42:1521–1531. doi: 10.1021/ar900052y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Flynn-Charlebois A, Wang Y, Prior TK, Rashid I, Hoadley KA, Coppins RL, Wolf AC, Silverman SK. J. Am. Chem. Soc. 2003;125:2444–2454. doi: 10.1021/ja028774y. [DOI] [PubMed] [Google Scholar]; (c) Wang Y, Silverman SK. J. Am. Chem. Soc. 2003;125:6880–6881. doi: 10.1021/ja035150z. [DOI] [PubMed] [Google Scholar]
- (11).In related efforts, we attempted to select for transfer to Tyr of the γ-thiophosphoryl group from ATPγS, GTPγS, or 5′-γ-thiotriphosphorylated RNA using APM (Hg2+) gels (Saran D, Nickens DG, Burke DH. Biochemistry. 2005;44:15007–15016. doi: 10.1021/bi051086h.; Biondi E, Burke DH. Methods Mol. Biol. 2012;883:111–120. doi: 10.1007/978-1-61779-839-9_8.). However, these experiments were unsuccessful.
- (12).Sachdeva A, Chandra M, Chandrasekar J, Silverman SK. ChemBioChem. 2012;13:654–657. doi: 10.1002/cbic.201200048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Velez TE, Singh J, Xiao Y, Allen EC, Wong O, Chandra M, Kwon SC, Silverman SK. ACS Comb. Sci. 2012;14:680–687. doi: 10.1021/co300111f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Becher I, Savitski MM, Savitski MF, Hopf C, Bantscheff M, Drewes G. ACS Chem. Biol. 2013;8:599–607. doi: 10.1021/cb3005879. [DOI] [PubMed] [Google Scholar]
- (15).Chandrasekar J, Silverman SK. Proc. Natl. Acad. Sci. USA. 2013;110:5315–5320. doi: 10.1073/pnas.1221946110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Nowakowski J, Shim PJ, Prasad GS, Stout CD, Joyce GF. Nat. Struct. Biol. 1999;6:151–156. doi: 10.1038/5839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.