

Abstract
Spinal muscular atrophy (SMA) is the leading genetic cause of infant mortality. SMA results from deletions or mutations of survival motor neuron 1 (SMN1), an essential gene. SMN2, a nearly identical copy, can compensate for SMN1 loss if SMN2 exon 7 skipping is prevented. Among the many cis-elements involved in the splicing regulation of SMN exon 7, intronic splicing silencer N1 (ISS-N1) has emerged as the most effective target for an antisense oligonucleotide (ASO)-mediated splicing correction of SMN2 exon 7. Blocking of ISS-N1 by an ASO has been shown to fully restore SMN2 exon 7 inclusion in SMA patient cells as well as in vivo. Here we review how ISS-N1 targeting ASOs that use different chemistries respond differently in the various SMA mouse models. We also compare other ASO-based strategies for therapeutic splicing correction in SMA. Given that substantial progress on ASO-based strategies to promote SMN2 exon 7 inclusion in SMA has been made, and that similar approaches in a growing number of genetic diseases are possible, this report has wide implications.
Keywords: Antisense oligonucleotide, splicing, ISS-N1, ASO, SMA, SMN, PMO, MOE
INTRODUCTION
Spinal muscular atrophy (SMA) is a leading neurodegenerative disease of children and infants (1-7). SMA is characterized by the progressive loss of α-motor neurons in the spinal cord, which leads to paralysis of the trunk and limbs and respiratory insufficiency (2-7). SMA is caused by the homozygous functional loss of the survival motor neuron 1 (SMN1) gene due to deletion, subtle mutation, or gene conversion (2-5). SMN2, a nearly identical copy of SMN1, fails to compensate for SMN1 loss due to a critical C to T transition at the 6th position (C6U in transcript) in exon 7 (8). C6U leads to exon 7 skipping during pre-mRNA splicing of SMN2 (9); as a consequence, a truncated, dysfunctional and rapidly degraded protein (SMNΔ7) is produced (10,11). The defects caused by SMN1 deficiency can be compensated by increased copies of SMN2, which produces low levels of full-length SMN (12). Considering all SMA patients retain at least one functional copy of SMN2, it is reasonable to assume that strategies aimed at correcting SMN2 exon 7 splicing hold the promise for a treatment. Indeed, the postnatal increase of SMN levels through SMN2 exon 7 splicing correction—by both small compounds and antisense oligonucleotides (ASOs)—provides substantial therapeutic benefits in animal models of SMA (13-16). Several recent reviews summarized the general progress in the rapidly evolving field of oligonucleotide research (17-21). This review is inspired by several independent reports in which different ASO chemistries against a single intronic target have shown unprecedented therapeutic benefits in various mouse models of SMA. Here we focus on challenges and lessons drawn from the recent ASO-based therapeutic approaches in SMA, and their implications for nucleic-acid-based therapy of a growing number of genetic diseases.
Developing ASO-based splicing correction therapeutics is not a straight-forward task. Splicing modulation using an ASO-mediated approach requires sequestration of a splicing cis-element (18). Since the majority of the human genome is transcribed (22), there is a substantial risk that an ASO could anneal to analogous sequences through mismatch base pairing and have off-target effects. Furthermore, transcribed RNAs form secondary structures within microseconds of emerging from polymerase II (23-25). Thus, low annealing efficiency of ASOs with targets located in the structured region of transcripts results in reduced antisense response (26). Most RNA-interacting proteins recognize small sequence motifs. Therefore, unwanted interactions of proteins with ASOs could also contribute to reduced antisense efficacy. Hence, designing an ASO that does not interact with protein factors and anneals to a desired cis-element located within a structurally accessible region of RNA remains the most challenging aspect of developing an ASO-based therapeutic strategy.
The degree of antisense response is dependent upon the strength of the targeted splicing cis-element as well as the nature of the context created by the duplex formed between the ASO and its target. Splicing is a dynamic process that involves numerous RNA-protein and RNA-RNA interactions (27-30), so it is safe to assume that permanent annealing of an ASO to its target affects several interactions and enforces remodeling of the RNA structure of the target and associated sequences. Therefore, the location of the target, ASO length and ASO chemistry play critical roles in determining the extent of antisense response. In vivo efficacy of an ASO is dependent upon additional factors including but not limited to dose, frequency, route and method of administration. The specific animal model and immune tolerance for the administered ASO also critically influence the phenotype of the ASO-treated animals.
ISS-N1 as a promising therapeutic target
To restore SMN2 exon 7 inclusion in SMA, it is essential that splice-switching ASOs target an inhibitory cis-element. It is also desirable that the inhibitory cis-element is located within an intronic sequence in order to avoid interfering with the translation and transport of mRNA. Discovery of ISS-N1 (intronic splicing silencer N1) in 2006 provided a major breakthrough for an ASO-mediated splicing correction approach in SMA (31). ISS-N1 is a 15-nucleotide sequence spanning the 10th to 24th positions of intron 7 (Fig. 1A). ISS-N1 creates a strong negative context at the 5′ splice site (5’ ss) of exon 7, and ISS-N1 deletion fully restores SMN2 exon 7 inclusion (31). Remarkably, the deletion of ISS-N1 confers such a stimulatory effect on SMN2 exon 7 splicing that all known positive cis-elements within exon 7 become dispensable. Therefore, ISS-N1 is described as the master checkpoint of SMN2 exon 7 splicing regulation (32). The therapeutic efficacy of ISS-N1 was first assessed by blocking ISS-N1 using a 20-mer ASO (Anti-N1) carrying phosphorothioate backbone and 2′-O-methyl modification (2′-OMe). Even at a very low concentration (5 nM), Anti-N1 substantially elevated SMN levels in type I SMA patient cells (31). These unprecedented results established that ASO-mediated sequestration of an intronic sequence could fully correct a splicing defect linked to an exonic mutation.
Figure 1.
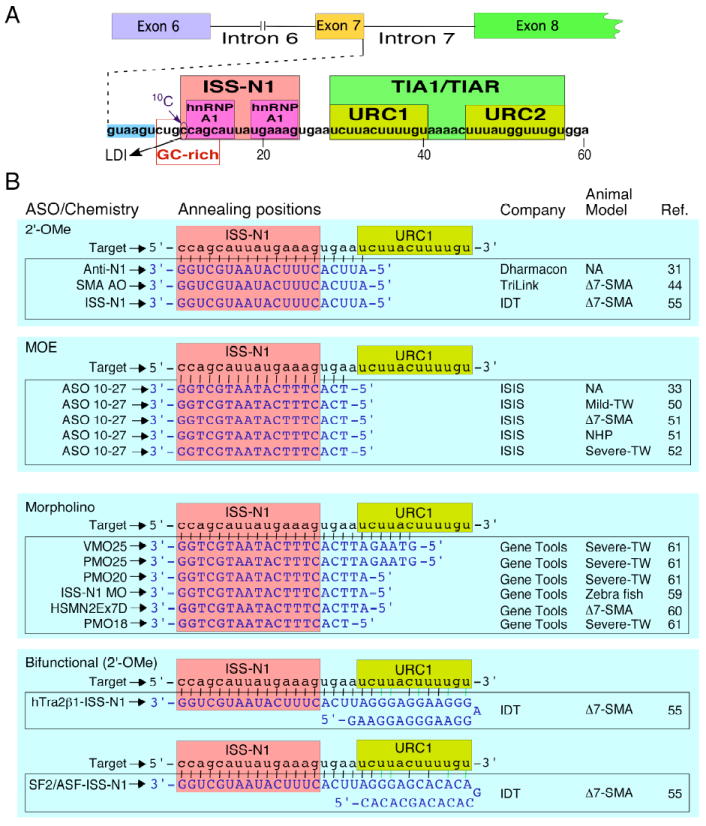
ASO-mediated splicing correction in SMA. (A) Diagrammatic representation of splicing cis elements. Splicing cis-elements are indicated by boxes. Numbering starts from the first position of SMN2 intron 7. Cis-elements shown here are described in detail in a recent review by Singh and Singh (9). ISS-N1 is negative cis-element that has emerged as the leading therapeutic target for an ASO-mediated splicing correction in SMA (31-33, 50-52, 55,60,61). Binding sites of inhibitory factor hnRNP A1 (33) and stimulatory factors TIA1 and TIAR (39) have been indicated. GC-rich sequence constitutes the target for splicing correction by the shortest ASO (35). A cytosine residue at the 10th intronic position (10C) associates with a long-distance interaction (LDI) with the downstream intronic sequences (36). Sequestration of ISS-N1 or GC-rich sequence appears to abrogate the inhibitory LDI associated with 10C. (B) Diagrammatic representation of annealing positions of ISS-N1 targeting ASOs. All ISS-N1 targeting ASOs sequestered the first residue of ISS-N1. Reference numbers of studies using different ASOs targeting ISS-N1 are shown. Abbreviations: ISS-N1, Intronic splicing silencer N1; LDI, long-distance interaction; URC, U-rich cluster; ASO, antisense oligonucleotide; 2′-OMe, an ASO with phosphorothioate backbone and 2′-O-methyl modification; MOE, an ASO with phosphorothioate backbone and 2′-O-methoxyethyl modification; Δ7-SMA, severe SMA model (45); NHP, non-human primate; Mild-TW, mild Taiwanese model of SMA (53); Severe-TW, severe Taiwanese model of SMA (56,57).
Two years after the discovery of ISS-N1, the Krainer group in collaboration with ISIS Pharmaceuticals conducted a systematic antisense microwalk employing ASOs carrying phosphorothioate backbone and 2′-O-methoxyethyl modification (MOE) chemistry (33). MOE chemistry, a proprietary product of ISIS Pharmaceuticals, is considered to confer increased nuclease resistance and decrease non-specific protein interactions (34). The authors concluded that the stimulatory effect of ISS-N1 targeting MOE ASO was higher than other MOE ASOs targeting different regions of SMN2 (33). They independently validated the strong inhibitory nature of ISS-N1 and generated interest in an ASO-mediated therapy for SMA.
ISS-N1 encompasses two hnRNP A1/A2 motifs and overlaps with a unique GC-rich motif (Fig.1A; 33,35). A cytosine residue at the 10th position (10C) of intron 7 occupies the first position of ISS-N1. Sequestration of the first five residues of ISS-N1, including 10C, was necessary for the stimulatory effect of the ISS-N1-targeting ASOs (36). Research data supports that ISS-N1-targeting ASOs stimulate SMN2 exon 7 inclusion by reconstructing a positive context in addition to displacing inhibitory factors (i.e. hnRNP A1/A2) at the 5′ ss of SMN2 exon 7 (9). The 5′ ss of exon 7 is weakened by the presence of a number of inhibitory cis-elements (31, 35-38). The only intronic positive cis-element in the vicinity of the 5′ ss of SMN2 exon 7 is the U-rich clusters (URCs) immediately downstream of ISS-N1 (Fig. 1A) (39). In particular, URC1 and URC2 have been shown to interact with T-cell-restricted intracellular antigen 1 (TIA1) and TIA1-related (TIAR) proteins that are splicing stimulatory factors (39). Recently, a point mutation within the glutamine-rich domain of TIA1 has been shown to affect SMN2 exon 7 splicing in the patients of Welander distal myopathy (WDM), an adult onset autosomal-dominant disorder characterized by distal limb weakness (40). These findings are significant as they reveal for the first time that a single point mutation within a splicing factor could have adverse effect on SMN2 exon 7 splicing.
It has been suggested that the ASO-mediated sequestration of ISS-N1 increases the interaction efficiency of TIA1/TIAR and/or other members of the glutamine-rich-RNA-binding proteins that stimulate SMN2 exon 7 inclusion by binding to U-rich sequences immediately downstream of ISS-N1 (39). Such interaction is necessary to recruit U1 snRNP at the weak 5′ ss of exon 7. Recruitment of U1 snRNP at the 5′ ss is the first and most critical step of spliceosomal assembly, which is a multistep dynamic process of intron removal (41). Recent reports suggest U1 snRNP recruitment plays a role in regulating isoform expression and polyadenylation (42,43). Considering intron 7 is the last intron of SMN2, it is possible that the enhanced recruitment of U1 snRNP upon sequestration of ISS-N1 provides additional benefits unrelated to splicing.
In vivo studies with ISS-N1 targeting ASOs
The first in vivo study of an ISS-N1 targeting ASO was conducted by Williams et al. with a 20-mer 2′-OMe ASO (SMA AO) employing the Δ7-SMA mouse model (44). The median survival age of Δ7-SMA mice is ~13 days, and this model has been widely used for understanding the pathogenesis of SMA as well as pre-clinical testing of potential SMA drugs (45-49). This particular study administered ASOs on P1, 3, 5, 7, and 10 by bilateral intracerebroventricular (ICV) injections (1 μg per lateral ventricle), and tissues were harvested for analysis at P12. SMA AO increased SMN levels in brain and spinal cord, whereas control AO had no appreciable effect. In addition, SMA AO increased body weight and improved righting reflex of the Δ7-SMA mice, whereas control AO provided no phenotypic benefit. Despite the limited nature of this study, the findings were encouraging for subsequent studies that employed different oligonucleotide chemistries, routes of oligonucleotide delivery and mouse models.
The Krainer group at Cold Spring Harbor Laboratory in collaboration with ISIS Pharmaceuticals conducted a series of in vivo studies with ISS-N1 targeting MOE ASOs (50-52). In particular, they used an 18-mer MOE ASO (ASO-10-27) that sequesters the entire ISS-N1 and three residues downstream of ISS-N1 (Fig. 1B). The initial study employed a mild SMA model (Taiwanese SMA model; four copies of SMN2) that displays tail and ear necroses (53). As expected, ICV injections of ASO-10-27 in adult mice stimulated SMN2 exon 7 inclusion in brain and spinal cord (50), and immunostaining confirmed the ASO-induced upregulation of SMN in brain and spinal cord. Single embryonic or neonatal ICV injection of ASO-10-27 successfully rescued phenotype. For instance, tail and ear necroses were either completely prevented or substantially delayed by treatment with ASO-10-27. Embryonic injections were more effective than neonatal injections. Overall, the study demonstrated the efficacy of MOE ASO in treating mild SMA mice when given very early.
The authors also compared the MOE ASO (ASO-10-27) with the 2′-OMe ASO used by Williams et al. (44,50). In contrast to the results obtained by Williams et al. in Δ7-SMA mice (44), ICV injection of 2′-OMe ASO in the Taiwanese SMA model did not stimulate SMN2 exon 7 inclusion (50). In addition, the 2′-OMe ASO, but not the MOE ASO, produced proinflammatory effects. The discrepancy between these two studies is not entirely unexpected since they used different sources of 2′-OMe oligonucleotide synthesis and different mouse models (Fig. 1B). A recent study that involved repeated long-term subcutaneous treatment of Duchenne muscular dystrophy (DMD) mice with a 2′-OMe ASO produced tangible therapeutic benefits without any safety concerns (54). Therefore, it is possible that the proinflammatory effect of 2′-OMe ASO is a characteristic of the Taiwanese SMA model, and/or this model is more tolerant to MOE chemistry than to 2′-OMe chemistry. Recently, the Lorson group used a third source of oligonucleotide synthesis and independently confirmed upregulation of SMN levels in brain and spinal cord upon ICV administration of an ISS-N1-targeting 2′-OMe ASO in Δ7-SMA mice (55).
The Passini group at Genzyme Corporation, in collaboration with the Krainer group and ISIS Pharmaceuticals, tested the efficacy of ASO-10-27 in Δ7-SMA mice (51). A single ICV injection of ASO-10-27 (4 μg) at P0 increased median life expectancy from 16 to 26 days. Treated mice had increased numbers of motor neurons within the spinal cord and improved motor function. Interestingly, higher and lower doses of ASO delivered by ICV were less effective. The authors also performed preliminary experiments in nonhuman primates (NHP) and demonstrated that 1) intrathecal infusion over a 24 hr period of a fixed dose of ASO 10-27 (3 mg) was well tolerated and could achieve therapeutically relevant levels of ASO 10-27 and 2) intrathecal infusion is an effective delivery method for global distribution of ASOs to the spinal cord of NHPs. The latter finding will be important for the future therapeutic development of other ASOs for SMA, and more broadly for other therapies that will require delivery to the spinal cord (51).
Encouraged by the results of ICV injections of MOE ASOs in two SMA mouse models (50,51), the Krainer group in collaboration with ISIS Pharmaceuticals performed an elaborate study using the Taiwanese type I SMA mice (two copies of SMN2), a more severe SMA mouse model (52). This model was generated from mild Taiwanese SMA mice and has a median lifespan of ~10 days (56,57). The major finding of this study was an unexpectedly high efficacy of ASO-10-27 when delivered peripherally (52). The animals tolerated a very high dose of subcutaneously (SC) administered ASO-10-27. The best survival outcome was observed when animals received two SC administrations (one at P0-P1 and the other at P2-P3) each at 160 μg/g body weight (52). The median life expectancy of treated mice increased ~25 fold (from ~10 days to ~273 days). Interestingly, ICV delivery was substantially less efficacious than SC delivery. For instance, ICV delivery of ASO-10-27 (20 μg) at P1 only increased median life expectancy of Taiwanese type 1 SMA mice from 10 days to 16 days (52). Although somewhat controversial, these results underscore the peripheral requirement of SMN protein and demonstrate the long-term benefits of early ASO administration in severe SMA mice.
In vivo studies with ISS-N1 targeting morpholinos
Phosphorodiamidate morpholino oligomers (PMOs) are a class of compounds in which the ribose sugar moiety is replaced with a morpholino ring and the phosphorothioate group is replaced with an uncharged phosphorodiamidate group. PMOs and their derivatives have been widely used for therapeutic splicing correction in animal models (18,58,59). Porensky and coworkers from the Burghes laboratory employed Δ7-SMA mice to test the efficacy of an ISS-N1 targeting PMO (HSMN2Ex7D) that sequestered the entire ISS-N1 and 5 residues downstream of ISS-N1 (60; Fig. 1B). ICV administration of HSMN2Ex7D corrected SMN2 exon 7 splicing and increased levels of SMN in brain and spinal cord. A single ICV administration of 54 μg HSMN2Ex7D at P0 prolonged median life expectancy of Δ7-SMA mice from ~15 days to more than 100 days. Interestingly, peripheral delivery of HSMN2Ex7D produced no therapeutic benefit in Δ7-SMA mice. These results are consistent with the prevailing view that brain and spinal cord are the primary tissues in which high levels of SMN are required for an effective SMA therapy. The results also demonstrate PMO as superior to MOE chemistry when delivered through ICV injections.
A recent study by the Muntoni group and coworkers utilizing Taiwanese type I SMA mice independently confirmed the high therapeutic efficacy of ISS-N1 targeting PMOs (61). The authors used PMOs of three different sizes and showed 25-mer PMO (PMO25) that sequestered the entire ISS-N1 and 10 residues downstream of ISS-N1 conferred better therapeutic efficacy than the 18-mer or 20-mer PMO (Fig. 1B). A single ICV administration of PMO25 (40 μg/g body weight) at P0 increased median life expectancy of Taiwanese type I SMA mice from ~10 days to more than 200 days. Contrary to findings in the Δ7-SMA mouse model (60), the authors found no significant difference in therapeutic efficacy of PMOs when delivered peripherally or ICV (61). These results again underscore the difference between the Δ7-SMA and Taiwanese type I SMA models with regards to the efficacy of an identical ASO. The observation that peripheral delivery of MOE and PMO25 has therapeutic benefit in the Taiwanese type I SMA model but not in the Δ7-SMA model supports that timings of BBB formation between these two mouse models could be distinct.
In vivo studies with ISS-N1 targeting bifunctional ASOs
Bifunctional ASOs anneal to specific RNA sequences and recruit additional factors through hanging tails (62). The Lorson group used bifunctional 2′-OMe ASOs to block ISS-N1 and recruit either Tra2-β1 or SF2/ASF in the vicinity of ISS-N1 (55). Upon ICV administration (6 μg/day) at P1, P3 and P5, these ASOs increased weight and gross motor functions of Δ7-SMA mice. Treatment also increased the median life expectancy of Δ7-SMA mice from ~16 days to 18-20 days. The efficacy of the ASO carrying a Tra2-β1 motif was higher than the ASO carrying a SF2/ASF motif; however, the therapeutic benefit of these bifunctional ASOs was substantially lower than ISS-N1-targeting MOEs and PMOs (52,60,61).
The low efficacy of ISS-N1 targeting bifunctional ASOs could result from a limited ability of Tra2-β1 and/or SF2/ASF to activate the 5′ ss of SMN2 exon 7. Since these bifunctional ASOs are larger, they may fold into secondary and/or higher order structures that could sequester critical residues responsible for annealing and/or protein recruitment. In addition, large oligonucleotides are notorious for non-specific trapping of protein factors. Some of these problems could be overcome through trial and error by testing different tailed sequences in the context of different chemistries. However, since a bifunctional ASO is designed to trap an arbitrary chosen splicing factor, there is a risk of unwanted secondary effect on splicing of other genes.
Other ASO-based approaches
Several other ASO-based studies independent of ISS-N1 have been used for splice correction in SMA. A bifunctional ASO targeting element 1 located within intron 6 exhibited efficacy in SMA mice (63,64). Other bifunctional ASOs that anneal to SMN2 exon 7 and recruit splicing factors either at the 3′ or 5′ ss of exon 7 have shown encouraging results in vitro, ex vivo and in SMA mice (65-68). Blocking the 3′ ss of exon 8 by an ASO embedded in U7 snRNAs was shown to prevent exon 7 skipping and increase SMN levels in SMA patient-derived cells (69). These results underscore that the transcripts retaining SMN2 intron 7 are capable of being polyadenylated, exported out of the nucleus, and translated in the cytoplasm. Recently, an 8-mer ASO (3UP8) that sequesters a GC-rich sequence within intron 7 was shown to have a strong stimulatory effect on SMN2 exon 7 inclusion in SMA patient fibroblast cell lines (35,70). In addition to being cost effective, small ASOs offer the advantage of being less tolerant towards non-canonical and mismatch base pairing, a major cause for the nonspecific effect associated with large ASOs. Consistently, 3UP8 showed zero tolerance for a single mismatch base pair mutation at the target site (35). However, it remains to be seen whether the expected benefit of a small ASO is realized in a mouse model of SMA.
Conclusions and future directions
SMA is a rare disease in which a correctable copy of the gene (SMN2) is universally present in all SMA patients. In addition, SMA is unique in that all patients have the opportunity for an ASO-based therapy to increase SMN levels by correcting the pre-mRNA splicing of one exon. In the absence of structure-specific targets, an ASO-based approach remains the best option for gene-specific splicing correction. In general, the success of an ASO-based approach for splicing correction depends on the potency of the target. Characterizing an efficient intronic target for inducing exon inclusion remains a challenging endeavor.
The discovery of ISS-N1 provided a major breakthrough as it demonstrated for the first time that a large intronic motif could serve as a master regulator of SMN2 exon 7 splicing. The finding that an ASO-mediated sequestration of ISS-N1 fully restores SMN2 exon 7 inclusion led to several in vivo studies in mouse models of SMA. Consistently, ISS-N1 stands as the most scrutinized antisense target for the potential treatment of a genetic disease. The last few years have witnessed remarkable progress towards the development of ISS-N1-based therapy. For instance, ISIS Pharmaceuticals successfully concluded a phase 1 clinical trial of ISIS-SMNRx, an ISS-N1 targeting ASO with MOE chemistry. This is the first clinical trial for an ASO-mediated restoration of exon inclusion in a human disease. Consistent with the results in NHP (51), intrathecal infusion of ISIS-SMNRx was well tolerated in SMA patients during phase 1 clinical trial. If successful, this would be the first mechanism-based therapy for SMA.
The great promise of an ASO based therapy is derived from the limitless number of compounds that could be tested against the same target. Two recent pre-clinical studies conducted independently support ISS-N1 targeting PMOs as an entirely different class of very effectives ASOs (60,61). When delivered peripherally, ISS-N1 targeting PMOs appear to offer an advantage of being active at a substantially lower dose than a MOE ASO (52,61). In a recent Duchenne muscular dystrophy clinical trial, PMO backbone was well tolerated when delivered systemically (71). However, it remains to be seen whether intrathecal administration of PMOs are tolerated in NHPs. It will be also tempting to determine if systemic delivery of PMOs is a viable alternative in SMA. Although in preliminary stages, bifunctional and small ASOs hold potential to offer additional antisense compounds. With the ongoing progress in clinical trials and advancement in oligonucleotide chemistry and delivery schemes, it is only a matter of time until an array of effective ASO-based drugs becomes available for the treatment of SMA, a devastating disease of children and infants. The lessons learned from ASO-based therapy development in SMA will be extremely informative for other diseases that could utilize similar approaches.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 NS055925, R21 NS072259 and R21 NS080294) and Salsbury Endowment (Iowa State University, Ames, IA, USA) to RNS. CJD is supported by Families of SMA grant DID1214 and the Muscular Dystrophy Association grant 255785. The authors acknowledge Dr. Natalia N. Singh and Joonbae Seo for providing critical comments on the manuscript.
Footnotes
Disclosures and competing interests: ISS-N1 target (US patent # 7,838,657) was discovered in the Singh lab at UMASS Medical School (Worcester, MA, USA). Inventors, including RNS and UMASS Medical School, are currently benefiting from licensing of ISS-N1 target (US patent # 7,838,657) to ISIS Pharmaceuticals. A GC-rich target for a small ASO (Patent# US 20110269820) was discovered in the Singh lab at Iowa State University (Ames, IA, USA). Therefore, inventors including RNS and Iowa State University could potentially benefit from any future commercial exploitation of the above-mentioned target.
References
- 1.Pearn J. Classification of spinal muscular atrophies. Lancet. 1980;8174:919–922. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- 2.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, LePaslier D, Frezal F, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:1–5. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 3.McAndrew PE, Parsons DW, Simard LR, Rochette C, Ray PN, Mendell JR, Prior TW, Burghes AH. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997;60:1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prior TW. Spinal muscular atrophy diagnostics. J Child Neurol. 2007;22:952–956. doi: 10.1177/0883073807305668. [DOI] [PubMed] [Google Scholar]
- 5.Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum Mutat. 2000;15:228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 6.Lannaccone ST. Modern management of spinal muscular atrophy. J Child Neurol. 2007;22:974–980. doi: 10.1177/0883073807305670. [DOI] [PubMed] [Google Scholar]
- 7.Araujo AP, Araujo M, Swoboda KJ. Vascular perfusion abnormalities in infants with spinal muscular atrophy. J Pediatr. 2009;155:292–294. doi: 10.1016/j.jpeds.2009.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lorson CL, Hahnen E, Androphy EJ, Wirth BA. Single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh NN, Singh RN. Alternative splicing in spinal muscular atrophy underscores the role of an intron definition model. RNA Biol. 2011;8:600–606. doi: 10.4161/rna.8.4.16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vitte J, Fassier C, Tiziano FD, Dalard C, Soave S, Roblot N, Brahe C, Saugier-Veber P, Bonnefont JP, Melki J. Refined characterization of the expression and stability of the SMN gene products. Am J Pathol. 2007;171:1269–1280. doi: 10.2353/ajpath.2007.070399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho S, Dreyfuss G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 2010;24:438–442. doi: 10.1101/gad.1884910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossoll W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 13.Lorson CL, Rindt H, Shababi M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet. 2010;19:R111–118. doi: 10.1093/hmg/ddq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Meerbeke JP, Sumner CJ. Progress and promise: the current status of spinal muscular atrophy therapeutics. Discov Med. 2011;12:291–305. [PubMed] [Google Scholar]
- 15.Kolb SJ, Kissel JT. Spinal muscular atrophy: a timely review. Arch Neurol. 2011;68:979–984. doi: 10.1001/archneurol.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markowitz JA, Singh P, Darras BT. Spinal muscular atrophy: a clinical and research update. Pediatr Neurol. 2012;46:1–12. doi: 10.1016/j.pediatrneurol.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Hammond SM, Wood MJ. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011;27:196–205. doi: 10.1016/j.tig.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Southwell AL, Skotte NH, Bennett CF, Hayden MR. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med. 2012;18:634–643. doi: 10.1016/j.molmed.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 20.Deleavey GF, Damha MJ. Designing chemically modified oligonucleotides for targeted gene silencing. Chem Biol. 2012;19:937–954. doi: 10.1016/j.chembiol.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 21.Aartsma-Rus A. Overview on AON design. Methods Mol Biol. 2012;867:117–129. doi: 10.1007/978-1-61779-767-5_8. [DOI] [PubMed] [Google Scholar]
- 22.ENCODE Project Consortium. Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gralla J, Crothers DM. Free energy of imperfect nucleic acid helices. II. Small hairpin loops. J Mol Biol. 1973;73:497–502. doi: 10.1016/0022-2836(73)90096-x. [DOI] [PubMed] [Google Scholar]
- 24.Pörschke D. Thermodynamic and kinetic parameters of an oligonucleotide hairpin helix. Biophys Chem. 1974;1:381–386. doi: 10.1016/0301-4622(74)85008-8. [DOI] [PubMed] [Google Scholar]
- 25.Xu Z, Almudevar A, Mathews DH. Statistical evaluation of improvement in RNA secondary structure prediction. Nucleic Acids Res. 2012;40:e26. doi: 10.1093/nar/gkr1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freier SM, Watt AT. Basic principles of antisense drug discovery, In Antisense Drug Technologies. 2. Taylor & Francis; 2007. pp. 118–138. [Google Scholar]
- 27.Xing Y, Lee C. Relating alternative splicing to proteome complexity and genome evolution. Adv Exp Med Biol. 2007;623:36–49. doi: 10.1007/978-0-387-77374-2_3. [DOI] [PubMed] [Google Scholar]
- 28.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2007;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matlin AJ, Moore MJ. Spliceosome assembly and composition. Adv Exp Med Biol. 2007;623:14–35. doi: 10.1007/978-0-387-77374-2_2. [DOI] [PubMed] [Google Scholar]
- 30.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buratti E, Baralle M, Baralle FE. Defective splicing, disease and therapy: searching for master checkpoints in exon definition. Nucleic Acids Res. 2006;34:3494–3510. doi: 10.1093/nar/gkl498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teplova M, Minasov G, Tereshko V, Inamati GB, Cook PD, Manoharan M, Egli M. Crystal structure and improved antisense properties of 2’-O-(2-methoxyethyl)-RNA. Nat Struct Biol. 1999;6:535–539. doi: 10.1038/9304. [DOI] [PubMed] [Google Scholar]
- 35.Singh NN, Shishimorova M, Cao LC, Gangwani L, Singh RN. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009;6:341–350. doi: 10.4161/rna.6.3.8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh NN, Hollinger K, Bhattacharya D, Singh RN. An antisense microwalk reveals critical role of an intronic position linked to a unique long-distance interaction in pre-mRNA splicing. RNA. 2010;16:1167–1181. doi: 10.1261/rna.2154310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh NN, Androphy EJ, Singh RN. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA. 2004;10:1291–1305. doi: 10.1261/rna.7580704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007;35:371–389. doi: 10.1093/nar/gkl1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh NN, Seo J, Ottesen EW, Shishimorova M, Bhattacharya D, Singh RN. TIA1 prevents skipping of a critical exon associated with spinal muscular atrophy. Mol Cell Biol. 2011;31:935–954. doi: 10.1128/MCB.00945-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klar J, Sobol M, Melberg A, Mäbert K, Ameur A, Johansson AC, Feuk L, Entesarian M, Orlén H, Casar-Borota O, Dahl N. Welander Distal Myopathy Caused by an Ancient Founder Mutation in TIA1 Associated with Perturbed Splicing. Hum Mutat. 2013 Jan 24; doi: 10.1002/humu.22282. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 41.Hicks MJ, Mueller WF, Shepard PJ, Hertel KJ. Competing upstream 5’-splice sites enhance the rate of proximal splicing. Mol Cell Biol. 2010;30:1878–1886. doi: 10.1128/MCB.01071-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, Zhang Z, Cho S, Sherrill-Mix S, Wan L, Dreyfuss G. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012;150:53–64. doi: 10.1016/j.cell.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, Dreyfuss G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams JH, Schray RC, Patterson CA, Ayitey SO, Tallent MK, Lutz GJ. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J Neurosci. 2009;29:7633–7638. doi: 10.1523/JNEUROSCI.0950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;4:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 46.Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butchbach ME, Singh J, Thorsteinsdóttir M, Saieva L, Slominski E, Thurmond J, Andrésson T, Zhang J, Edwards JD, Simard LR, Pellizzoni L, Jarecki J, Burghes AH, Gurney ME. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum Mol Genet. 2010;19:454–467. doi: 10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O’Donovan MJ. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69:453–467. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bebee TW, Dominguez CE, Chandler DS. Mouse models of SMA: tools for disease characterization and therapeutic development. Hum Genet. 2012;131:1277–1293. doi: 10.1007/s00439-012-1171-5. [DOI] [PubMed] [Google Scholar]
- 50.Hua Y, Sahashi K, Hung D, Rigo F, Passini MA, Bennett CF, Krainer AR. Antisense correction of SMN splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Passini MA, Bu J, Richards AM, Kinnecom C, Sardi S, Stanek LM, Hua Y, Rigo F, Matson J, Hung G, Kaye EM, Shihabuddin LS, Krainer AR, Bennett CF, Cheng SH. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 54.Tanganyika-de Winter CL, Heemskerk H, Karnaoukh TG, van Putten M, de Kimpe SJ, van Deutekom J, Aartsma-Rus A. Long-term Exon Skipping Studies With 2’-O-Methyl Phosphorothioate Antisense Oligonucleotides in Dystrophic Mouse Models. Mol Ther Nucleic Acids. 2012;1:e44. doi: 10.1038/mtna.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osman EY, Yen PF, Lorson CL. Bifunctional RNAs targeting the intronic splicing silencer N1 increase SMN levels and reduce disease severity in an animal model of spinal muscular atrophy. Mol Ther. 2012;20:119–126. doi: 10.1038/mt.2011.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gogliotti RG, Hammond SM, Lutz C, Didonato CJ. Molecular and phenotypic reassessment of an infrequently used mouse model for spinal muscular atrophy. Biochem Biophys Res Commun. 2010;391:517–522. doi: 10.1016/j.bbrc.2009.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Riessland M, Ackermann B, Förster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, Wirth B. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010;19:1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- 58.Ellett F, Lieschke GJ. Zebrafish as a model for vertebrate hematopoiesis. Curr Opin Pharmacol. 2010;10:563–570. doi: 10.1016/j.coph.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Hao le T, Burghes AH, Beattie CE. Generation and Characterization of a genetic zebrafish model of SMA carrying the human SMN2 gene. Mol Neurodegener. 2011;6:24. doi: 10.1186/1750-1326-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Porensky PN, Mitrpant C, McGovern VL, Bevan AK, Foust KD, Kaspar BK, Wilton SD, Burghes AH. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou H, Janghra N, Mitrpant C, Dickinson RL, Anthony K, Price L, Eperon IC, Wilton SD, Morgan J, Muntoni F. A novel morpholino oligomer targeting ISS-N1 improves resuce of severe SMA transgenic mice. Hum Gene Ther. doi: 10.1089/hum.2012.211. [Epub ahead of print 22 Jan 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gendron D, Carriero S, Garneau D, Villemaire J, Klinck R, Elela SA, Damha MJ, Chabot B. Modulation of 5’ splice site selection using tailed oligonucleotides carrying splicing signals. BMC Biotechnol. 6:5. doi: 10.1186/1472-6750-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miyajima H, Miyaso H, Okumura M, Kurisu J, Imaizumi K. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J Biol Chem. 2002;277:23271–23277. doi: 10.1074/jbc.M200851200. [DOI] [PubMed] [Google Scholar]
- 64.Baughan TD, Dickson A, Osman EY, Lorson CL. Delivery of bifunctional RNAs that target an intronic repressor and incease SMN levels in an animal model of spinal muscular atrophy. Hum Mol Genet. 2009;18:1600–1611. doi: 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003;10:120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 66.Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci U S A. 2003;100:4114–4119. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Owen N, Zhou H, Malygin AA, Sangha J, Smith LD, Muntoni F, Eperon IC. Design principles for bifunctional targeted oligonucleotide enhancers of splicing. Nucleic Acids Res. 2011;39:7194–7208. doi: 10.1093/nar/gkr152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meyer K, Marquis J, Trüb J, Nlend Nlend R, Verp S, Ruepp MD, Imboden H, Barde I, Trono D, Schümperli D. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum Mol Genet. 2009;18:546–555. doi: 10.1093/hmg/ddn382. [DOI] [PubMed] [Google Scholar]
- 69.Geib T, Hertel KJ. Restoration of full-length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS One. 2009;4:e8204. doi: 10.1371/journal.pone.0008204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Singh NN, Seo J, Rahn SJ, Singh RN. A multi-exon-skipping detection assay reveals surprising diversity of splice isoforms of spinal muscular atrophy genes. PLoS One. 2012;7:e49595. doi: 10.1371/journal.pone.0049595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]