

Abstract
The prokaryotic cell was once thought of as a “bag of enzymes” with little or no intracellular compartmentalization. In this view, most reactions essential for life occurred as a consequence of random molecular collisions involving substrates, cofactors and cytoplasmic enzymes. Our current conception of a prokaryote is far from this view. We now consider a bacterium or an archaeon as a highly structured, non-random collection of functional membrane-embedded and proteinaceous molecular machines, each of which serves a specialized function. In this article we shall present an overview of such microcompartments including (i) the bacterial cytoskeleton and the apparati allowing DNA segregation during cells division, (ii) energy transduction apparati involving light-driven proton pumping and ion gradient-driven ATP synthesis, (iii) prokaryotic motility and taxis machines that mediate cell movements in response to gradients of chemicals and physical forces, (iv) machines of protein folding, secretion and degradation, (v) metabolasomes carrying out specific chemical reactions, (vi) 24 hour clocks allowing bacteria to coordinate their metabolic activities with the daily solar cycle and (vii) proteinaceous membrane compartmentalized structures such as sulfur granules and gas vacuoles. Membrane-bounded prokaryotic organelles were considered in a recent JMMB written symposium concerned with membraneous compartmentalization in bacteria [Saier and Bogdanov, 2013]. By contrast, in this symposium, we focus on proteinaceous microcompartments. These two symposia, taken together, provide the interested reader with an objective view of the remarkable complexity of what was once thought of as a simple non-compartmentalized cell.
Keywords: Microcompartments, protein complexes, hyperstructures, protein machines, bacterial cytoskeleton, motility, chemotaxis, bacteriorhodopsin, F-type ATPase, circadian clocks, prokaryotic evolution
Introduction
The classical view of bacterial cell structure is that of a “bag of enzymes” that mediates the essential catalytic functions of life. However, for several decades, this description of the prokaryotic cell has been known to be inadequate [Mathews, 1993]. Many prokaryotic cells possess membrane-bounded structures that can be considered to be organelles as discussed in detail in a recent JMMB symposium [Saier and Bogdanov, 2013]. These organelles are usually specialized for a specific function such as photosynthesis [Drews, 2013; Woronowicz et al., 2013], magnetic field detection [Lower and Bazylinski, 2013; Murat, 2013] and ammonium oxidation [van Teeseling et al., 2013; see also the article in this symposium entitled “Focus on membrane differentiation and membrane domains in the prokaryotic cell” by Boekema, Scheffers, Bezouwen, Bolhuis and Folea.]. However, bacteria are known which appear to possess membranous nuclear envelopes superficially resembling eukaryotic nuclei [Fuerst and Sagulenko, 2013]. Further, outer membrane vesicles of Gram-negative bacteria are now known to function in communication, protection and pathogenesis although such vesicular structures were thought to be largely limited to eukaryotes just a few years ago [Manning and Kuehn, 2013; Schertzer and Whiteley, 2013; Vasilyeva et al., 2013]. Finally, some intracellular organelles such as acidocalcisomes are ubiquitous, being present in bacteria, archaea and eukaryotes [Caetano-Anolles and Seufferheld, 2013]. These observations have led to new hypotheses regarding the evolution of eukaryotic organelles which may have had prokaryotic origins [Seufferheld and Caetano-Anolles, 2013]. It is now crystal clear that prokaryotes as well as eukaryotes possess the abilities to synthesize a large variety of membrane-bounded organelles, many not found in eukaryotes [Arechaga, 2013; Bogdanov et al., 2013].
Considerations of prokaryotic compartmentalization have prompted the formulation of a second JMMB written symposium, one dealing with proteinaceous machines and microcompartments. The result of this effort is the present compendium. We do not try to provide a comprehensive survey of such structures. Indeed, there are far too many such structures to allow inclusion of them all in a single volume. Instead, we have selected some well-characterized examples of bacterial protein machines/microcompartments that serve the cell in diverse capacities. Some of these will be discussed in detail in the contributions included in this symposium while others will be briefly considered in this introductory article with reference to recent research and review articles. As hundreds of such machines actually occur in Nature, these topics will provide rich sources of information as guidelines for future research.
The “bag of enzymes” concept serves as grossly inaccurate picture of a prokaryotic cell. We now recognize that few enzymes “float” around in the cytoplasm, and that most enzymes form “hyperstructures,” some of them transiently, others permanently, allowing the direct flow of metabolites from one enzyme active site to another [Amar et al., 2008; Norris et al., 2007b]. This concept applies, for example, to the sugar-transporting phosphotransferase system and glycolysis [Barabote and Saier, 2005], collections of transport/metabolic systems including both biosynthetic and catabolic protein complexes [Norris et al., 2007a; Norris et al., 1999] and the bacterial flagellar and chemotaxis systems [Cabin-Flaman et al., 2005]. Indeed, most biologists now accept the notion that a majority of cellular reactions occur in microcompartments.
If the hyperstructural view of a bacterium, and thus of any living cell, is accepted, then a written symposium on microcompartments and protein apparati is both timely and relevant. It is our intent in organizing this symposium to be selective in describing some of the better characterized representative proteinaceous compartments. Others, yet to be studied in detail, can be examined using similar molecular biological approaches.
The Bacterial Cytoskeleton
The cytoskeleton is a key regulator and central organizer of many eukaryotic cellular processes including cell shape determination (morphogenesis), cell polarity, DNA segregation, cell division, phagocytosis, movement, and macromolecular trafficking [Fuesler and Li, 2012]. Most cytoskeletal elements known from eukaryotic cell studies appear to be present in bacteria, where they perform vital tasks relevant to many aspects of cell physiology (Table 1). Bacterial tubulins (FtsZ; BtubAB; TubZ; PhuZ), actins (MreB; ParM; MamK; AlfA; Alp7A), and intermediate filaments (IF; crescentin) are key elements in cell division, chromosome and plasmid segregation, and maintenance of proper cell shape, as well as in maintenance of cell polarity and assembly of intracellular organelle-like structures [Cabeen and Jacobs-Wagner, 2007; Madkour and Mayer, 2007; Wang and Shaevitz, 2013]. Although similar tasks are performed by eukaryotic cytoskeletal elements, the individual functions of specific bacterial tubulins, actins and IFs are in general different from those performed by their eukaryotic homologues, revealing a striking evolutionary plasticity of cytoskeletal proteins. However, similar to the functions of their eukaryotic counterparts, the functions conferred by bacterial cytoskeletal proteins are driven by their abilities to form dynamic filamentous structures [Pilhofer and Jensen, 2013]. Cytoskeletal elements were undoubtedly a prokaryotic invention, and additional bacterium-specific filament-forming elements such as MinD-type ATPases provide functions apparently not found in eukaryotes. Similarly, bacteriofilins and several other novel cytoskeletal proteins that seem to lack homologues in eukaryotes have been identified in prokaryotes [Kuhn et al., 2010]. Modern imaging approaches are revealing their functional roles [Celler et al., 2013]. The study of these proteinaceous compartments provides fundamental information relevant to all types of cells. They can reveal the molecular modes of action of cytoskeletal filament-forming proteins found universally [Graumann, 2007].
Table 1.
Superfamilies of filamentous proteins in bacteria
| 1. P-loop ATPases | 2. Actins | 3. Tubulins |
|---|---|---|
|
| ||
| ParA (bacteria & archaea) (e.g., plasmid Pl) | MreB: cell shape; chromosome segregation | BtubA/B of Prosthecobacter(function unknown) |
| SopA (e.g., F-plasmids) | FtsA: cell division | Archaeal tubulins (cell division) |
| Soj (in sporulation; with SpoOJ) | Various actin like protein (ALP) systems function independently of each other in plasmid segreation. e.g., ParM (with ParR-parC), R1. AlfA (with AlfB-alfC), pLS32. Alp7A (with Alp7R-alpC), pLS20. |
FtsZ (cell division) |
| Various ParA systems function independently of each other in plasmid and chromosomal segregation. | TubZ in Bacillus species (plasmid partitioning) PhuZ in Pseudomonas bacteriophage forms a spindle that centers replicating phage DNA |
|
The tubulin-like protein, FtsZ, is a highly conserved cytosolic GTPase, present in most bacteria and in many archaea. It forms a ring (the Z ring) at the future site of cytokinesis and plays an essential role in cell division. Although the E. coli FtsZ’s primary amino acid sequence identity to eukaryotic tubulin is low (~17%), its three-dimensional structure and assembly properties are remarkably similar to the eukaryotic homologues. Three other families of tubulin homologues have been described in bacteria (TubZ, BtubA/B, PhuZ). TubZ is a highly divergent tubulin relative that plays a role in plasmid segregation in many Bacillus species [Larsen et al., 2007]. Another divergent tubulin like protein, PhuZ is encoded by a bacteriophage that infects Pseudomonas species [Kraemer et al., 2012]. PhuZ assembles dynamic polymers in vivo that position replicating phage DNA in the center of the cell. Unlike most other tubulins, where the role of the C-terminal tail is to interact with other proteins, the tail of PhuZ plays an essential role in polymerization. Acidic amino acids within the tail make direct contact with a patch of basic amino acids in the next monomer within a filament. BtubA and BtubB from the bacterial genus Prosthecobacter, display higher sequence identity (~35%) and closer structural homology to eukaryotic tubulins. FtsZ tubulin diverged from a common ancestor early in evolution, whereas BtubA/B likely split from eukaryotic tubulin more recently. Their structural and organizational features have been reviewed [Souza, 2012], but the functions of BtuBa and BtubB are unknown.
The MreB and Mbl (MreB-like) proteins of Bacillus subtilis are required for different aspects of cell morphogenesis. They assemble into helical structures that run along the length of the cell. However they seldom act as single filaments and instead form complex superstructures [Popp and Robinson, 2012]. In addition to the highly conserved MreB subfamily of actins, bacteria contain many highly divergent actin-like proteins (ALPs) [Derman et al., 2009]. Most of the ALPs are present on mobile elements, typically plasmids, where they likely participate in plasmid DNA segregation.
The Caulobacter crescentus coiled-coil-rich protein crescentin has been shown to assemble into filaments that play a key role in determining the curved and helical shapes of this bacterium [Ausmees, 2006]. The protein has biochemical properties and a domain structure similar to those of eukaryotic IFs. Crescentin localizes to the concave side of the bacterium along its inner surface, associating with the cell membrane [Souza, 2012]. Coiled coil-rich proteins are also required for the maintenance of helical cell shape in the human pathogen Helicobacter pylori [Waidner et al., 2009], revealing a conserved function for this type of proteins.
In addition to homologues of eukaryotic cytoskeletal proteins (actins, tubulins, and IFs), a subclass of filament-forming Walker A ATPases, belonging to the large MinD-ParA superfamily, has been categorized as a unique class of bacterial cytoskeletal proteins [Gitai, 2007]. They form ATP-induced dynamic filaments in vivo and play important organizing roles during cell division (the MinD subgroup) and plasmid/chromosome DNA segregation (the ParA/Soj subgroup) [Kalliomaa-Sanford et al., 2012]. Although Walker A cytoskeletal ATPases are not obviously homologous to known eukaryotic cytoskeletal elements, they appear to be an additional component of the prokaryotic repertoire of cytoskeletal elements [Dye and Shapiro, 2007]. In many bacteria, the constituents of the cytoskeleton appear to form a single interconnected dynamic network that coordinates many cellular functions [Barry and Gitai, 2011].
Chromosomal and Plasmid Segregation in Bacteria
Chromosome segregation in eukaryotes is mediated by dynamic proteins such as tubulins, which polymerize into filamentous structures that are capable of rapid reorganization by means of cycles of depolymerization and repolymerization. The mitotic spindle, a network of microtubules, forms during prophase, attaches to the chromosomes and draws them apart. There seems to be a single segregation mechanism common to all eukaryotes [Wan et al., 2012].
In prokaryotes, segregation is much less well understood, but evidence suggests that there is not just one mechanism. There are at least three. Each one is mediated by a different family of filamentous proteins that can undergo polymerization/depolymerization in different ways in response to different signals. Some of these proteins are tabulated with their functions in Table 1 [Hazan and Ben-Yehuda, 2006; Jun and Wright, 2010].
Among the proteins listed in Table 1, the SopA P-loop ATPase segregates F-plasmids in E. coli [Komai et al., 2011]. It functions in plasmid positioning and segregation in a process involving its ATP-dependent polymerization (demonstrated both in vitro and in vivo). A SopB-sopC complex nucleates polymerization and co-localizes with SopA in the cell. The process is dynamic (as measured by fluorescence recovery after photobleaching (FRAP)), and mutants that lose their dynamic properties also can not segregate plasmids. The mechanisms by which SopA and other ParA systems segregate DNA are still not fully understood. One current view suggests that ParA proteins form a self-organizing system that uses chromosomal DNA as a scaffold [Hwang et al., 2013]. It has been observed that filaments emanate from SopB-sopC in radial asters (spindle-like). Moreover, SopA polymerization occurs at the same rate as chromosomal or plasmid segregation. It may be that SopA polymerizes and depolymerizes continuously and moves plasmids across the nucleoid so that in general, plasmids are well separated from each other. This ensures that each daughter cell obtains at least one plasmid. A schematic view of one model by which the segregation process occurs is presented in Figure 1 [Bouet et al., 2007; Hatano et al., 2007; Hwang et al., 2013; Pillet et al., 2011].
Figure 1.
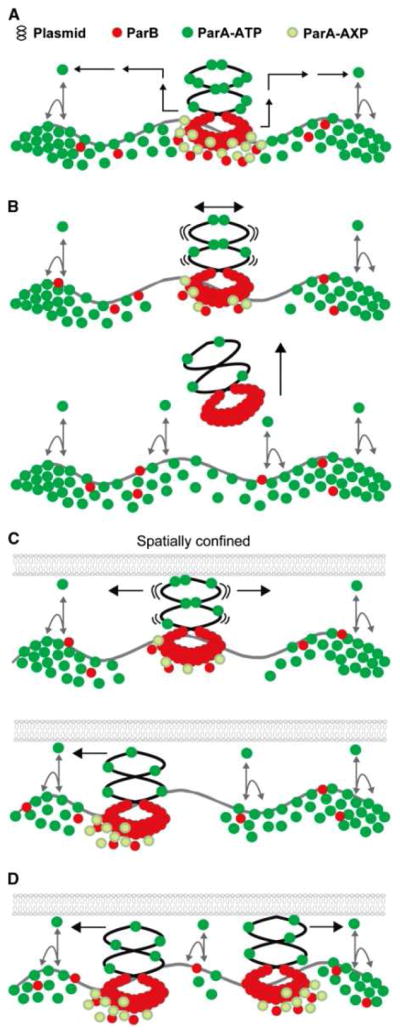
A diffusion-ratchet model for ParA-mediated plasmid partitioning. (A) ParB loads onto the plasmid at parS to form the partition complex. ParA-ATP dynamically binds the DNA carpet. ParA is converted to a specific state (ParA-AXP) on the DNA carpet (and plasmid) upon ParB interaction, which tethers the partition complex to the DNA carpet. ParA-AXP converts to ParA-ADP and releases from the DNA carpet in the vicinity of the partition complex. ADP to ATP nucleotide exchange and a series of ATP-dependent transitions (black arrows) delay ParA reactivation for DNA binding, thus allowing ParA to diffuse before rebinding the DNA carpet. (B) ParB-stimulated release of ParA decreases the anchor points between plasmid and the DNA carpet, and the plasmid begins to ‘wiggle’. This tethered Brownian motion accelerates the removal of ParA from the surrounding DNA carpet, creating a ParA-depletion zone. In the absence of spatial confinement, the partition complex releases from the DNA carpet when the last anchor point disassembles, and the ParA-depletion zone refills. (C) In a bacterial cell, the small gap between the inner membrane and nucleoid surface provides spatial confinement. Once a partition complex loses all anchor points and forms a ParA-depletion zone, its stochastic drift towards one direction would be reinforced by the high concentration of ParA contacts at its movement front. A reversal in direction would be suppressed by the ParA-depletion zone in the wake of movement. (D) A loss of inter-plasmid bridging within a partition complex leads to splitting of the complex. The split complexes would chase opposite slopes of the ParA gradient and separate. When two partition complexes approach each other, the converging motion would be stalled by the depletion of ParA between the two complexes, and their directions of motion would reverse. This figure was reproduced with permission from: Hwang et al. (2013) ParA-mediated plasmid partition driven by protein pattern self-organization. EMBO J. doi: 10.1038/emboj.2013.34
Bacterial actin-like proteins (ALPs) have been described that are involved in plasmid segregation and are divergent in both sequence and biochemical properties. The AlfA protein segregates plasmid LS32 in Bacillus subtilis (2 copies/chromosome). It functions with AlfB (encoded by the downstream gene), which is its adaptor bound to the plasmid, and during sporulation, with RacA (a sporulation chromosome remodeling protein). Segregation during asymmetric cell division in sporulating cells is not dependent on SpoIIIE, the septal DNA translocase, and plasmid segregation requires that AlfA assembles into filaments. The turnover rate is < 45 sec (as measured by FRAP). Mutants that do not turn over can not segregate plasmids, and thus, dynamic turnover is apparently required for segregation [Becker et al., 2006; Tanaka, 2010]. Alp7A from plasmid pLS20 forms polymers that display both dynamic instability and treadmilling [Derman et al., 2009].
Plasmid segregation into the forespore of the B. subtilis differentiating sporangium provides an interesting case. The forespore is about 1/8th of the volume of the mother cell, and therefore, precise segregation is required. Segregation into the forespore compartment is 2x slower in a racA mutant. The chromosomal Par proteins, Soj or Spo0J, as well as SpoIIIE, are apparently not involved, even though all 3 are required for chromosome segregation [Becker et al., 2006].
By analogy to AlpA, TubZ pushes low copy number plasmids towards opposite poles. Directional filamentous polymerization of a tubulin homologue, TubZ, in Bacillus species, is GTP-dependent and exhibits a rate of ~1 μ/min. Treadmilling is observed; subunits are added at the + end, and removed from the − end of the filament. This process, called “end assembly” [Larsen et al., 2007], is rapid and reversible. Depolymerization can occur without energy at a rate of > 2 μ/min. In this way, filaments are actively translocated unidirectionally through the cytoplasm. The same behavior is observed in many other bacteria. In elongated cells, the filaments can become > 10 μm in length with little or no depolymerization. Polymerization and depolymerization apparently occur independently of each other.
The Bacteriorhodopsin Light-driven Proton Pump
Bacteriorhodopsin is a homologue of mammalian rhodopsin (Yee et al., 2013, submitted), but their functions are very different. While bacteriorhodopsin is present in archaeal membranes in a 2-dimensional crystalline lattice called the “purple membrane,” smaller aggregates such as trimers may be present in bacterial membranes. In either case, bacteriorhodopsin pumps protons out of the cell in response to light absorption to generate a proton-motive force (pmf) [Lanyi, 2006]. By contrast, mammalian rhodopsin serves as a light receptor for the activation of nerves involved in vision [Palczewski, 2012]. Ion pumping is the primary purpose of bacteriorhodopsin and its closely related halorhodopsin, a chloride pump [Essen, 2002; Hirai et al., 2009; Schobert and Lanyi, 1982]. However, bacterial and archaeal homologues include sensory rhodopsins which activate phototactic systems through methylated chemotaxis proteins (MCPs) [Kawanabe et al., 2011; Klare et al., 2011]. Other homologues include channelrhodopsins which mediate ion fluxes in response to light-activated channel opening [Klare et al., 2008; Nagel et al., 2005]. These proteins are all derived from a common ancestral protein of 4 TMSs that duplicated internally to give an 8 TMS protein, and then, in many cases, an N-terminal TMS (or more rarely, a C-terminal TMS) was lost to give present-day 7 TMS proteins [Shlykov et al., 2012; Yee et al., 2013 (submitted)]. Included in this superfamily are several families of transporters, receptors and integral membrane chaperone proteins [Shlykov et al., 2012; Zhai et al., 2001; Yee et al., 2013 (submitted)]. Their high degree of functional divergence is particularly unusual and noteworthy.
The bacteriorhodopsin (BR) 3-D structure is known to 1.43 Å resolution [Schobert et al., 2002; PDB 1M0L]. This is the highest resolution structure currently available for a transmembrane transport protein. The structure, together with biochemical studies, has revealed the following details [Hirai et al., 2009; Lanyi, 2007]. Retinal (VitA aldehyde or retinaldehyde; one of the 3 forms of VitA, in the all-trans form) is nearly parallel to the plane of the membrane, bound to lysine216 in the middle of helix G (helix 7). The internal cavity is divided into half channels, cytoplasmic and external half channels that comprise the two parts of the transmembrane H+ pathway. In the resting state, the cytoplasmic half channel is more hydrophobic while the extracellular half channel is more hydrophilic with numerous bound water molecules. The N-terminus of the protein is outside while the C-terminus is inside. The high resolution structures revealed the exact hydrogen-bonding patterns as well as the bound water molecules, both essential in understanding how the proton is transferred from donor to acceptor in each photocycle step.
In bovine rhodopsin, Lysine296 (out of 348 aas) in TMS 7 is equivalent to K229 (out of 262 aas) in bacteriorhodopsin and is covalently linked to 11-cis retinal via a Schiff’s base (SB), essentially the same as in bacteriorhodopsin. As for bacteriorhodopsin, the N-terminus is outside while the C-terminus is inside. Although the chromophore and the covalent linkage are the same in the two proteins, their functions are surprisingly different. The structures of retinal, free and in Schiff’s base (SB) linkage to a lysine residue, are shown in Figure 2.
Figure 2.
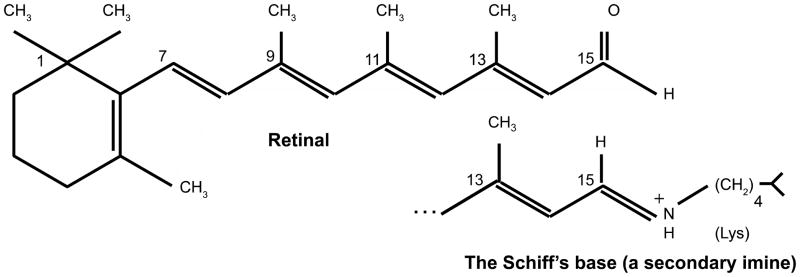
In mammalian rhodopsin (MR) and in bacteriorhodopsin (BR), a lysine residue K296 (of 348 aas) in MR, and K216 (of 262 aas) in BR is covalently linked to retinal (top figure) via a Schiff’s base (bottom figure). In both cases, the Schiff’s base is in the center of TMS 7, and the retinal separates the two half channels (see text).
Energy cycling in bacteriorhodopsin involves light (hυ) absorption which gives rise to photoisomerization of retinal from the all-trans configuration to the 13-cis isomer (Figures 2 and 3). This causes pKa changes in the SB and amino acyl residues of the protein. Sequential conformational changes yield differential access of the protonatable SB to the two half channels. Each step is fast but has its own characteristic half-life. The overall turn-over in the wild type protein is about 100 s−1, which is not fast compared to most enzymes, although it is comparable to several other pumps. In general, each step is faster than the following one. These changes allow access of the SB to deprotonation to the external medium and protonation only from the cytoplasm [Wang et al., 2013]. For a detailed model, see [Lanyi and Schobert, 2004] which presents a self-consistent but still somewhat controversial mechanistic model. H+ transport is unidirectional. In the final step, the slowest step, thermal reisomerization allows the 13-cis isoform to return to the all-trans isomer. Then the cycle repeats upon absorption of another photon. Steps in the photocycle have been defined as suggested from the stepwise scheme presented in Figure 4.
Figure 3.
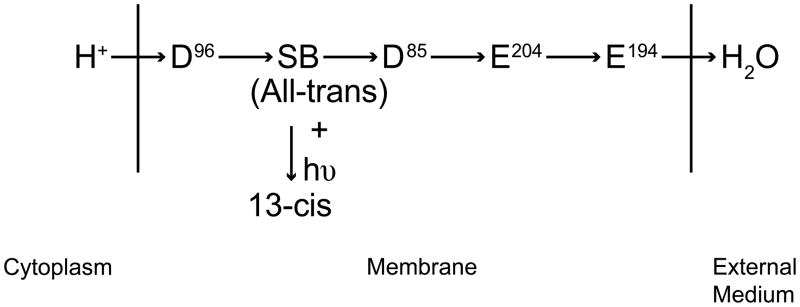
The proposed residue pathway for light (hυ)-driven bacteriorhodopsin proton pumping. A proton from the cytoplasm protonates aspartate 96 (D96) which transfers the H+ to the Schiff’s base (SB) which separates the two half channels. From the SB, the proton is sequentially passed to aspartate 85 (D85), glutamate 204 (E204), E194, and finally to water in the external medium.
Figure 4.

Proposed steps in the bacteriorhodopsin (BR) photocycle [see Fluhr et al., 2007; Hirai et al., 2009; Lanyi, 2004; Lanyi, 2006 for comprehensive reviews]. There is still some controversy about this proposal, such as the distances between D85 and the SB at various steps in the photocycle. For questions of reproducibility, signal/noise, controls, and an appreciation of the epistemology of the BR field, see [Lanyi, 2004].
F-type ATPases: A Rotary Motor
F-type ATPases are found in the membranes of virtually all prokaryotes as well as the mitochondria (α-proteobacterium-derived) and chloroplasts (cyanobacterium-derived) of eukaryotes. These molecular machines reversibly catalyze the hydrolysis of ATP while pumping protons or sodium ions out of the cell or organelle to generate a proton or sodium electrochemical gradient, negative inside, the pmf or the smf, respectively. In the reverse reaction, ATP is synthesized, driven by the pmf or smf. Thus, these enzyme transporters interconvert chemical and chemiosmotic energy.
Figure 5 presents structural and mechanistic aspects of F-type ATPases. These enzyme complexes are rotary machines wherein the subunits of the rotor rotate relative to the subunits of the stator. The stator includes the α3β3δab2 subunits (numbers refer to stoichiometries) while the rotor includes the c12γε subunits. Clockwise rotation results in ATP hydrolysis and ion pumping, while counterclockwise rotation is driven by transmembrane ion electrophoresis through the membrane sector of the ATPase with concomitant ATP synthesis [Kinosita et al., 1998].
Figure 5.

F-type ATPase constituents and structure. (A) The subunits and their properties. The membrane sector (F0) includes subunits indicated by arabic letters while the cytoplasmic sector (F1) includes subunits indicated by Greek letters. Subscripted numbers indicate the subunit stoichiometries. [Pedersen, 2007; Saroussi and Nelson, 2009]. TMS, transmembrane α-helical segment. (B) Structural depiction of the catalytic ATPase (F1 part showing arrangements of the 3 α and 3 β subunits. Numbers 1–3 refer to the three steps involved in the synthesis of ATP (or its hydrolysis, the reverse reaction)), the 3-step mechanism of action. (C) Overall schematized structure of the F-type ATPase complex showing association of subunits with the rotor (top) and stator (bottom; refer to (A) for rotor and stator subunit compositions).
Within the proton-conducting channel of the E. coli ATPase, an aspartate residue (D61) in c and an arginine residue (R210) in a, which together form parts of the channel, are essential. A histidine (H245) and a glutamate (E219) in a also play important roles. D61 is on the proton pathway and is reversibly protonated although R210 is always protonated. The stator component (a) and the rotor component (c) comprise the two half channels through which protons flow. Possibly the dipole of the protonated D61 interacts with the positive charge in R210 to create the torque for rotation. Thus, details of the molecular mechanism for energy interconversion are emerging.
The Bacterial Flagellum: Another Rotary Machine
A typical bacterial flagellum is about 10 microns long with a helical period of about two microns. The size of the flagellum is about 1 billion Da! It is an amazingly complex rotary machine. In E. coli, there are about 40 flagellum genes (flg, flh, fli), about 20 of which are structural genes [Apel and Surette, 2008]. Additionally, there are 2 mot (motor), 6 che (chemotaxis) and 5 primary receptor genes encoding methylated chemotaxis proteins (MCPs). There are also many secondary receptors. An E. coli cell has, on average, about 10 flagella. They rotate coordinately at roughly 20,000 rpm, driven by the pmf although some bacteria use the smf instead of the pmf, and still others, such as Vibrio parahaemolyticus, have two distinct flagella, one which uses the smf, and one that uses the pmf (see below). E. coli can swim at a speed of about 30 μ/sec (~ 2 mm/min). However, other bacteria can have up to 1,000 flagella per cell while swarming, and they can move at speeds that are much greater than those of E. coli, > 300 μ/sec (~ 20 mm/min) [Berg, 2003].
If E. coli were the size of a car, and its velocity were to be proportional to its size, how fast would it go? E. coli is about 1 μ long and swims at about 2 mm/min. This equals 120 mm/h or 0.12 meters/h. A car is about 5 meters in length and moves at about 65 mph, or 100 km/h, maximally about 250 km/h. The size difference is thus 5 × 106. 0.12 meters/h x 5 × 106 = 0.6 × 106 meters/h = 600 km/h. Not bad for a “simple” single celled organism, but remember, some bacteria can swim 10-fold faster, equivalent to 6,000 km/h, faster than any jet airplane! Bacteria are truly remarkable.
As noted above, the flagellar motor is a pmf- or smf-driven machine [Terashima et al., 2008]. The stator, MotAB, is the ion selector (H+ or Na+) [Asai et al., 2003], while the rotor is the flagellum (Table 3). The FliFGMN complex in the basal region of the flagellum interacts with the motor (Figure 6). In fact, the C-terminal domain of FliG interacts directly with MotAB to produce the torque. A cluster of charged residues on a ridge of the rotor serves as the site of interaction. FligG, in complex with FliF, M and N, interacts with MotAB. Only their genes, when mutated, give rise to three distinct phenotypes depending on the site of mutation, (1) fla (flagellum-defective), (2) mot (motor-defective) and (3) che (chemotaxis-defective). Thus, the phenotypes observed for mutants in these four genes, and these genes alone, can correspond to (1) the absence of flagella, (2) intact flagella but no motor function, and (3) normal flagella with an active motor but lacking chemotactic responses. This last phenotype is due to the fact that this subcomplex of four proteins is part of the “switch” that determines the direction of flagellar rotation. These observations reveal the multifaceted functions of this flagellar complex. MotAB complexes have been reconstituted in artificial membranes yielding channels specific for H+ or Na+ [Yonekura et al., 2011].
Table 3.
Targets of e− flow in GEOBACTER
|
Electron donors:
|
Uses:
|
Figure 6.
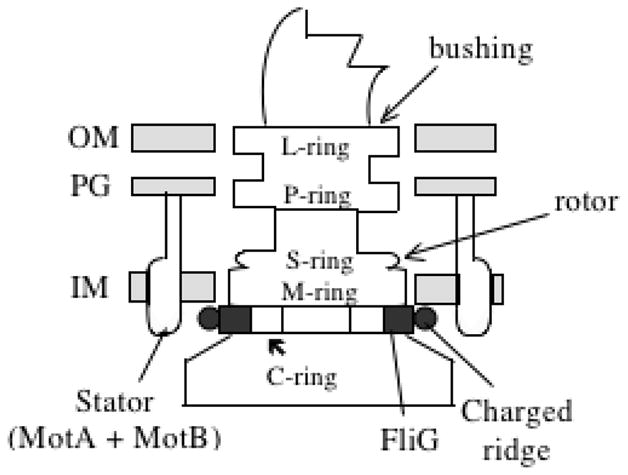
Structure of the flagellar basal region and its association with the bacterial cell envelope. OM, outer membrane; PG, peptidoglycan cell wall; IM, inner membrane. The rings visible in electron micrographs are: C - cytoplasmic; M - membrane; S - soluble (periplasmic) P - peptidoglycan (PG) and L - Lipopolysaccharide-containing outer membrane (OM). The letter in the name of each ring indicates the envelope structure with which it associates.
ExbBD-TonB and TolQR-TolA are sets of proteins that form complexes where ExbBD and TolQR are homologous to, but distantly related to MotAB stators (see Table 2). Their proteins of interaction are TonB and TolA, respectively, instead of FliG. However, few of these proteins function in motility [Bolam and Koropatkin, 2012; Krewulak and Vogel, 2011; Schalk et al., 2012]. Instead, they energize uptake of: (1) Cu2+, Ni2+ and Fe3+-siderophore complexes, (2) vitamins such as vitamin B12 and thiamin, (3) oligosaccharides, (4) phage DNA and (5) colicins (toxic proteins for enteric bacteria) across the outer membranes of Gram-negative bacteria. The uptake of (1–3) are the normal functions of the outer membrane receptors (OMR), but phage (4) and colicins (5) have parasitized some of these receptors to allow their uptake. Energization of TonB due to H+ fluxes via the H+ channel in ExbBD, and of TolA due to H+ fluxes via the H+ channel in TolQR, probably involves high energy conformations of these proteins that promote opening of the OMR porin channels, allowing translocation of the solute across the outer membrane and accumulation in the periplasm [Table 2; Zhang et al., 2011]. These solutes can be concentrated more than 1000 fold in the periplasm relative to the external medium. The energy source is the pmf across the cytoplasmic membrane. TonB and TolA are homologous proteins that interact directly with the OMR to control (energize) its conformation.
Table 2.
The Mot/Exb family of H+/Na+ energizers (motors)
| Process: | Flagellar Rotation
|
Outer Membrane Transport
|
||
|---|---|---|---|---|
| Energy: | pmf | smf | pmf | pmf |
| Motor: | MotAB | PomAB | ExbBD ↓ |
TolQR ↓ |
| Interact with: | FliG (rotor) | FliG (rotor) | TonB ↓ |
TolA ↓ |
| Many distinct OM receptors (OMRs) (substrate-specific porins) | ||||
| Topologies of homologous motors | |||
|---|---|---|---|
| Protein | #TMSs | Protein | #TMSs |
| MotA or PomA | 4 (N- and C-termini, cytoplasm) | ExbB or TolQ | 3 (N-terminus, periplasm, C-terminus, cytoplasm) |
| MotB or PomB | 1 | ExbD or TolR | 1 (interacts with TonB or TolA) |
| TonB or TolA | 1 (interacts with OMRs) | ||
The TolQRA complex also plays a role in the transport of lipopolysaccharide (LPS) to the outer surface of the outer membrane, and is thus indirectly involved in drug resistance [Gaspar et al., 2000]. An involvement in LPS export explains why TolQRA mutants have a “leaky” outer membrane and consequent increased drug sensitivities. Surprisingly, TolQRA systems in Myxobacteria have been implicated in motor function for adventurous gliding motility, a form of motility that is independent of flagella (see below) [Youderian et al., 2003].
MotB/PomB and ExbD/TolR have 1 TMS, with N-terminal cytoplasmic domains and C-terminal periplasmic peptidoglycan anchoring domains that are essential for their roles as parts of the stators of these systems (Table 2). These cell wall anchoring domains are found in many other proteins not functionally related to the homologous MotB/PomB/ExbD/TolR proteins. The conserved aspartate in MotB/ExbD is essential for transport and is involved in determining the cation (H+ vs Na+) specificity. Residues in MotA/ExbB are also H+/Na+ sites of recognition and binding that provide a pathway for cation translocation across the cytoplasmic membrane. Thus, as in the case of the F-type ATPase where the a- and c-subunits form the proton- or sodium-conduction pathway, two proteins in these systems (MotA and B, PomA and B, ExbB and D, or TolQ and R) comprise the cation channel.
Chemotaxis Involving Bacterial Flagella
Peritrichous flagella, as in E. coli, are essentially propellers. They form a bundle behind the bacterium when coordinately rotating counterclockwise (CCW) allowing smooth swimming, or they disperse when rotating clockwise [Larkin et al., 2007], allowing the bacteria to tumble and change direction. Negative cooperativity and adaptation allow responses to chemicals over a 105 fold range, from < 1 μM to > 10 mM. This is due to polar clustering of receptors and to receptor methylation [Eisenbach, 2007; Tindall et al., 2012; Webre et al., 2004]. There are five MCPs in E. coli: Tsr (transducer for serine), Tar (transducer for aspartate), Trg (transducer for ribose and galactose), Tap (transducer for peptides) and Aer (the aerotaxis MCP). Their relative proportions are about 12:6:1:1:1, respectively. All of these MCPs (except Aer) can sense multiple chemicals and physical conditions, not just the ones indicated by their names. The signaling pathway is illustrated in Figure 7. Six chemotaxis proteins are depicted in Figure 7A. A decrease in attractant (Att) binding, or an increase in repellent (Rep) binding to an MCP induces conformational changes that are transmitted via CheW to CheA, the histidine sensor kinase of the chemotaxis system. CheA phosphorylates CheY in a fast reaction that dictates the initial response. CheY-phosphate (CheY-P), but not CheY, binds to the flagellar switch (FliMNFG) to induce clockwise flagellar rotation and tumbling (change of direction) [Figure 7B; Larkin et al., 2007]. The opposite response is observed when the Att concentration increases or the Rep concentration decreases. The adaptation response to increases in attractant concentration involves MCP methylation by CheR, and to increases in repellent concentration, demethylation by CheB. Regulation occurs at the level of CheB while CheR acts constitutively. Thus, in a slow reaction, CheB is phosphorylated by CheA, promoting demethylation of the MCPs. This deactivates CheA and hence decreases the concentration of CheY-P. Restoration of normal swimming behavior, with the pre-stimulus rate of tumbling, ensues.
Figure 7.

(A) and (B), Signaling pathway and mechanism of chemotactic responses to attractant (Att) and repellent (Rep) in bacteria and archaea. The model is based largely on studies with E. coli methylated chemotaxis proteins (MCPs). For explanations, see the text. CheW (W): signal transduction protein connecting the MCP signal generation with the CheA (A) sensor kinase to control its activity. CheA phosphorylates CheY (Y), and CheY phosphorylation determines the direction of flagellar rotation during activation by binding to the basal region of the flagellum, FliM(NFG). CheR (R) is the methylating S-adenosyl methionine:MCP methyltransferase while CheB (B) is the MCP methylesterase, both involved in adaptation (see also Figure 8) [see Baker and Stock, 2007; Kojadinovic et al., 2013 for reviews and comparative analyses]. CheB is phosphorylated, and thereby activated, by CheA in a slow step. CheB catalyzes deamidation, converting certain glutamines (Gln) to glutamates (Glu) so they can be methylated by CheR. Minus Att, weaker interactions are observed between intersubunit periplasmic domains and intersubunit cytoplasmic domains as compared with when Att is present. Subunit exchange occurs – Att but not + Att. Almost all amino acid replacements in TMS1 → CCW; i.e., weaker TM1:TM2 interactions, stronger intersubunit interactions, and poorer signaling. Intrasubunit movements (TM1 relative to TM2) may initiate signaling, i.e., Att binding → relative displacement of the two TMs in the membrane. CheZ is a CheY-P phosphatase. (C) Schematized view of the structure of an MCP. (D) Proposed “piston” mechanism to explain the responses of an MCP to attractant (Att➂) binding and glutamate methylation (methylated glutamates, ▲). See text for details.
In Figures 7C and D, the schematized structure of an MCP (C) and a postulated mechanism of regulation (D) are presented. The extracytoplasmic receptor domain receives a signal from the external medium (chemical binding) that induces a conformational change in the MCP (C). This may give rise to a piston-like action as illustrated in (D).
Att binding to an MCP induces a conformational change so that the MCP becomes a poor substrate of CheB and a good substrate of CheR. Increased methylation using S-adenosylmethionine (SAM) as the methyl donor reduces the affinity for Att. Maybe Att binding induces conformational changes that render the glutamate residues more readily available for CheR-mediated methylation. Methylation removes the negative charge, and the protein returns to its previous more folded conformation (Figure 8).
Figure 8.
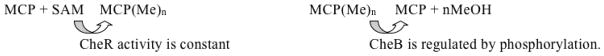
Both the MCP conformation and the relative activities of CheR and CheB determine the MCP methylation state. Conversion of the methylatable glutamates in an MCP to alanines by site specific mutagenesis blocks adaptation, but some adaptation still occurs due to cooperativity within the MCP lattice that results from methylation changes in other MCPs. SAM, S-adenosylmethionine; MeOH, methanol.
The overall response, including both excitation and adaptation (see below) can be described as follows: Prior to adding attractant, flagellar rotation reverses from CCW to CW with a certain frequency (the pre-stimulus CW/CCW ratio). Addition of attractant rapidly increases the tendency for CCW rotation yielding smooth swimming. This is the excitation phase. Slow increases in MCP methylation also results, over a period of many seconds, and as long as a couple of minutes. This gradually causes the CW/CCW ratio to increase, approaching the pre-stimulus CW/CCW ratio. Then the system is ready to respond to another change in the attractant (or repellent) concentration.
The receptor complexes are built of trimers of dimers giving rise to a 2-dimensional hexagonal lattice (built from trigonal units). The hexagonal unit consists of 6 MCPs (dimers) + 2 Ws (monomers) + 2 half As (dimers). Each CheA dimer is shared by two units, and each unit differs with respect to its composition containing variable numbers of the 5 MCPs. Minor MCPs show amplification. A proposed mechanism of MCP action [Kojadinovic et al., 2013] is shown in Figure 7D and involves several steps. A piston-like displacement of one TMS in the monomer relative to the other is the primary signal. Subunit interactions play a role as documented for MCPs such as Tar: counterclockwise (CCW) rotation correlates with formation of oligomers while clockwise rotation correlates with the opposite. Thus, Att promotes supramolecular complex formation. Lateral signaling within the lattice gives rise to sigmoidal kinetic responses by the kinase, CheA. The decrease in complex formation caused by Rep gives rise to broader than hyperbolic responses. Following the excitation phase in response to the addition of an attractant that causes CCW rotation and smooth swimming, the slower adaptation response occurs as follows:
![]()
Finally, after completion of the adaptation phase, the near pre-stimulus distribution of CW/CCW rotation, and hence the near pre-stimulus frequency of tumbling, ensues.
CheR has two domains, N-terminal and C-terminal, where N is for MCP binding only, while C is for MCP binding and catalysis. Some MCPs such as the major MCPs, Tar and Tsr, but not the minor MCPs such as Trg, have C-terminal binding sites (the “docking sites”) for CheR and CheB, necessary for regulation by methylation (Figure 7C). From these sites, CheR can methylate all MCPs, and CheB can demethylate them because the trimers of dimers are mixed. The methylation sites (Glutamate (Glu) residues) in all five MCPs are physically near the docking sites. Mutation of the docking site on Tsr slows methylation of all MCPs.
CheB also has two domains: a phosphorylatable receiver (regulatory) domain and a catalytic (methylesterase) domain. Without phosphorylation, the receiver binds the active site and the enzyme is inactive. With phosphorylation, the active site becomes free and is therefore activated for methylester hydrolysis. CheB docks to the CheR binding site.
The switch/motor complex is perceived as follows: Free CheY binds CheA but not FliM, but CheY-P binds FliM but not CheA. The frequency of CW rotation may increase sigmoidally with the concentration of CheY-P in a cooperative fashion, but this suggestion has been disputed [Alon et al., 1998; Liu et al., 1997]. The FliM(NFG) complex comprises the switch (gear box) as well as part of the motor. This complex has a stoichiometry of ~1 FliF, ~1 FliG, ~1 FliM, and ~3 FliN, + an unknown stoichiometry of MotAB complexes. Many models for the motor and switch have been proposed, but the C-terminus of FliG clearly binds MotA. The two directions (CW and CCW) correlate with and may be determined by various charged residue interactions that influence the protein complex’s conformation.
Swimming and swarming in Vibrio parahaemolyticus (a causative agent of gastroenteritis) results from the presence of two motility systems that are independent of each other (NOT like E. coli). Polar flagella are for swimming. They are sheathed and use the smf. These flagellar filaments are made of 6 flagellins, all of which are important; 3 represent major subunits while 3 are minor. Surprisingly this constitutively synthesized polar flagellum also serves as a mechanosensor. The lateral flagella are for swarming. They are non-sheathed, use the pmf and are made of a single flagellin. Their synthesis responds to and is induced by increased viscosity or surface growth using the polar flagellum as the sensor. Induction of lateral flagella occurs when: (1) ↑ viscosity → ↓ rotation of polar flagella. (2) Growth on surfaces → ↓ rotation. (3) Antibody binding to polar flagellin → ↓ rotation. (4) Na+ channel blockers are present (induction is proportional to the percent inhibition of rotation) → ↓ rotation. In other words, anything that decreases the rotation rate of the polar flagellum induces lateral flagellar synthesis. Induction also occurs when mutations are introduced in flagellar constituents that block flagellar assembly (and hence block rotation). Mutations that arise in the motor similarly induce lateral flagellar production, although this is not true for all swarmer cell types.
Induction of swarming is a complex process involving several coordinated events. These include: (1) Cessation of cell division. (2) Elongation of the cells, and (3) induction of all swarmer cell genes in four stages. The sets of genes sequentially induced are: (A.) Regulatory genes (flhDC). (B.) Basal body and hook genes. (C.) Motor and hook-associated protein, as well as the stage 4 flagellar sigma factor, σ28, and its anti-sigma factor, FlgM genes. (D.) Flagellin + chemotaxis genes. Induction of lipopeptide surfactin genes also occurs, facilitating swarming over solid surfaces. While E. coli has just one CheY and five MCPs, another Vibrio species, Vibrio cholera, has 5 CheYs and > 40 MCPs, indicating a much higher level of complexity.
Spirochetes exhibit a variation on the flagellar motility theme. Their flagella are periplasmic, and they contain: (1) FlaA sheath proteins (secreted and processed via the General Secretory (Sec) Pathway), (2) FlaB flagellins which are not processed, (3) all other flagellar constituents, and (4) some motility/chemotaxis genes unique to spriochetes. The flagella are attached at both ends, but as for E. coli, rotation is pmf-driven. As a result of periplasmic flagellar rotation, the spiral shaped cells “screw” through the medium by a still poorly understood mechanism.
Other types of bacterial motility are recognized and involve: (1) Twitching or gliding (due to retraction/extension of type IV pili) as occurs in Pseudomonas species and Myxobacteria; (2) Gliding, involving cell surface macromolecular translocation as occurs in Flavobacteria; (3) Gliding, dependent on mucopolysaccharide (slime) secretion (Myxobacteria and Cyanobacteria) and (4) gliding involving contractile cytoskeleton-mediated cell morphological changes (Mycoplasma). Some of these will be briefly discussed in the next section.
Poorly Characterized Types of Bacterial Motility
I. Archaeal Flagella are Rotary Type IV Pili
Archaeal flagella are rotatory and pmf-driven [Ghosh and Albers, 2011]. They have a “hook” but no observable basal body with rings, comparable to those of bacterial flagella although cell envelope anchoring proteins may be present. Multiple (2–6) pilins are secreted, processed by a prepilin peptidase, glycosylated, and incorporated into the base of the growing pilus. The hook protein may be one of the pilins. A single operon with about 12 genes usually codes for the complete structure. An ATPase and an integral membrane protein common to bacterial type IV pili are always present [Peabody et al., 2003]. Chemotaxis proteins like those in bacteria (including CheY) are present in archaea, although a FliM homologue has not been found. Thus, although bacterial flagella are absent in archaea, the chemosensory apparatus appears to be essentially the same.
II. Social Gliding Motility via Retractable Type IV Pili in Myxobacteria
Type IV pili mediate “twitching” motility in Pseudomonas aeruginosa [Burrows, 2012] and “social” gliding motility in Myxococcus xanthus [Kaiser, 2003]. Both depend on cell-cell or cell-surface contacts. The pilus fiber consists of pilin subunits (PilA plus minor pilins) with an N-terminal α-helical core and a C-terminal β-sheet surface structure. There are 5 pilin monomers per helical turn. In P. aeruginosa, over 35 genes are involved in motility and taxis. Three polar ATPases may energize pilus assembly and extension (PilB) and retraction, respectively (PilU and PilT) [Chiang et al., 2005; Chiang et al., 2008]. An adhesin (PilY1) is at the tip of the pilus. PilQ is a channel-forming “secretin” in the outer membrane that allows the pilus to cross this membrane. Type IV bacterial pili, in addition to adherence and motility, are involved in biofilm formation, competence for DNA uptake and pathogenesis. In P. aeruginosa, pilus retraction pulls the cell forward; no movement is observed during pilus extension. As chemotaxis proteins control pilus assembly and activity, CheY-like proteins may interact with the PilB and/or PilT ATPases [Bertrand et al., 2010].
Type IV pilus-mediated motility in M. xanthus is smooth and does not display twitching. However, like twitching motility in P. aeruginosa it requires extension and retraction of Type IV pili assembled at the leading cell pole. During cellular reversal, resulting in a change of direction, the pili disassemble at the old leading pole and reassemble at the opposite new leading pole [Bulyha et al., 2009]. An extracellular polysaccharide receptor, deposited on the surfaces of cells or the substratum to which the pilus adhesin attaches, is also required. A 3-component ABC transporter for macromolecular secretion may also be involved. One interesting difference between Pseudomonas and Myxococcus Type IV-mediated motility is that Pseudomonas cells generally produce pili from only a single cell pole and rarely reverse, while Myxococcus cells reverse periodically about every 7–10 min. During reversals, the cellular polarity axis becomes inverted, and the pili assemble at the opposite cell pole [Bulyha et al., 2013; Bulyha et al., 2009].
The chemotaxis proteins control cellular reversals; A CheY homolog (called FrzZ), when phosphorylated, localizes to the leading cell pole where it interacts with MglA, a cell polarity regulator with GTPase activity, triggering reversals [Kaimer and Zusman, 2013]. Further, two additional small GTPases act in concert with the bacteriofilin cytoskeleton to regulate the dynamic polarity properties of the cell upon which directional motility and reversal depend [Bulyha et al., 2013]. A dependency of cell shape and motility on the cytoskeleton is reminiscent of similar evidence for wall-less Mycoplasma and Spiroplasma cells [Calisto et al., 2012; Cohen-Krausz et al., 2011].
III. Adventurous Gliding Motility in Myxobacteria
Both δ-proteobacteria and cyanobacteria exhibit gliding motility [Wolgemuth and Oster, 2004]. In M. xanthus, “adventurous” gliding motility, or A-motility, involves the movement of single cells rather than groups of cells as in the case of social motility [Hodgkin and Kaiser, 1977]. The mechanism of Myxococcus A-motility gliding has been difficult to understand since surface structures are apparently not involved. Adventurous motility allows velocities of ~3 μ/min, ~500-fold slower than flagellar motility in typical bacteria such as E. coli.
The work of Youdarian et al. [2003] showed that about 40–50 genes are responsible for A-motility. Recently, Mignot and co-workers [Ducret et al., 2013; Ducret et al., 2012; Sun et al., 2011] showed that A-motility is powered by the pmf and is sensitive to A22, an inhibitor of MreB, a bacterial cytoskeletal protein and actin homolog. They also showed that Myxococcus contains MotA/MotB homologues and that these are required for gliding. Nan et al. [2011; 2010] showed that these bacterial stator proteins function as motors in M. xanthus, although they lack the peptidoglycan binding motif responsible for anchoring stators in flagellated bacteria to the cell wall. They are therefore free to move within the membrane. Current data are consistent with a model in which A-motility is powered by large motility complexes containing the MotAB homologs. These complexes move within the membrane on a helical cytoskeletal filament, possibly consisting of an actin-like MreB homolog. The large clusters generate torque by accumulating as clusters on contacting a surface, exerting force on the surface. Evidence for this model is supported by single molecule studies with the MotA homolog using photoactivatable localization microscopy (PALM) [Nan et al., 2013]. Directionality is regulated by phosphorylation of myxobacterial chemotaxis system proteins [Kaimer and Zusman, 2013]. This model unifies elements of gliding and swimming motility.
IV. Ratchet Structures (Cytophaga/Flavobacteria)
Motility in Flavobacterium johnsoniae appears to be due to pmf-driven movement of cell surface components. Surface protein complexes may extend through the envelope. An ABC transporter with 3 components (GldA, F and G) seems to play a role although it is not present in all bacteria that apparently use this mechanism of gliding. Early studies suggested that slime is extruded at one cellular location and then taken up from another.
F. johnsoniae cells move rapidly over surfaces, and Gld proteins are thought to comprise the multicomponent motor that propels the cell surface adhesin SprB. Cells with mutations in the sprB gene are partially defective in motility. RemA is a cell surface protein that has a lectin domain and appears to interact with polysaccharides. Three other proteins, RemC, Wza, and Wzc, are predicted to be involved in polysaccharide synthesis and secretion [Shrivastava et al., 2012]. RemA localizes to the cell surface and is propelled rapidly along the cell at speeds of 1 to 2 μm/sec. Deletion of the gldN and gldO genes, which encode components of a protein secretion system, block the transport of RemA to the cell surface, and overproduction of RemA results in the formation of cell aggregates that are dispersed by the addition of galactose or rhamnose [Shrivastava et al., 2012]. Cells lacking RemC, Wza or Wzc fail to aggregate. Cells of a remC mutant and cells of a remA mutant, neither of which form aggregates in isolation, aggregate when they are mixed together. This fact suggests that polysaccharides secreted by one cell may interact with RemA on another cell. Fluorescently labeled lectin, Ricinus communis agglutinin I, detected polysaccharides secreted by F. johnsoniae. Polysaccharides associated with cells expressing RemA were rapidly propelled on the cell surface. RemA thus appears to be a mobile cell surface adhesin, and secreted polysaccharides may interact with the lectin domain of RemA to enhance motility [Shrivastava et al., 2012].
Bacteria of the phylum, Bacteroidetes, are diverse, with rapid gliding motility. A novel protein secretion system, the Por secretion system (PorSS) is present in F. johnsoniae, but the sequences of its constituents are not similar to those of other well-studied bacterial secretion systems. They are a subset of the gliding motility genes, suggesting a role in motility. PorSS is needed for assembly of the gliding motility apparatus as well as for secretion of a chitinase. Comparative analysis of 37 Bacteroidetes genomes revealed the widespread concurrent occurrence of gliding motility genes and porSS genes [McBride and Zhu, 2013].
Bioelectricity via Nanowires (Bug Juice)
Most bacteria use soluble cytoplasmic electron acceptors for respiration, but some use insoluble extracellular electron acceptors. There are multiple mechanisms that allow for such a use. One such mechanism relies on nanowires that conduct electrons to insoluble oxides of manganese and iron (e.g., rust; Fe2O3). Nanowires are type IV pili, but most type IV pili do not conduct electricity, so nanowires have special characteristics that are poorly understood at the molecular level. The bacteria reduce ferric oxide to magnetite (Fe3O4), a magnetic substance found in nature and in cells (including ancient bacterial fossils billions of years old on Earth and possibly on Mars) [Jones, 2006]. Bacteria that have been shown to use nanowires to transmit electrons to Fe2O3 include: Geobacter sulfurreducens (a δ-proteobacterium), Shewanella (a γ-proteobacterium), Synechococcus (a cyanobacterium) and Pelomaulum (a thermophilic firmicute). The identification of nanowires in such a wide range of bacterial phyla suggests that they are widespread in the prokaryotic world [Reguera et al., 2005; Reguera et al., 2006].
Geobacter was discovered by Derek Lovley in 1987 at the University of Massachusetts, Amherst [Lovley, 2006]. It is a strict anaerobe that respires using extracellular electron acceptors via type IV pilus-type nanowires. It oxidizes organic molecules to reduce external substances. Biofilms conduct electricity to external Fe2O3 more efficiently than cell suspensions. They have potential as tools in bioengineering (see Table 3). Electrons are believed to pass through a chain of electron carriers (mostly cytochrome c’s) as follows [Londer et al., 2006]: NADH → NADH DH → menaquinone (MQ) → MacA (inner membrane (IM)) → PpcA (periplasm) → OmeB (outer membrane (OM)) → OmeE (outer surface of the OM) → OmeS (outer surface of the OM) → pili → Fe2O3. In essence, Derek Lovley and independently, Ken Nealson, created the fields of geomicrobiology, bionanotechnology and microbiological electronics as a result of their discoveries of extracellular electron flow [Gorby et al., 2006]. Although it sounds like science fiction, it has been reported that live bacteria can use nanowires to energize “dead” bacteria, bringing them back to life while creating a membrane potential, negative inside.
Protein Secretion/Membrane Insertion Machines
We have previously reviewed protein secretion systems in bacteria, summarizing the results of thousands of research articles dealing with this topic [Saier, 2006; Saier et al., 2008]. All living organisms possess the essential rudiments of the General Secretory (Sec) Pathway, described in the Transporter Classification (TC) Database, TCDB (www.tcdb.org) under TC# 3.A.5. This pathway in E. coli is illustrated in Figure 9. Both soluble proteins and integral membrane proteins can be secreted and inserted into the cytoplasmic membrane, respectively, via the Sec system. The common constituents found in all living organisms comprise the SecYEG channel complex through which substrate proteins must pass to get to the external surface of the membrane or to be inserted into the membrane (Figure 9). However, in Gram-negative bacteria with two membranes separating the cytoplasm from the external environment, a plethora of protein exporting transport systems have evolved. In fact, at least sixteen independently evolving systems are known [see Table 4; Saier et al., 2008]. Export across the cytoplasmic membrane is usually energy-dependent, and the most common source of energy is ATP. In the symposium article by Peña and Arechaga entitled “Molecular Motors in Bacterial Secretion,” the ATPases that drive secretion via several of these systems are discussed and compared.
Figure 9.
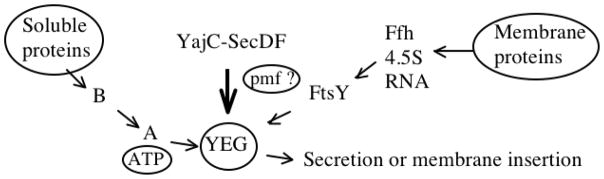
The ubiquitous general secretory (Sec) system for export of proteins from the cell (top left) as well as insertion of integral proteins into the membrane (top right). SecB, SecA, SecY, SecE and SecG are indicated by single letters. Steps involving energy (ATP or the pmf) are indicated. See [Cao and Saier, 2003] for a review.
Table 4.
Protein Secretory Pathways (PSP) in living organisms
| SP # | Type | Name (Pathway) | TC # | Bact1 | Arch | Euk | # Prots | Energy |
|---|---|---|---|---|---|---|---|---|
| I | ABC | ATP-binding cassette translocase | 3.A.1 | + | + | (+) | 1–2 | ATP |
| IIa | Sec | General secretory translocase | 3.A.5 | + | + | + | ~12 | GTP or ATP + pmf |
| IIb | MTB | Main terminal branch translocase (Gram- bacterial outer membrane translocase) | 3.A.15 | + | − | − | ~14 | ATP? |
| III | Fla/Path | Flagellum/pathogenesis-related translocase | 3.A.6 | + | − | − | >10 | ATP |
| IV | Conj/Vir | Conjugation/virulence-related translocase | 3.A.7 | + | − | − | >6 | ATP |
| V | Tat | Twin arginine targeting translocase | 2.A.64 | + | + | + (chloro)2 | 5 | pmf |
| VI | MPT | Mitochondrial protein translocase | 3.A.8 | − | − | + (mito) | ~20 | ATP |
| VII | CEPT | Chloroplast envelope protein translocase | 3.A.9 | + | − | + (chloro) | 3 | GTP |
| VIII | Oxa1 | Cytochrome oxidase biogenesis | 2.A.9 | + | + | + (mito; chloro) | 1? | pmf |
| IX | MscL | Mechanosensitive channel | 1.A.22 | + | + | + | 1 | None |
| X | Holins | Holins (inner bacterial membranes) (cytolytic) | 1.E.1–1.E.52 | + | − | − | 1 | None |
| XI | Toxins | Diphtheria, botulinum, tetanus toxins (target cell membrane) | 1.C.7; 1.C.8 | + | − | − | 1 | None |
| XII | OMPs | Bacterial outer membrane channel-forming translocase | ||||||
| (a) FUP | Fimbrial usher protein | 1.B.11 | + | − | − | 1 | None | |
| (b) AT-1 | Autotransporter-1 | 1.B.12 | + | − | − | 1 | None | |
| (c) OMR | Outer membrane receptor | 1.B.14 | + | − | − | 1 | None | |
| (d) OMF | Outer-membrane factor | 1.B.17 | + | − | − | 1 | None | |
| (e) TPS | Two-partner secretion | 1.B.20 | + | − | − | 1 | None | |
| (f) Secretin | Secretin | 1.B.22 | + | − | − | 1 | None | |
| (g) OmpIP | Outer-membrane protein insertase | 1.B.33 | + | − | +? | 5 | ? | |
| (h) AT-2 | Autotransporter-2 | 1.B.40 | + | − | − | 1 | None |
Bact: bacteria; Arch: archaea; Euk: eukaryotes; # Prots; approximate number of dissimilar proteins in a system. Some such systems have variable numbers of proteins depending on the system and the conditions. Some of these proteins are auxiliary constituents that are not essential for activity.
chloro: chloroplast; mito: mitochondria.
A few protein secretion systems do not rely on ATPases and instead use the proton motive force. In order to utilize this energy source, any such system must allow the flow of protons down their electrochemical gradient. One well studied system is the Twin Arginine Targeting (Tat) system (TC# 2.A.64). Another type of system that uses the pmf is the YidC/Oxa1/Amber systems of prokaryotes, mitochondria and chloroplasts, respectively (TC# 2.A.9). These homologous systems function in integral membrane insertion rather than secretion, sometimes in conjunction with the SecYEG apparatus, but in other cases, independently of this complex.
Other bacterial secretion systems are known – types I [TC# 3.A.1; Holland et al., 2005], II [2.A.15; Korotkov et al., 2012], III [3.A.6; Chatterjee et al., 2013], IV [3.A.7; Voth et al., 2012] and VI [Silverman et al., 2012]. All of these systems are multicomponent, and all of them span the two membranes of the Gram-negative bacterial cell envelope. All except type II (also called the “Main Terminal Branch,” MTB, TC# 3.A.15) allow transport of proteins from the cytoplasm directly into the external milieu in a single energy-coupled step. In fact, type III and type IV systems allow injection of proteins, and in the case of type IV, DNA-protein complexes, directly from the donor bacterial cytoplasm into the cytoplasm of a recipient bacterium, or in the case of bacterial pathogens and symbionts, into that of a host plant or animal cell. These systems serve as complex molecular machines usually involving about a dozen different proteins. The reader is referred to Saier et al. (2008), the symposium article by Peña and Arechaga, and the system-specific reviews cited above for more detailed considerations of these systems.
Chaperonins and Protein Folding
The pathway for protein folding in E. coli is multistep, involving three chaperone complexes; DnaJK, GrpE and GroEL/ES [Houry, 2001; Langer et al., 1992; Mansell et al., 2008]. This proposed pathways is:
[P = the protein substrate; U = unfolded; PF1 and PF2 = partially folded intermediates; PF = fully folded protein].
Protein folding by GroEL/ES has been characterized in some detail [Azia et al., 2012; Chaudhuri et al., 2009]. The substrate protein, P, can exist in 3 or more states outside of GroEL: (a) PU, (b) PPF, and (c) PF. P can also exist in 3 or more states inside of the GroEL barrel: (a) PS bound to the inner surface of the lid or chamber lips near the opening, (b) Pi bound internally to the closed chamber in a partially folded state, and (c) PF, the fully folded state, ready for release. The GroES lid is a single domain protein (subunit MW = 10 kDa, 7 subunits) which forms a circular heptameric ring structure. The 3 domain GroEL barrel subunit (subunit MW = 56 kDa, 14 subunits) can exist in 3 states: (a) extended + ATP, (b) extended + ADP and (c) contracted. A half of sites mechanism is operative where protein folding only occurs in the extended not the contracted chamber (see Figure 10).
Figure 10.
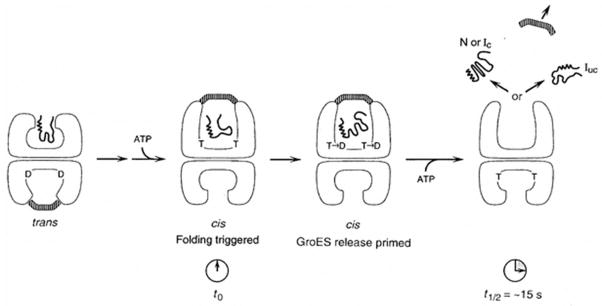
Schematic representation of the action of ATP in cis (top) and trans (bottom) compartments of the GroEL/ES complex during GroEL-GroES-mediated protein folding. The substrate polypeptide is initially bound in the trans compartment (bottom) of a GroEL-GroES complex, and a step of ATP-triggered substrate protein/GroES release then occurs. This results in formation of a cis complex (top) in which a second substrate polypeptide is sequestered in the GroEL central channel underneath GroES. The act of formation of the cis complex, through binding of ATP and GroES to the ring with bound polypeptide, triggers rapid release of the substrate polypeptide from the apical binding sites into the central channel, and folding begins (t0). Hydrolysis of ATP (T) to ADP (D) in the cis ring weakens the high affinity interaction between GroES and (cis)ATP-bound GroEL. Binding of ATP in trans subsequently causes release of GroES and polypeptide from the cis chamber. N, native; L, non-native conformation committed to completing folding to the native form (no longer recognizable by chaperonin); tUC, non-native form that must be rebound by chaperonin or by a different chaperone to reach its native form [Figure adapted with permission from an article by Rye et al., 1997].
The specific steps proposed are as follows: (1) The GroESlid binds and closes the cis cavity with the substrate, a partially folded protein, inside. (2) GroES displaces PU (or PPF) to the central cis cavity. (3) Folding begins. (4) ATP is hydrolyzed to ADP in a slow process, during which folding continues; GroES loosens. (5) ATP7 binds to the trans ring; ADP, PF and the GroES lid are released from the cis compartment. (6) Another substrate protein binds to the lips of the trans compartment which now becomes expanded while the cis compartment assumes the contracted state. (7) The cycle repeats in the trans compartment [Grallert and Buchner, 2001; Walter, 2002].
Proteasomes and Regulated Protein Degradation
Proteasomes resemble chaperonins in some structural and functional respects. They consist of heptameric rings of α and β subunits. The similarities with chaperonins suggest that there could be an evolutionary link. However, the subunit arrangements are substantially different. There are two rings of α subunits and two rings of β subunits in an α- β- β- α vertical arrangement. Protein substrates are degraded within the proteasome compartment in an ATP hydrolysis-dependent fashion. These structures and their modes of action are discussed in detail by Humbard and Maupin-Furlow in the article entitled “Prokaryotic Proteasomes – Nanocompartments of Degradation” in this written symposium. These structures will therefore not be considered further here.
Not all cytoplasmic protein degradation occurs within proteasomes. Alternatives are discussed in the symposium article entitled “Control of Protein Function Through Regulated Degradation – Biotechnological and Biomedical Applications” by Nagpal, Tan, Heras, Truscott and Dougan. This article considers adaptor proteins and unfoldases that influence these protease activities and also discusses novel antimicrobial agents that target these machines.
Bacteria possess a variety of ATP-dependent and ATP-independent proteases. E. coli, for example has five AAA+ ATP-dependent proteases, FtsH, Lon, ClpXP, ClpAP and HslUV, which play roles in cell physiology and programmed cell death. All are self-compartmentalizing cytoplasmic or membrane anchored proteolytic complexes with the enzymatic active sites in ring shaped or barrel structures [Langklotz et al., 2012]. Other proteases such as the ubiquitous rhomboid and CAAX proteases are integral membrane proteins that hydrolyze peptide bonds in transmembrane proteins and newly synthesized lipoproteins, respectively [Pryor et al., 2013; Urban and Dickey, 2011]. They function in several capacities such as initiating cell signaling, facilitating quorum sensing and regulating homeostasis. Intracellular protein degradation has become recognized in recent years as a highly complex means of regulating hundreds of aspects of cell physiology.
Carboxysomes for CO2 Fixation
One of the best studied metabolic microcompartments is the carboxysome found in cyanobacteria and chemoautotrophs [Kerfeld et al., 2010; see symposium article by Yeats, Jorda and Bobik entitled “The Shells of BMC-Type Microcompartment Organelles in Bacteria”]. These structures house the enzyme D-ribulose 1,5 bisphosphate carboxylase/oxygenase (RuBisCO) within assembled icosahedral protein shells, superficially similar to some viral particles [Kerfeld et al., 2005]. Their primary functions are to dehydrate bicarbonate (HCO3−) and to concentrate CO2 for fixation by RuBisCO [Cot et al., 2008]. Carboxysomes concentrate bicarbonate and carbon dioxide using transport systems in the cell membrane as well as the microcompartment. A carboxysomal “shell” protein can open and close, allowing selective permeability by diffusion. The different types of carboxysomes and their structures, physiology, mechanism of action and evolution are the subjects of the intriguing symposium article by Rae, Long, Badger and Price entitled “Cyanobacterial Carboxysomes: Micro-compartments that Facilitate CO2 Fixation.”
Ethanolamine Utilization (Eut) Nanocompartments
Related bacterial microcompartments for the utilization of 1,2-propanediol (Pdu), ethanol (Etu) and ethanolamine (Eut) have been identified and characterized (see symposium article by Yeats, Jorda and Bobik entitled “The Shells of BMC-Type Microcompartment Organelles in Bacteria”). The symposium article by Held, Quin and Schmidt-Dannert entitled “Eut Bacterial Micro-compartments: Insight into their Function, Structure and Bioengineering Potential” provides detailed information about one of these machines. These structures sequester volatile and toxic metabolites and therefore provide the foundations of metabolic efficiency and cellular protection. The high resolution X-ray crystallographic structures of four homologous shell constituents of the E. coli Eut complex have been solved [Tanaka et al., 2010].
One of these (EutC) apparently undergoes conformational transitions that promote gating of metabolites through pores in the protein shell. These microcompartments are found in a range of bacteria from proteobacteria to firmicutes, and therefore are presumed to be ancient structures. Crystal structures of clostridial shell proteins revealed details of their hexameric quaternary 3-D permutated structures following self association into arrays [Pitts et al., 2012]. In addition to the symposium article by Held et al, the interested reader is referred to the recent publication of Choudhary et al [2012], detailing the engineering of these nanocompartments for biosynthesis and catalysis. The use of engineered protein-based nano-bioreactors has tremendous technological and environmental remediative potential.
Circadian Clocks in Cyanobacteria
As for several other molecular machines discussed in this symposium, it was once thought that 24 hour circadian clocks were characteristic strictly of eukaryotes, but we now know this also to be incorrect [Mackey et al., 2011]. In general, any circadian clock exhibits the following three characteristics: (1) It has a cycle of about 24 hr that continues several cycles without external stimulus input; (2) it is subject to entrainment by cycles of light/dark, warm/cold, humidity change and/or redox variations and (3) it is temperature compensated so that its 24 hour cycle continues regardless of the temperature. At the molecular level, it is subject to feedback/forward controls that are genetically determined. In cyanobacteria, the oscillator of the clock is a multiprotein molecular machine [Nakajima et al., 2005]. This 3-component clock is capable of keeping time outside of the bacterium in an in vitro environment as well as in the cell. Homologues of the components of this clock have not been found in eukaryotes, and hence, prokaryotic and eukaryotic clocks may have evolved independently.
The advantages to an organism of having a clock include the opportunity to optimize activities with light and temperature. It can be utilized for predation, mating, differentiation and many other biological processes. Disadvantages for higher organisms include delayed adaptability as observed in cases of jet lag and sleep disorders in humans. Not only at the molecular level, but also at the phenomenological level, it seems that different organisms use very different types of clocks, suggesting that pressure to evolve clocks has been considerable. Eukaryotes use transcription/translation coupled feedback loops and sense light as a primary signal, but cyanobacteria primarily use redox signals as environmental input, and at least some apparently don’t sense light directly other than through the photosynthetic apparatus. The best characterized prokaryotic clock is in Synechococcus elongatus [Dong and Golden, 2008].
In some Cyanobacteria the temporal separation of photosynthesis and molecular dinitrogen (N2) fixation is accomplished by rhythmic gene expression [Vinh et al., 2013]. Photosynthesis occurs mostly in the day; N2 fixation mostly at night, but there can be overlap. In an early experiment, 30,000 random bioluminescent luciferase (luxAB of V. harveyi) fusions were made to genes within the whole genome. 800 were bright enough to assay, and almost all of these manifested circadian rhythmicity. Different genes exhibited different waveforms, amplitudes and phase relationships (see Figure 11; [Liu et al., 1995]). In a second experiment, a photosystem II (PSII) promoter (psbAI)-luxAB fusion was used in mutagenesis studies. From 150,000 clones of chemically mutagenized cells, about 100 mutants were isolated exhibiting no growth defect, but with altered periods, shorter or longer (14–60 hrs) than normal, and/or abnormal patterns with arrhythmia and/or altered amplitudes or waveforms [Kondo et al., 1994].
Figure 11.

Circadian patterns of expression for 3 representative genes in Synechococcus elongatus.
In a third experiment, the clock-specific genes were cloned by complementation [Ishiura et al., 1998]. Clock mutations were found predominantly in the three kaiABC genes which together comprise an operon and encode the central oscillator. All mutants that exhibited some rhythmicity were due to missense mutations; most proved to be recessive. The name Kai, derived from the Japanese word meaning cycle or rotation, was given to these genes and their encoded proteins. Deletion of the entire kaiABC operon, or individually, just one of the kaiA, B or C genes, gave rise to total arrhythmicity. Two promoters were identified, one in front of the kaiA gene, and one before the kaiBC genes.
The sizes of these three clock proteins in Synechococcus elongatus in numbers of amino acyl residues (aas) were KaiA, 284 aas; KaiB, 102 aas and KaiC, 519 aas. KaiC proved to be an ATP/GTP binding protein, and all proved to be cytoplasmic proteins with no transmembrane segments. They form homotypic and heterotypic interactions, existing in a large complex that interacts with many other cellular regulatory elements and targets of regulation. Nevertheless, they alone form the multimeric protein machine, the oscillator of the clock. Homologues are present in other cyanobacteria, some other bacteria, and several archaea. Thus, it seems that some archaea may also have rhythm [Whitehead et al., 2009].
Increased kaiC expression causes kaiBC repression (feedback control) and alters the phases of expression rhythms of many genes. Increased kaiA expression, on the other hand, causes kaiBC activation (feedforward control). One long period mutant showed defective KaiA-KaiB interactions suggesting that intermolecular interactions must determine the period.
KaiC is an auto-kinase and an auto-phosphatase. It phosphorylates itself on two sites. It burns about 15 ATPs per day, less than one ATP per hour, an exceptionally slow turnover rate. The C-terminus of KaiA stimulates KaiC autophosphorylation and facilitates attenuation of KaiC autophosphorylation by KaiB [Murakami et al., 2012]. The N-terminal domain of KaiA is a pseudoreceiver domain, a conclusion that is based on the 3-D structure. The pseudoreceiver is a timing input device that usually senses light and regulates KaiA stimulation of KaiC autophosphorylation, synchronizing the oscillator with environmental cycles [Kim et al., 2012]. Crystal structures are available for each of these three proteins, but not for the intact complex.
CikA is another important constituent of the circadian system, probably functioning in entrainment. It is a redox sensor, not a light sensor, that resets the clock [Narikawa et al., 2008; Schmitz et al., 2000]. CikA derives its name from the fact that it is a Circadian Input Kinase. Inactivation of cikA results in (1) a shorter period (by 2 hours), (2) changes in the phasing, (3) abolition of the resetting of the phases by light/dark pulses via the redox cycle, and (4) elongated cells. Surprisingly, cell division is inhibited, indicating a link (perhaps indirect) between the clock and cell division. CikA is 754 aas long, soluble, and homologous to many sensor kinases.
CikA is not only a histidine sensor kinase (SK), it contains a cryptic response regulator (RR) domain (PsR). The domain structure of CikA is shown in Figure 12 where GAF is a small molecule binding domain that in many other proteins binds bilin chromophores and serves as a light sensor, while PsR is a pseudoreceiver domain with a binding site for quinones. This protein provides input to the oscillator for entrainment. Somehow, CikA affects FtsZ polymerization via the clock, and therefore influences cell division.
Figure 12.
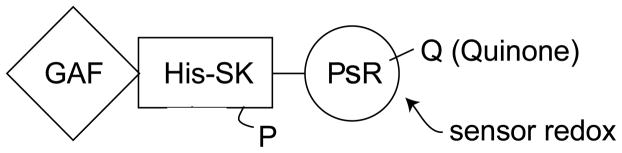
Schematic view of the domain structure of CikA. The GAF domain in some other proteins binds a bilin chromophore. This histidine sensor kinase (His-SK) can be autophosphorylated on a histidyl residue as shown. The pseudoreceiver domain (PsR) contains a quinone (Q) and serves as a sensor of the oxidoreduction (redox) state of the cell.
Another essential constituent that impinges on the clock, SasA, is a KaiC-interacting sensor kinase required for sustained, robust oscillations [Takai et al., 2006]. It acts directly on the response regulator, RpaA. SasA bears a KaiB-like domain for KaiC interaction. Deletion of the sasA gene (sasA) causes (1) lower kaiBC expression, (2) reduced amplitude of kai gene expression, and (3) a shorter period. sasA mutants give rise to attenuated circadian expression patterns for all genes tested. In fact, some genes become totally arrhythmic, and continuous sasA overexpression also eliminates all rhythms. Temporal overexpression of sasA changes the phase of the kaiBC expression rhythm.
sasA− and/or rpaA− mutants show lower levels of gene expression with decreased amplitudes, but there is no cell division defect, and metabolism continues unabated. As implied above, SasA and RpaA comprise a two-component sensor kinase/response regulator system for energy transfer from phycobilisomes to photosystems [Iwasaki et al., 2000] and mediate circadian timing. Together, they influence global transcriptional rhythms in response to cycles of KaiC phosphorylation [Takai et al., 2006; Valencia et al., 2012].
Gutu and O’Shea (2013) investigated how temporal information determined by KaiC phosphorylation is transduced to RpaA, the transcription factor mentioned above required for circadian gene expression. Phosphorylation of RpaA proved to be regulated by the two antagonistic histidine kinases, SasA and CikA, which are sequentially activated at distinct times by the Kai clock complex. SasA acts as a kinase toward RpaA, whereas CikA, previously implicated in clock input, acts as a phosphatase that dephosphorylates RpaA-phosphate. CikA and SasA thus antagonistically cooperate to generate an oscillation of RpaA activity that is distinct from that which could be generated by either enzyme alone. These observations suggest how circadian clocks can precisely control the timing of output pathways via the concerted action of two oppositely acting enzymes [Gutu and O’Shea, 2013].
Another protein, LabA, is required for negative feedback regulation of KaiBC expression. The phenotype of a knockout mutation in labA is low amplitude and bright. It acts in parallel with SasA, upstream of RpaA [Taniguchi et al., 2007]. However surprisingly, a cikA− sasA− double mutant has normal cell division. To determine the relationship between circadian timing and cell division, another set of experiments revealed that: (a.) cell division is regulated by the clock. (b.) division occurs only in the light phase. (c.) in constant light, cells still divide in the “subjective” day. (d.) ΔkaiC abolishes rhythms of both gene expression and cell division. (e.) ftsZ is expressed with a circadian rhythmic pattern. (f.) overexpression of ftsZ prevents cell division and therefore causes filament formation. However, these filaments retain robust, unaltered circadian rhythms. It therefore appears that although division is dependent on the clock, the clock is not dependent on cell division.
In further experiments, an involvement of sigma factors in circadian regulation of the expression of genes in the global cyanobacterial genome was demonstrated [Nair et al., 2002; Nair et al., 2001]. Four sigma factors (RpoD2, D3, D4 and SigC), when inactivated, singly or pairwise, changed the amplitude, phase angle, waveform and/or period of the expression of a test promoter, psbAI, one of three genes encoding a photosystem II (PSII) reaction center (RC). psbAI is regulated both transcriptionally and post-transcriptionally at the level of mRNA degradation. Apparently, the psbAI promoter can be read by multiple sigma factors. Most of the sigma factor mutants show normal kaiB promoter activity. Evidently, separate timing circuits with different periods can coexist in a cell. Sigma factors apparently work as a consortium to convey circadian information to “downstream” genes.
What is the mechanism for light input for entrainment? Potential answers to this question were provided by Katayama et al. [2003]. Random transposon insertions were used to identify a gene involved in the light input pathway. This gene was called ldpA where Ldp stood for light-dependent period. Mutations in ldpA conditionally altered the period but did not alter the ability to phase shift the gene expression rhythm by dark pulses. The shorter period phenotype apparently resulted from insensitivity to light gradients that normally modulate the period. LdpA has a 7Fe-8S cluster binding motif expected to be involved in redox reactions. Possibly LdpA modulates the clock by sensing changes in redox potential, thereby indirectly sensing light intensity. A proposed model is presented in Figure 13.
Figure 13.

An overview of the proposed molecular mechanism of the circadian clock in S. elongatus. The central oscillator is composed of KaiA, KaiB and KaiC. KaiA stimulates KaiC phosphorylation, and KaiB inactivates KaiA when KaiC reaches a certain phosphorylation state. In the input pathway, both LdpA and CikA sense the cellular redox state, which is influenced by light and cell metabolism. LdpA affects the stabilities of CikA and KaiA through an unknown mechanism. Through its PsR domain, CikA binds quinones directly, which destabilizes CikA. CikA affects the phosphorylation states of KaiC, but where and how it works in the signal transduction pathway is not known. Pex is a transcriptional repressor of KaiA, and its abundance is sensitive to light. However, it is not clear whether the pathway that regulates pex senses light directly or does it through cellular redox. In the output pathway, SasA interacts physically with KaiC and autophosphorylates, and then transfers the phosphoryl group to RpaA, a response regulator with a DNA-binding domain. The target of RpaA has not been identified. LabA works upstream of RpaA and downstream of KaiC, but its exact function is not clear. A SasA-independent, RpaA-independent output pathway might exist. The output pathway controls DNA topology, which is proposed to regulate global gene expression. A transcription/translation rhythm could interact with and reinforce the post-translational rhythm of KaiC activities. A solid line indicates a direct effect whereas a dotted line indicates an indirect effect or an effect whose mechanism is unknown. Arrows indicate the direction of the information flow or a stimulation of activity or both. Blunt ends represent inhibition of protein activity or abundance, whereas an end with a filled circle suggests regulation of unspecified direction. Reproduced with permission for Dong and Golden, 2008.
Sulfur Granules for Redox Processes
An example of a prokaryotic protein bound compartment used for redox-dependent energy generation and sulfur storage is the sulfur granule or sulfur globule, found in photosynthetic and chemolithotrophic bacteria under certain conditions [Konkol et al., 2010; Reigstad et al., 2011]. The sulfur housed in these granules is often derived from the oxidation of hydrogen sulfide and other compounds, and then it can be used as an electron donor or acceptor in subsequent redox processes, for example, when energy, or reduced or oxidized sulfur is needed. These protective organelles almost exclusively house elemental sulfur, which if in direct contact with the cytoplasm, can be toxic. Bacteria therefore protect themselves while storing large quantities of sulfur by surrounding the particles with a protein membrane. This topic is reviewed by Maki in the fascinating symposium article entitled “Bacterial Intracellular Sulfur Globules: Structure and Function.” In this article the properties of the proteinaceous membrane and its protein subunits are described. The generation and degradation of these globules is also considered. Striking superficial similarities between sulfur granule and gas vacuole protein membranes has been noted.
Gas Vesicles for Flotation
In addition to sulfur globules, other proteinaceous assemblies serve to compartmentalize specific agents. Perhaps the best studied example is the gas vacuoles which houses air in cytoplasmic compartments that provide the function of flotation [van Keulen et al., 2005]. These structures are described in the symposium article entitled “Gas Vesicles Across Kingdoms: A Comparative Solid State NMR Study,” by Daviso, Belenky, Griffin and Herzfeld. See also [Bayro et al., 2012; Ramsay and Salmond, 2012; Sivertsen et al., 2010]. These structures are found in a range of aquatic bacteria and archaea. In the case of some gas vesicles, a single small hydrophobic protein forms the membrane, although other proteins play roles. Biogenesis of these vesicles and its regulation have been the topics of recent publications [Pfeifer, 2012; Ramsay et al., 2011].
Cellulosomes for Cellulose Degradation
All of the examples discussed above are either cytoplasmic, membrane integrated or transenvelope machines, but some protein machines are extracellular, being assembled from collections of secreted protein constituents. A well studied example is the cellulosome for the degradation of plant and fungal cell walls, reviewed in the symposium article entitled “Cellulosome Complexes – Natural Biocatalysts as Arming Microcompartments of Enzymes,” by Bae, Morisaka, Kuroda and Ueda. The structural diversity, functional significance and biotechnological potential of these assemblages are remarkable and particularly worth noting. These aspects are discussed by Bae et al. (2013) in a fascinating treatment that the reader will find enlightening and entertaining.
Concluding Remarks
The “bag of enzymes” view of a living cell may have been a reasonable view of a young primordial cells, before extensive evolution gave rise to structural and functional refinement. However “evolution tends towards complexity,” when complexity benefits the organism, and complexity of molecular construction increases metabolic efficiency, allowing for novel functions such as morphogenesis, cellular motility, intracellular compartmentalization, intercellular interactions and novel mechanisms of energy generation [Diekmann and Pereira-Leal, 2013; Fares et al., 2011; Mulkidjanian et al., 2009; Wong et al., 2007]. We currently recognize that at the molecular level, few if any enzymes act alone. Instead, they interact with other enzymes, structural constituents of the cell, and regulatory proteins to increase efficiency and minimize waste. What we now see in any living cell is the end product of billions of years of evolution creating and perfecting thousands of exquisite molecular machines in an interconnected network [Marsh et al., 2013]. Thus, what we describe in this JMMB written symposium provides a glimpse of ongoing temporal processes that have no obvious end. While we can marvel at the current status of biological apparati that allow living organisms to defy entropy, we can only dream of the results of the ongoing evolutionary process given another billion years. Are we currently at evolutionary equilibrium, or are we still striving for perfection? Perhaps both views are valid for different systems. Bacteria, having evolved for a couple of billion years before the advent of eukaryotes, may be at a much higher level of perfection, therefore evolving more slowly than their eukaryotic grandchildren. On the other hand, their need to adapt to a continuously changing environment may require that they too must maintain a substantial rate of evolutionary change. As noted by Heraclitus (535-475 BC), “this universe has not been made by a god or man but has always been, is, and will be an ever-living fire, kindling itself and going out by regular measures. There is nothing permanent but change. Everything flows, nothing stands still.”
Acknowledgments
I thank Professors Susan Golden, Peter Graumann, Janos Lanyi, George Ordal, Joe Pogliano, Jeff Stock and Dave Zusman for critically reading the manuscript and for their valuable suggestions. This work was supported by NIH grants GM077402 and GM094610.
References
- Alon U, Camarena L, Surette MG, Aguera y Arcas B, Liu Y, Leibler S, Stock JB. Response regulator output in bacterial chemotaxis. Embo J. 1998;17:4238–4248. doi: 10.1093/emboj/17.15.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amar P, Legent G, Thellier M, Ripoll C, Bernot G, Nystrom T, Saier MH, Jr, Norris V. A stochastic automaton shows how enzyme assemblies may contribute to metabolic efficiency. BMC Syst Biol. 2008;2:27. doi: 10.1186/1752-0509-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apel D, Surette MG. Bringing order to a complex molecular machine: the assembly of the bacterial flagella. Biochim Biophys Acta. 2008;1778:1851–1858. doi: 10.1016/j.bbamem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Arechaga I. Membrane Invaginations in Bacteria and Mitochondria: Common Features and Evolutionary Scenarios. J Mol Microbiol Biotechnol. 2013;23:13–23. doi: 10.1159/000346515. [DOI] [PubMed] [Google Scholar]
- Asai Y, Yakushi T, Kawagishi I, Homma M. Ion-coupling determinants of Na+-driven and H+-driven flagellar motors. J Mol Biol. 2003;327:453–463. doi: 10.1016/s0022-2836(03)00096-2. [DOI] [PubMed] [Google Scholar]
- Ausmees N. Intermediate filament-like cytoskeleton of Caulobacter crescentus. J Mol Microbiol Biotechnol. 2006;11:152–158. doi: 10.1159/000094051. [DOI] [PubMed] [Google Scholar]
- Azia A, Unger R, Horovitz A. What distinguishes GroEL substrates from other Escherichia coli proteins? Febs J. 2012;279:543–550. doi: 10.1111/j.1742-4658.2011.08458.x. [DOI] [PubMed] [Google Scholar]
- Baker MD, Stock JB. Signal transduction: networks and integrated circuits in bacterial cognition. Curr Biol. 2007;17:R1021–1024. doi: 10.1016/j.cub.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Barabote RD, Saier MH., Jr Comparative genomic analyses of the bacterial phosphotransferase system. Microbiol Mol Biol Rev. 2005;69:608–634. doi: 10.1128/MMBR.69.4.608-634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry RM, Gitai Z. Self-assembling enzymes and the origins of the cytoskeleton. Curr Opin Microbiol. 2011;14:704–711. doi: 10.1016/j.mib.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayro MJ, Daviso E, Belenky M, Griffin RG, Herzfeld J. An amyloid organelle, solid-state NMR evidence for cross-beta assembly of gas vesicles. J Biol Chem. 2012;287:3479–3484. doi: 10.1074/jbc.M111.313049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker E, Herrera NC, Gunderson FQ, Derman AI, Dance AL, Sims J, Larsen RA, Pogliano J. DNA segregation by the bacterial actin AlfA during Bacillus subtilis growth and development. Embo J. 2006;25:5919–5931. doi: 10.1038/sj.emboj.7601443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg HC. The rotary motor of bacterial flagella. Annu Rev Biochem. 2003;72:19–54. doi: 10.1146/annurev.biochem.72.121801.161737. [DOI] [PubMed] [Google Scholar]
- Bertrand JJ, West JT, Engel JN. Genetic analysis of the regulation of type IV pilus function by the Chp chemosensory system of Pseudomonas aeruginosa. J Bacteriol. 2010;192:994–1010. doi: 10.1128/JB.01390-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Aboulwafa M, Saier MH., Jr Subcellular Localization and Logistics of Integral Membrane Protein Biogenesis in Escherichia coli. J Mol Microbiol Biotechnol. 2013;23:24–34. doi: 10.1159/000346517. [DOI] [PubMed] [Google Scholar]
- Bolam DN, Koropatkin NM. Glycan recognition by the Bacteroidetes Sus-like systems. Curr Opin Struct Biol. 2012;22:563–569. doi: 10.1016/j.sbi.2012.06.006. [DOI] [PubMed] [Google Scholar]
- Bouet JY, Ah-Seng Y, Benmeradi N, Lane D. Polymerization of SopA partition ATPase: regulation by DNA binding and SopB. Mol Microbiol. 2007;63:468–481. doi: 10.1111/j.1365-2958.2006.05537.x. [DOI] [PubMed] [Google Scholar]
- Bulyha I, Lindow S, Lin L, Bolte K, Wuichet K, Kahnt J, van der Does C, Thanbichler M, Sogaard-Andersen L. Two Small GTPases Act in Concert with the Bactofilin Cytoskeleton to Regulate Dynamic Bacterial Cell Polarity. Dev Cell. 2013 doi: 10.1016/j.devcel.2013.02.017. [DOI] [PubMed] [Google Scholar]
- Bulyha I, Schmidt C, Lenz P, Jakovljevic V, Hone A, Maier B, Hoppert M, Sogaard-Andersen L. Regulation of the type IV pili molecular machine by dynamic localization of two motor proteins. Mol Microbiol. 2009;74:691–706. doi: 10.1111/j.1365-2958.2009.06891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows LL. Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu Rev Microbiol. 2012;66:493–520. doi: 10.1146/annurev-micro-092611-150055. [DOI] [PubMed] [Google Scholar]
- Cabeen MT, Jacobs-Wagner C. Skin and bones: the bacterial cytoskeleton, cell wall, and cell morphogenesis. J Cell Biol. 2007;179:381–387. doi: 10.1083/jcb.200708001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabin-Flaman A, Ripoll C, Saier MH, Jr, Norris V. Hypothesis: chemotaxis in Escherichia coli results from hyper-structure dynamics. J Mol Microbiol Biotechnol. 2005;10:1–14. doi: 10.1159/000090343. [DOI] [PubMed] [Google Scholar]
- Caetano-Anolles G, Seufferheld MJ. The Coevolutionary Roots of Biochemistry and Cellular Organization Challenge the RNA World Paradigm. J Mol Microbiol Biotechnol. 2013;23:152–177. doi: 10.1159/000346551. [DOI] [PubMed] [Google Scholar]
- Calisto BM, Broto A, Martinelli L, Querol E, Pinol J, Fita I. The EAGR box structure: a motif involved in mycoplasma motility. Mol Microbiol. 2012;86:382–393. doi: 10.1111/j.1365-2958.2012.08200.x. [DOI] [PubMed] [Google Scholar]
- Cao TB, Saier MH., Jr The general protein secretory pathway: phylogenetic analyses leading to evolutionary conclusions. Biochim Biophys Acta. 2003;1609:115–125. doi: 10.1016/s0005-2736(02)00662-4. [DOI] [PubMed] [Google Scholar]
- Celler K, Koning RI, Koster AJ, van Wezel GP. A multi-dimensional view of the bacterial cytoskeleton. J Bacteriol. 2013 doi: 10.1128/JB.02194-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Chaudhury S, McShan AC, Kaur K, De Guzman RN. Stucture and biophysics of Type III secretion in bacteria. Biochemistry. 2013 doi: 10.1021/bi400160a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri TK, Verma VK, Maheshwari A. GroEL assisted folding of large polypeptide substrates in Escherichia coli: Present scenario and assignments for the future. Prog Biophys Mol Biol. 2009;99:42–50. doi: 10.1016/j.pbiomolbio.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Chiang P, Habash M, Burrows LL. Disparate subcellular localization patterns of Pseudomonas aeruginosa Type IV pilus ATPases involved in twitching motility. J Bacteriol. 2005;187:829–839. doi: 10.1128/JB.187.3.829-839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang P, Sampaleanu LM, Ayers M, Pahuta M, Howell PL, Burrows LL. Functional role of conserved residues in the characteristic secretion NTPase motifs of the Pseudomonas aeruginosa type IV pilus motor proteins PilB, PilT and PilU. Microbiology. 2008;154:114–126. doi: 10.1099/mic.0.2007/011320-0. [DOI] [PubMed] [Google Scholar]
- Choudhary S, Quin MB, Sanders MA, Johnson ET, Schmidt-Dannert C. Engineered protein nano-compartments for targeted enzyme localization. PLoS One. 2012;7:e33342. doi: 10.1371/journal.pone.0033342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Krausz S, Cabahug PC, Trachtenberg S. The monomeric, tetrameric, and fibrillar organization of Fib: the dynamic building block of the bacterial linear motor of Spiroplasma melliferum BC3. J Mol Biol. 2011;410:194–213. doi: 10.1016/j.jmb.2011.04.067. [DOI] [PubMed] [Google Scholar]
- Cot SS, So AK, Espie GS. A multiprotein bicarbonate dehydration complex essential to carboxysome function in cyanobacteria. J Bacteriol. 2008;190:936–945. doi: 10.1128/JB.01283-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derman AI, Becker EC, Truong BD, Fujioka A, Tucey TM, Erb ML, Patterson PC, Pogliano J. Phylogenetic analysis identifies many uncharacterized actin-like proteins (Alps) in bacteria: regulated polymerization, dynamic instability and treadmilling in Alp7A. Mol Microbiol. 2009;73:534–552. doi: 10.1111/j.1365-2958.2009.06771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekmann Y, Pereira-Leal JB. Evolution of intracellular compartmentalization. Biochem J. 2013;449:319–331. doi: 10.1042/BJ20120957. [DOI] [PubMed] [Google Scholar]
- Dong G, Golden SS. How a cyanobacterium tells time. Curr Opin Microbiol. 2008;11:541–546. doi: 10.1016/j.mib.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews G. The Intracytoplasmic Membranes of Purple Bacteria - Assembly of Energy-Transducing Complexes. J Mol Microbiol Biotechnol. 2013;23:35–47. doi: 10.1159/000346518. [DOI] [PubMed] [Google Scholar]
- Ducret A, Theodoly O, Mignot T. Single cell microfluidic studies of bacterial motility. Methods Mol Biol. 2013;966:97–107. doi: 10.1007/978-1-62703-245-2_6. [DOI] [PubMed] [Google Scholar]
- Ducret A, Valignat MP, Mouhamar F, Mignot T, Theodoly O. Wet-surface-enhanced ellipsometric contrast microscopy identifies slime as a major adhesion factor during bacterial surface motility. Proc Natl Acad Sci U S A. 2012;109:10036–10041. doi: 10.1073/pnas.1120979109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye NA, Shapiro L. The push and pull of the bacterial cytoskeleton. Trends Cell Biol. 2007;17:239–245. doi: 10.1016/j.tcb.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Eisenbach M. A hitchhiker’s guide through advances and conceptual changes in chemotaxis. J Cell Physiol. 2007;213:574–580. doi: 10.1002/jcp.21238. [DOI] [PubMed] [Google Scholar]
- Essen LO. Halorhodopsin: light-driven ion pumping made simple? Curr Opin Struct Biol. 2002;12:516–522. doi: 10.1016/s0959-440x(02)00356-1. [DOI] [PubMed] [Google Scholar]
- Fares MA, Ruiz-Gonzalez MX, Labrador JP. Protein coadaptation and the design of novel approaches to identify protein-protein interactions. IUBMB Life. 2011;63:264–271. doi: 10.1002/iub.455. [DOI] [PubMed] [Google Scholar]
- Fluhr JW, Miteva M, Primavera G, Ziemer M, Elsner P, Berardesca E. Functional assessment of a skin care system in patients on chemotherapy. Skin Pharmacol Physiol. 2007;20:253–259. doi: 10.1159/000104423. [DOI] [PubMed] [Google Scholar]
- Fuerst JA, Sagulenko E. Nested Bacterial Boxes: Nuclear and Other Intracellular Compartments in Planctomycetes. J Mol Microbiol Biotechnol. 2013;23:95–103. doi: 10.1159/000346544. [DOI] [PubMed] [Google Scholar]
- Fuesler JA, Li HJ. Dynamic instability--a common denominator in prokaryotic and eukaryotic DNA segregation and cell division. Cell Mol Biol Lett. 2012;17:542–548. doi: 10.2478/s11658-012-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar JA, Thomas JA, Marolda CL, Valvano MA. Surface expression of O-specific lipopolysaccharide in Escherichia coli requires the function of the TolA protein. Mol Microbiol. 2000;38:262–275. doi: 10.1046/j.1365-2958.2000.02094.x. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Albers SV. Assembly and function of the archaeal flagellum. Biochem Soc Trans. 2011;39:64–69. doi: 10.1042/BST0390064. [DOI] [PubMed] [Google Scholar]
- Gitai Z. Diversification and specialization of the bacterial cytoskeleton. Curr Opin Cell Biol. 2007;19:5–12. doi: 10.1016/j.ceb.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Gorby YA, Yanina S, McLean JS, Rosso KM, Moyles D, Dohnalkova A, Beveridge TJ, Chang IS, Kim BH, Kim KS, Culley DE, Reed SB, Romine MF, Saffarini DA, Hill EA, Shi L, Elias DA, Kennedy DW, Pinchuk G, Watanabe K, Ishii S, Logan B, Nealson KH, Fredrickson JK. Electrically conductive bacterial nanowires produced by Shewanella oneidensis strain MR-1 and other microorganisms. Proc Natl Acad Sci U S A. 2006;103:11358–11363. doi: 10.1073/pnas.0604517103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grallert H, Buchner J. Review: a structural view of the GroE chaperone cycle. J Struct Biol. 2001;135:95–103. doi: 10.1006/jsbi.2001.4387. [DOI] [PubMed] [Google Scholar]
- Graumann PL. Cytoskeletal elements in bacteria. Annu Rev Microbiol. 2007;61:589–618. doi: 10.1146/annurev.micro.61.080706.093236. [DOI] [PubMed] [Google Scholar]
- Gutu A, O’Shea EK. Two Antagonistic Clock-Regulated Histidine Kinases Time the Activation of Circadian Gene Expression. Mol Cell. 2013 doi: 10.1016/j.molcel.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano T, Yamaichi Y, Niki H. Oscillating focus of SopA associated with filamentous structure guides partitioning of F plasmid. Mol Microbiol. 2007;64:1198–1213. doi: 10.1111/j.1365-2958.2007.05728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan R, Ben-Yehuda S. Resolving chromosome segregation in bacteria. J Mol Microbiol Biotechnol. 2006;11:126–139. doi: 10.1159/000094049. [DOI] [PubMed] [Google Scholar]
- Hirai T, Subramaniam S, Lanyi JK. Structural snapshots of conformational changes in a seven-helix membrane protein: lessons from bacteriorhodopsin. Curr Opin Struct Biol. 2009;19:433–439. doi: 10.1016/j.sbi.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin J, Kaiser D. Cell-to-cell stimulation of movement in nonmotile mutants of Myxococcus. Proc Natl Acad Sci U S A. 1977;74:2938–2942. doi: 10.1073/pnas.74.7.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland IB, Schmitt L, Young J. Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway (review) Mol Membr Biol. 2005;22:29–39. doi: 10.1080/09687860500042013. [DOI] [PubMed] [Google Scholar]
- Houry WA. Chaperone-assisted protein folding in the cell cytoplasm. Curr Protein Pept Sci. 2001;2:227–244. doi: 10.2174/1389203013381134. [DOI] [PubMed] [Google Scholar]
- Hwang LC, Vecchiarelli AG, Han YW, Mizuuchi M, Harada Y, Funnell BE, Mizuuchi K. ParA-mediated plasmid partition driven by protein pattern self-organization. Embo J. 2013 doi: 10.1038/emboj.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiura M, Kutsuna S, Aoki S, Iwasaki H, Andersson CR, Tanabe A, Golden SS, Johnson CH, Kondo T. Expression of a gene cluster kaiABC as a circadian feedback process in cyanobacteria. Science. 1998;281:1519–1523. doi: 10.1126/science.281.5382.1519. [DOI] [PubMed] [Google Scholar]
- Iwasaki H, Williams SB, Kitayama Y, Ishiura M, Golden SS, Kondo T. A kaiC-interacting sensory histidine kinase, SasA, necessary to sustain robust circadian oscillation in cyanobacteria. Cell. 2000;101:223–233. doi: 10.1016/S0092-8674(00)80832-6. [DOI] [PubMed] [Google Scholar]
- Jones S. Microbial physiology: new electricigens get wired. Nat Rev Microbiol. 2006;4:642. [Google Scholar]
- Jun S, Wright A. Entropy as the driver of chromosome segregation. Nat Rev Microbiol. 2010;8:600–607. doi: 10.1038/nrmicro2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaimer C, Zusman DR. Phosphorylation-dependent localization of the response regulator FrzZ signals cell reversals in Myxococcus xanthus. Mol Microbiol. 2013 doi: 10.1111/mmi.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser D. Coupling cell movement to multicellular development in myxobacteria. Nat Rev Microbiol. 2003;1:45–54. doi: 10.1038/nrmicro733. [DOI] [PubMed] [Google Scholar]
- Kalliomaa-Sanford AK, Rodriguez-Castaneda FA, McLeod BN, Latorre-Rosello V, Smith JH, Reimann J, Albers SV, Barilla D. Chromosome segregation in Archaea mediated by a hybrid DNA partition machine. Proc Natl Acad Sci U S A. 2012;109:3754–3759. doi: 10.1073/pnas.1113384109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama M, Kondo T, Xiong J, Golden SS. ldpA encodes an iron-sulfur protein involved in light-dependent modulation of the circadian period in the cyanobacterium Synechococcus elongatus PCC 7942. J Bacteriol. 2003;185:1415–1422. doi: 10.1128/JB.185.4.1415-1422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanabe A, Furutani Y, Jung KH, Kandori H. An inward proton transport using Anabaena sensory rhodopsin. J Microbiol. 2011;49:1–6. doi: 10.1007/s12275-011-0547-x. [DOI] [PubMed] [Google Scholar]
- Kerfeld CA, Greenleaf WB, Kinney JN. The carboxysome and other bacterial microcompartments. Microbe. 2010;5:257–262. [Google Scholar]
- Kerfeld CA, Sawaya MR, Tanaka S, Nguyen CV, Phillips M, Beeby M, Yeates TO. Protein structures forming the shell of primitive bacterial organelles. Science. 2005;309:936–938. doi: 10.1126/science.1113397. [DOI] [PubMed] [Google Scholar]
- Kim YI, Vinyard DJ, Ananyev GM, Dismukes GC, Golden SS. Oxidized quinones signal onset of darkness directly to the cyanobacterial circadian oscillator. Proc Natl Acad Sci U S A. 2012;109:17765–17769. doi: 10.1073/pnas.1216401109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinosita K, Jr, Yasuda R, Noji H, Ishiwata S, Yoshida M. F1-ATPase: a rotary motor made of a single molecule. Cell. 1998;93:21–24. doi: 10.1016/s0092-8674(00)81142-3. [DOI] [PubMed] [Google Scholar]
- Klare JP, Bordignon E, Engelhard M, Steinhoff HJ. Transmembrane signal transduction in archaeal phototaxis: the sensory rhodopsin II-transducer complex studied by electron paramagnetic resonance spectroscopy. Eur J Cell Biol. 2011;90:731–739. doi: 10.1016/j.ejcb.2011.04.013. [DOI] [PubMed] [Google Scholar]
- Klare JP, Chizhov I, Engelhard M. Microbial rhodopsins: scaffolds for ion pumps, channels, and sensors. Results Probl Cell Differ. 2008;45:73–122. doi: 10.1007/400_2007_041. [DOI] [PubMed] [Google Scholar]
- Kojadinovic M, Armitage JP, Tindall MJ, Wadhams GH. Response kinetics in the complex chemotaxis signalling pathway of Rhodobacter sphaeroides. J R Soc Interface. 2013;10:20121001. doi: 10.1098/rsif.2012.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai M, Umino M, Hanai R. Mode of DNA binding by SopA protein of Escherichia coli F plasmid. J Biochem. 2011;149:455–461. doi: 10.1093/jb/mvq151. [DOI] [PubMed] [Google Scholar]
- Kondo T, Tsinoremas NF, Golden SS, Johnson CH, Kutsuna S, Ishiura M. Circadian clock mutants of cyanobacteria. Science. 1994;266:1233–1236. doi: 10.1126/science.7973706. [DOI] [PubMed] [Google Scholar]
- Konkol NR, Bruckner JC, Aguilar C, Lovalvo D, Maki JS. Dominance of epiphytic filamentous Thiothrix spp. on an aquatic macrophyte in a hydrothermal vent flume in Sedge Bay, Yellowstone Lake. Wyoming Microb Ecol. 2010;60:528–538. doi: 10.1007/s00248-010-9656-z. [DOI] [PubMed] [Google Scholar]
- Korotkov KV, Sandkvist M, Hol WG. The type II secretion system: biogenesis, molecular architecture and mechanism. Nat Rev Microbiol. 2012;10:336–351. doi: 10.1038/nrmicro2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer JA, Erb ML, Waddling CA, Montabana EA, Zehr EA, Wang H, Nguyen K, Pham DS, Agard DA, Pogliano J. A phage tubulin assembles dynamic filaments by an atypical mechanism to center viral DNA within the host cell. Cell. 2012;149:1488–1499. doi: 10.1016/j.cell.2012.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krewulak KD, Vogel HJ. TonB or not TonB: is that the question? Biochem Cell Biol. 2011;89:87–97. doi: 10.1139/o10-141. [DOI] [PubMed] [Google Scholar]
- Kuhn J, Briegel A, Morschel E, Kahnt J, Leser K, Wick S, Jensen GJ, Thanbichler M. Bactofilins, a ubiquitous class of cytoskeletal proteins mediating polar localization of a cell wall synthase in Caulobacter crescentus. Embo J. 2010;29:327–339. doi: 10.1038/emboj.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer T, Lu C, Echols H, Flanagan J, Hayer MK, Hartl FU. Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature. 1992;356:683–689. doi: 10.1038/356683a0. [DOI] [PubMed] [Google Scholar]
- Langklotz S, Baumann U, Narberhaus F. Structure and function of the bacterial AAA protease FtsH. Biochim Biophys Acta. 2012;1823:40–48. doi: 10.1016/j.bbamcr.2011.08.015. [DOI] [PubMed] [Google Scholar]
- Lanyi JK. What is the real crystallographic structure of the L photointermediate of bacteriorhodopsin? Biochim Biophys Acta. 2004;1658:14–22. doi: 10.1016/j.bbabio.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Lanyi JK. Proton transfers in the bacteriorhodopsin photocycle. Biochim Biophys Acta. 2006;1757:1012–1018. doi: 10.1016/j.bbabio.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Lanyi JK. Studies of the bacteriorhodopsin photocycle without the use of light: clues to proton transfer coupled reactions. J Mol Microbiol Biotechnol. 2007;12:210–217. doi: 10.1159/000099642. [DOI] [PubMed] [Google Scholar]
- Lanyi JK, Schobert B. Local-global conformational coupling in a heptahelical membrane protein: transport mechanism from crystal structures of the nine states in the bacteriorhodopsin photocycle. Biochemistry. 2004;43:3–8. doi: 10.1021/bi035843s. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2. 0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Larsen RA, Cusumano C, Fujioka A, Lim-Fong G, Patterson P, Pogliano J. Treadmilling of a prokaryotic tubulin-like protein, TubZ, required for plasmid stability in Bacillus thuringiensis. Genes Dev. 2007;21:1340–1352. doi: 10.1101/gad.1546107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Levit M, Lurz R, Surette MG, Stock JB. Receptor-mediated protein kinase activation and the mechanism of transmembrane signaling in bacterial chemotaxis. Embo J. 1997;16:7231–7240. doi: 10.1093/emboj/16.24.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tsinoremas NF, Johnson CH, Lebedeva NV, Golden SS, Ishiura M, Kondo T. Circadian orchestration of gene expression in cyanobacteria. Genes Dev. 1995;9:1469–1478. doi: 10.1101/gad.9.12.1469. [DOI] [PubMed] [Google Scholar]
- Londer YY, Pokkuluri PR, Orshonsky V, Orshonsky L, Schiffer M. Heterologous expression of dodecaheme “nanowire” cytochromes c from Geobacter sulfurreducens. Protein Expr Purif. 2006;47:241–248. doi: 10.1016/j.pep.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Lovley DR. Bug juice: harvesting electricity with microorganisms. Nat Rev Microbiol. 2006;4:497–508. doi: 10.1038/nrmicro1442. [DOI] [PubMed] [Google Scholar]
- Lower BH, Bazylinski DA. The Bacterial Magnetosome: A Unique Prokaryotic Organelle. J Mol Microbiol Biotechnol. 2013;23:63–80. doi: 10.1159/000346543. [DOI] [PubMed] [Google Scholar]
- Mackey SR, Golden SS, Ditty JL. The itty-bitty time machine genetics of the cyanobacterial circadian clock. Adv Genet. 2011;74:13–53. doi: 10.1016/B978-0-12-387690-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madkour MH, Mayer F. Intracellular cytoskeletal elements and cytoskeletons in bacteria. Sci Prog. 2007;90:73–102. doi: 10.3184/003685007X215913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AJ, Kuehn MJ. Functional Advantages Conferred by Extracellular Prokaryotic Membrane Vesicles. J Mol Microbiol Biotechnol. 2013;23:131–141. doi: 10.1159/000346548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansell TJ, Fisher AC, DeLisa MP. Engineering the protein folding landscape in gram-negative bacteria. Curr Protein Pept Sci. 2008;9:138–149. doi: 10.2174/138920308783955243. [DOI] [PubMed] [Google Scholar]
- Marsh JA, Hernandez H, Hall Z, Ahnert SE, Perica T, Robinson CV, Teichmann SA. Protein Complexes Are under Evolutionary Selection to Assemble via Ordered Pathways. Cell. 2013;153:461–470. doi: 10.1016/j.cell.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews CK. The cell-bag of enzymes or network of channels? J Bacteriol. 1993;175:6377–6381. doi: 10.1128/jb.175.20.6377-6381.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride MJ, Zhu Y. Gliding motility and Por secretion system genes are widespread among members of the phylum bacteroidetes. J Bacteriol. 2013;195:270–278. doi: 10.1128/JB.01962-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkidjanian AY, Galperin MY, Koonin EV. Co-evolution of primordial membranes and membrane proteins. Trends Biochem Sci. 2009;34:206–215. doi: 10.1016/j.tibs.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami R, Mutoh R, Iwase R, Furukawa Y, Imada K, Onai K, Morishita M, Yasui S, Ishii K, Valencia Swain JO, Uzumaki T, Namba K, Ishiura M. The roles of the dimeric and tetrameric structures of the clock protein KaiB in the generation of circadian oscillations in cyanobacteria. J Biol Chem. 2012;287:29506–29515. doi: 10.1074/jbc.M112.349092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat D. Magnetosomes: How do They Stay in Shape? J Mol Microbiol Biotechnol. 2013;23:81–94. doi: 10.1159/000346655. [DOI] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Kateriya S, Adeishvili N, Hegemann P, Bamberg E. Channelrhodopsins: directly light-gated cation channels. Biochem Soc Trans. 2005;33:863–866. doi: 10.1042/BST0330863. [DOI] [PubMed] [Google Scholar]
- Nair U, Ditty JL, Min H, Golden SS. Roles for sigma factors in global circadian regulation of the cyanobacterial genome. J Bacteriol. 2002;184:3530–3538. doi: 10.1128/JB.184.13.3530-3538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair U, Thomas C, Golden SS. Functional elements of the strong psbAI promoter of Synechococcus elongatus PCC 7942. J Bacteriol. 2001;183:1740–1747. doi: 10.1128/JB.183.5.1740-1747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M, Imai K, Ito H, Nishiwaki T, Murayama Y, Iwasaki H, Oyama T, Kondo T. Reconstitution of circadian oscillation of cyanobacterial KaiC phosphorylation in vitro. Science. 2005;308:414–415. doi: 10.1126/science.1108451. [DOI] [PubMed] [Google Scholar]
- Nan B, Bandaria JN, Moghtaderi A, Sun IH, Yildiz A, Zusman DR. Flagella stator homologs function as motors for myxobacterial gliding motility by moving in helical trajectories. Proc Natl Acad Sci U S A. 2013;110:E1508–1513. doi: 10.1073/pnas.1219982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan B, Chen J, Neu JC, Berry RM, Oster G, Zusman DR. Myxobacteria gliding motility requires cytoskeleton rotation powered by proton motive force. Proc Natl Acad Sci U S A. 2011;108:2498–2503. doi: 10.1073/pnas.1018556108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan B, Mauriello EM, Sun IH, Wong A, Zusman DR. A multi-protein complex from Myxococcus xanthus required for bacterial gliding motility. Mol Microbiol. 2010;76:1539–1554. doi: 10.1111/j.1365-2958.2010.07184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narikawa R, Kohchi T, Ikeuchi M. Characterization of the photoactive GAF domain of the CikA homolog (SyCikA, Slr1969) of the cyanobacterium Synechocystis sp. PCC 6803 Photochem Photobiol Sci. 2008;7:1253–1259. doi: 10.1039/b811214b. [DOI] [PubMed] [Google Scholar]
- Norris V, den Blaauwen T, Cabin-Flaman A, Doi RH, Harshey R, Janniere L, Jimenez-Sanchez A, Jin DJ, Levin PA, Mileykovskaya E, Minsky A, Saier M, Jr, Skarstad K. Functional taxonomy of bacterial hyperstructures. Microbiol Mol Biol Rev. 2007a;71:230–253. doi: 10.1128/MMBR.00035-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris V, den Blaauwen T, Doi RH, Harshey RM, Janniere L, Jimenez-Sanchez A, Jin DJ, Levin PA, Mileykovskaya E, Minsky A, Misevic G, Ripoll C, Saier M, Jr, Skarstad K, Thellier M. Toward a hyperstructure taxonomy. Annu Rev Microbiol. 2007b;61:309–329. doi: 10.1146/annurev.micro.61.081606.103348. [DOI] [PubMed] [Google Scholar]
- Norris V, Gascuel P, Guespin-Michel J, Ripoll C, Saier MH., Jr Metabolite-induced metabolons: the activation of transporter-enzyme complexes by substrate binding. Mol Microbiol. 1999;31:1592–1595. doi: 10.1046/j.1365-2958.1999.01275.x. [DOI] [PubMed] [Google Scholar]
- Palczewski K. Chemistry and biology of vision. J Biol Chem. 2012;287:1612–1619. doi: 10.1074/jbc.R111.301150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peabody CR, Chung YJ, Yen MR, Vidal-Ingigliardi D, Pugsley AP, Saier MH., Jr Type II protein secretion and its relationship to bacterial type IV pili and archaeal flagella. Microbiology. 2003;149:3051–3072. doi: 10.1099/mic.0.26364-0. [DOI] [PubMed] [Google Scholar]
- Pedersen PL. Transport ATPases into the year 2008: a brief overview related to types, structures, functions and roles in health and disease. J Bioenerg Biomembr. 2007;39:349–355. doi: 10.1007/s10863-007-9123-9. [DOI] [PubMed] [Google Scholar]
- Pfeifer F. Distribution, formation and regulation of gas vesicles. Nat Rev Microbiol. 2012;10:705–715. doi: 10.1038/nrmicro2834. [DOI] [PubMed] [Google Scholar]
- Pilhofer M, Jensen GJ. The bacterial cytoskeleton: more than twisted filaments. Curr Opin Cell Biol. 2013;25:125–133. doi: 10.1016/j.ceb.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillet F, Sanchez A, Lane D, Anton Leberre V, Bouet JY. Centromere binding specificity in assembly of the F plasmid partition complex. Nucleic Acids Res. 2011;39:7477–7486. doi: 10.1093/nar/gkr457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts AC, Tuck LR, Faulds-Pain A, Lewis RJ, Marles-Wright J. Structural insight into the Clostridium difficile ethanolamine utilisation microcompartment. PLoS One. 2012;7:e48360. doi: 10.1371/journal.pone.0048360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp D, Robinson RC. Supramolecular cellular filament systems: how and why do they form? Cytoskeleton (Hoboken) 2012;69:71–87. doi: 10.1002/cm.21006. [DOI] [PubMed] [Google Scholar]
- Pryor EE, Jr, Horanyi PS, Clark KM, Fedoriw N, Connelly SM, Koszelak-Rosenblum M, Zhu G, Malkowski MG, Wiener MC, Dumont ME. Structure of the integral membrane protein CAAX protease Ste24p. Science. 2013;339:1600–1604. doi: 10.1126/science.1232048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay JP, Salmond GP. Quorum sensing-controlled buoyancy through gas vesicles. Intracellular bacterial microcompartments for environmental adaptation. Commun Integr Biol. 2012;5:96–98. doi: 10.4161/cib.18532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay JP, Williamson NR, Spring DR, Salmond GP. A quorum-sensing molecule acts as a morphogen controlling gas vesicle organelle biogenesis and adaptive flotation in an enterobacterium. Proc Natl Acad Sci U S A. 2011;108:14932–14937. doi: 10.1073/pnas.1109169108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguera G, McCarthy KD, Mehta T, Nicoll JS, Tuominen MT, Lovley DR. Extracellular electron transfer via microbial nanowires. Nature. 2005;435:1098–1101. doi: 10.1038/nature03661. [DOI] [PubMed] [Google Scholar]
- Reguera G, Nevin KP, Nicoll JS, Covalla SF, Woodard TL, Lovley DR. Biofilm and nanowire production leads to increased current in Geobacter sulfurreducens fuel cells. Appl Environ Microbiol. 2006;72:7345–7348. doi: 10.1128/AEM.01444-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reigstad LJ, Jorgensen SL, Lauritzen SE, Schleper C, Urich T. Sulfur-oxidizing chemolithotrophic proteobacteria dominate the microbiota in high arctic thermal springs on Svalbard. Astrobiology. 2011;11:665–678. doi: 10.1089/ast.2010.0551. [DOI] [PubMed] [Google Scholar]
- Rye HS, Burston SG, Fenton WA, Beechem JM, Xu Z, Sigler PB, Horwich AL. Distinct actions of cis and trans ATP within the double ring of the chaperonin GroEL. Nature. 1997;388:792–798. doi: 10.1038/42047. [DOI] [PubMed] [Google Scholar]
- Saier MH., Jr Protein secretion and membrane insertion systems in gram-negative bacteria. J Membr Biol. 2006;214:75–90. doi: 10.1007/s00232-006-0049-7. [DOI] [PubMed] [Google Scholar]
- Saier MH, Jr, Bogdanov M. Membranous Organelles in Bacteria. J Mol Microbiol Biotechnol. 2013;23:5–12. doi: 10.1159/000346496. [DOI] [PubMed] [Google Scholar]
- Saier MH, Ma CH, Rodgers L, Tamang DG, Yen MR. Protein secretion and membrane insertion systems in bacteria and eukaryotic organelles. Adv Appl Microbiol. 2008;65:141–197. doi: 10.1016/S0065-2164(08)00606-0. [DOI] [PubMed] [Google Scholar]
- Saroussi S, Nelson N. The little we know on the structure and machinery of V-ATPase. J Exp Biol. 2009;212:1604–1610. doi: 10.1242/jeb.025866. [DOI] [PubMed] [Google Scholar]
- Schalk IJ, Mislin GL, Brillet K. Structure, function and binding selectivity and stereoselectivity of siderophore-iron outer membrane transporters. Curr Top Membr. 2012;69:37–66. doi: 10.1016/B978-0-12-394390-3.00002-1. [DOI] [PubMed] [Google Scholar]
- Schertzer JW, Whiteley M. Bacterial Outer Membrane Vesicles in Trafficking, Communication and the Host-Pathogen Interaction. J Mol Microbiol Biotechnol. 2013;23:118–130. doi: 10.1159/000346770. [DOI] [PubMed] [Google Scholar]
- Schmitz O, Katayama M, Williams SB, Kondo T, Golden SS. CikA, a bacteriophytochrome that resets the cyanobacterial circadian clock. Science. 2000;289:765–768. doi: 10.1126/science.289.5480.765. [DOI] [PubMed] [Google Scholar]
- Schobert B, Cupp-Vickery J, Hornak V, Smith S, Lanyi J. Crystallographic structure of the K intermediate of bacteriorhodopsin: conservation of free energy after photoisomerization of the retinal. J Mol Biol. 2002;321:715–726. doi: 10.1016/s0022-2836(02)00681-2. [DOI] [PubMed] [Google Scholar]
- Schobert B, Lanyi JK. Halorhodopsin is a light-driven chloride pump. J Biol Chem. 1982;257:10306–10313. [PubMed] [Google Scholar]
- Seufferheld MJ, Caetano-Anolles G. Phylogenomics Supports a Cellularly Structured Urancestor. J Mol Microbiol Biotechnol. 2013;23:178–191. doi: 10.1159/000346552. [DOI] [PubMed] [Google Scholar]
- Shlykov MA, Zheng WH, Chen JS, Saier MH., Jr Bioinformatic characterization of the 4-Toluene Sulfonate Uptake Permease (TSUP) family of transmembrane proteins. Biochim Biophys Acta. 2012;1818:703–717. doi: 10.1016/j.bbamem.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava A, Rhodes RG, Pochiraju S, Nakane D, McBride MJ. Flavobacterium johnsoniae RemA is a mobile cell surface lectin involved in gliding. J Bacteriol. 2012;194:3678–3688. doi: 10.1128/JB.00588-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JM, Brunet YR, Cascales E, Mougous JD. Structure and regulation of the type VI secretion system. Annu Rev Microbiol. 2012;66:453–472. doi: 10.1146/annurev-micro-121809-151619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivertsen AC, Bayro MJ, Belenky M, Griffin RG, Herzfeld J. Solid-state NMR characterization of gas vesicle structure. Biophys J. 2010;99:1932–1939. doi: 10.1016/j.bpj.2010.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza W. Prokaryotic cells: structural organisation of the cytoskeleton and organelles. Mem Inst Oswaldo Cruz. 2012;107:283–293. doi: 10.1590/s0074-02762012000300001. [DOI] [PubMed] [Google Scholar]
- Sun M, Wartel M, Cascales E, Shaevitz JW, Mignot T. Motor-driven intracellular transport powers bacterial gliding motility. Proc Natl Acad Sci U S A. 2011;108:7559–7564. doi: 10.1073/pnas.1101101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai N, Nakajima M, Oyama T, Kito R, Sugita C, Sugita M, Kondo T, Iwasaki H. A KaiC-associating SasA-RpaA two-component regulatory system as a major circadian timing mediator in cyanobacteria. Proc Natl Acad Sci U S A. 2006;103:12109–12114. doi: 10.1073/pnas.0602955103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Sawaya MR, Yeates TO. Structure and mechanisms of a protein-based organelle in Escherichia coli. Science. 2010;327:81–84. doi: 10.1126/science.1179513. [DOI] [PubMed] [Google Scholar]
- Tanaka T. Functional analysis of the stability determinant AlfB of pBET131, a miniplasmid derivative of bacillus subtilis (natto) plasmid pLS32. J Bacteriol. 2010;192:1221–1230. doi: 10.1128/JB.01312-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y, Katayama M, Ito R, Takai N, Kondo T, Oyama T. labA: a novel gene required for negative feedback regulation of the cyanobacterial circadian clock protein KaiC. Genes Dev. 2007;21:60–70. doi: 10.1101/gad.1488107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima H, Kojima S, Homma M. Flagellar motility in bacteria structure and function of flagellar motor. Int Rev Cell Mol Biol. 2008;270:39–85. doi: 10.1016/S1937-6448(08)01402-0. [DOI] [PubMed] [Google Scholar]
- Tindall MJ, Gaffney EA, Maini PK, Armitage JP. Theoretical insights into bacterial chemotaxis. Wiley Interdiscip Rev Syst Biol Med. 2012;4:247–259. doi: 10.1002/wsbm.1168. [DOI] [PubMed] [Google Scholar]
- Urban S, Dickey SW. The rhomboid protease family: a decade of progress on function and mechanism. Genome Biol. 2011;12:231. doi: 10.1186/gb-2011-12-10-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia SJ, Bitou K, Ishii K, Murakami R, Morishita M, Onai K, Furukawa Y, Imada K, Namba K, Ishiura M. Phase-dependent generation and transmission of time information by the KaiABC circadian clock oscillator through SasA-KaiC interaction in cyanobacteria. Genes Cells. 2012;17:398–419. doi: 10.1111/j.1365-2443.2012.01597.x. [DOI] [PubMed] [Google Scholar]
- van Keulen G, Hopwood DA, Dijkhuizen L, Sawers RG. Gas vesicles in actinomycetes: old buoys in novel habitats? Trends Microbiol. 2005;13:350–354. doi: 10.1016/j.tim.2005.06.006. [DOI] [PubMed] [Google Scholar]
- van Teeseling MCF, Neumann S, van Niftrik L. The Anammoxosome Organelle is Crucial for the Energy Metabolism of Anaerobic Ammonium Oxidizing Bacteria. J Mol Microbiol Biotechnol. 2013;23:104–117. doi: 10.1159/000346547. [DOI] [PubMed] [Google Scholar]
- Vasilyeva NV, Tsfasman IM, Kudryakova IV, Suzina NE, Shishkova NA, Kulaev IS, Stepnaya OA. The Role of Membrane Vesicles in Secretion of Lysobacter sp. Bacteriolytic Enzymes. J Mol Microbiol Biotechnol. 2013;23:142–151. doi: 10.1159/000346550. [DOI] [PubMed] [Google Scholar]
- Vinh NX, Chetty M, Coppel R, Gaudana S, Wangikar PP. A model of the circadian clock in the cyanobacterium Cyanothece sp. ATCC 51142 BMC. Bioinformatics. 2013;14 (Suppl 2):S14. doi: 10.1186/1471-2105-14-S2-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, Broederdorf LJ, Graham JG. Bacterial Type IV secretion systems: versatile virulence machines. Future Microbiol. 2012;7:241–257. doi: 10.2217/fmb.11.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waidner B, Specht M, Dempwolff F, Haeberer K, Schaetzle S, Speth V, Kist M, Graumann PL. A novel system of cytoskeletal elements in the human pathogen Helicobacter pylori. PLoS Pathog. 2009;5:e1000669. doi: 10.1371/journal.ppat.1000669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter S. Structure and function of the GroE chaperone. Cell Mol Life Sci. 2002;59:1589–1597. doi: 10.1007/PL00012485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Subramonian D, Zhang XD. SUMOylation in control of accurate chromosome segregation during mitosis. Curr Protein Pept Sci. 2012;13:467–481. doi: 10.2174/138920312802430563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Shaevitz JW. The mechanics of shape in prokaryotes. Front Biosci (Schol Ed) 2013;5:564–574. doi: 10.2741/s390. [DOI] [PubMed] [Google Scholar]
- Wang T, Sessions AO, Lunde CS, Rouhani S, Glaeser RM, Duan Y, Facciotti MT. Deprotonation of D96 in bacteriorhodopsin opens the proton uptake pathway. Structure. 2013;21:290–297. doi: 10.1016/j.str.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webre DJ, Wolanin PM, Stock JB. Modulated receptor interactions in bacterial transmembrane signaling. Trends Cell Biol. 2004;14:478–482. doi: 10.1016/j.tcb.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Whitehead K, Pan M, Masumura K, Bonneau R, Baliga NS. Diurnally entrained anticipatory behavior in archaea. PLoS One. 2009;4:e5485. doi: 10.1371/journal.pone.0005485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolgemuth CW, Oster G. The junctional pore complex and the propulsion of bacterial cells. J Mol Microbiol Biotechnol. 2004;7:72–77. doi: 10.1159/000077871. [DOI] [PubMed] [Google Scholar]
- Wong T, Amidi A, Dodds A, Siddiqi S, Wang J, Yep T, Tamang D, Saier MH., Jr Evolution of the bacterial flagellum. Microbe. 2007;2:335–340. [Google Scholar]
- Woronowicz K, Harrold JW, Kay JM, Niederman RA. Structural and Functional Proteomics of Intracytoplasmic Membrane Assembly in Rhodobacter sphaeroides. J Mol Microbiol Biotechnol. 2013;23:48–62. doi: 10.1159/000346520. [DOI] [PubMed] [Google Scholar]
- Yonekura K, Maki-Yonekura S, Homma M. Structure of the flagellar motor protein complex PomAB: implications for the torque-generating conformation. J Bacteriol. 2011;193:3863–3870. doi: 10.1128/JB.05021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youderian P, Burke N, White DJ, Hartzell PL. Identification of genes required for adventurous gliding motility in Myxococcus xanthus with the transposable element mariner. Mol Microbiol. 2003;49:555–570. doi: 10.1046/j.1365-2958.2003.03582.x. [DOI] [PubMed] [Google Scholar]
- Zhai Y, Heijne WH, Smith DW, Saier MH., Jr Homologues of archaeal rhodopsins in plants, animals and fungi: structural and functional predications for a putative fungal chaperone protein. Biochim Biophys Acta. 2001;1511:206–223. doi: 10.1016/s0005-2736(00)00389-8. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Goemaere EL, Seddiki N, Celia H, Gavioli M, Cascales E, Lloubes R. Mapping the interactions between Escherichia coli TolQ transmembrane segments. J Biol Chem. 2011;286:11756–11764. doi: 10.1074/jbc.M110.192773. [DOI] [PMC free article] [PubMed] [Google Scholar]