

Abstract
Objective: To study the effect of the transfected Breast cancer metastasis suppressor 1 (BRMS1) gene on the migration of breast cancer cells and the possible mechanisms involved. Methods: MDA-MB-231HM cells which have a high propensity of metastasize to lung was sieved from MDA-MB-231 and its derivative cells stable transfected with BRMS1 were used to study in vitro. Cell migratory ability was observed. The cellular cyclic adenylic acid (cAMP) concentration was tested by radioimmunoassay (RIA). The activity of adenylate cyclase (AC), phosphodiesterase (PDE) and protein kinase A (PKA) were measured by enzyme immunoassay (EIA) and (γ-32P) ATP incorporation. The effect of BRMS1 on connexins (Cx) expression was analyzed by by RT-PCR and Western blot. Results: Overexpression of BRMS1 significantly inhibited cell migration in MDA-MB-231HM cells in vitro. However, BRMS1’s effect on cell migration could be eliminated after pretreating with pertussis toxin (PTX). BRMS1 overexpression increased cellular cAMP and PKA activity by activating the activity of AC. Furthermore, BRMS1 overexpression up-regulated Cx26 expression, whereas Cx32, Cx43 expressions did not changed. Conclusion: The present study indicated G-protein-coupled cAMP signaling pathway was involved in BRMS1 related MDA-MB-231HM cells migration, and BRMS1 could change connexins (Cx) expression profiles through increasing expression of Cx26 in cells.
Keywords: Breast cancer, BRMS1, cAMP, migration, metastasis, G protein, connexin
Introduction
Metastasis suppressor genes are a class of genes that reduces the metastatic propensity of cancer cells in vivo without blocking these neoplastic cells growth at orthotopic or subcutaneous sites [1]. Breast cancer metastasis suppressor 1 (BRMS1) is a gene that was mapped to chromosome 11q13 and was originally identified as a metastasis suppressor gene [2]. BRMS1 mRNA expression has been shown to be markedly reduced in melanoma, breast cancer, ovarian cancer and non-small cell lung cancer (NSCLC) cell line, and stable over expression of BRMS1 in these cell lines significantly inhibited their metastatic potential [3-5]. Hedley BD et al. found BRMS1 expression significantly reduced the numbers of solitary single cells that survive after initial arrest within the lung microvasculature, and also inhibited the initiation of growth subsequent to arrest; in vitro, BRMS1 expression decreased cancer cell survival under stress conditions (hypoxia), increased anoikis, and decreased the ability of cancer cells to adhere [6]. However, Samant RS et al. reported there is no evidence that BRMS1 regulates metastatic movement through alterations in matrix metalloproteinase levels, cellular adhesion profiles, or alterations in tumor cell invasion [7]. Thus, the BRMS1 mechanisms of action remain unknown largely in breast cancer.
Maintenance of homeostasis is a leading role for tissue function and its disorder usually causes development of organ diseases including cancers. Gap junctional intercellular communication (GJIC) contributes to tissue homeostasis, is mediated largely by gap junction. Gap junctions are clusters of channels formed by proteins called connexins (Cx). They allow small signaling molecules and ions less than 1000Da, such as Ca2+ and cyclic adenylic acid (cAMP), to pass directly between the adjacent cells. cAMP is an important second messenger within cell which can have numerous effects. Many studies have so far proved that down-regulation of the GJ is involved in carcinogenic pathways and that connexin proteins can function as tumor-suppressors [8,9].
Given that metastasis involves a multitude of signaling changes in steps of the metastatic cascade. In the present study, we systematically up-regulated the expression of BRMS1 to explore the roles of BRMS1 on migration in breast cancer cells and tried to elucidate the mechanism. We therefore addressed in the present study the question of the ability of BRMS1 to regulate second messenger cAMP, in turn, could affect cell migration. Then we tried to determine whether BRMS1 regulate Cx expression including Cx26, Cx32, and Cx43 which mediate gap junction communication.
Materials and methods
Plasmid construction
The human BRMS1 full-length coding sequence was amplified by polymerase chain reaction (PCR) using the primers containing EcoRI and HindIII restriction sites. The sense primer of BRMS1 is 5’-ATCGAAGCTTACTATGCCTGTCCAGCCTCCAAGC-3’ and the antisense primer is 5’-ATGCGAATTCTCAAGGTCCATCCGATTTTC-3’, the restriction sites are underlined. The PCR product was sub cloned into the pcDNA3.1 (+) vector (Invitrogen, Carlsbad, CA) that contains neomycin resistant gene. The recombinant plasmid construct, pcDNA3.1/BRMS1 was confirmed by EcoRI/HindIII digestion (New England Biolabs, Beverly, MA) and DNA sequencing (Invitrogen, Carlsbad, CA).
Cell culture and stable cell line generation
The MDA-MB-231 human breast cancer cell line was obtained from the American Type Culture Collection (Manassas, VA). The cells were cultured in DMEM supplemented with 1% penicillin/streptomycin (P/S) and 10% fetal bovine serum (FBS) at 37°C in a humidified 5% CO2 incubator. MDA-MB-231HM with a high propensity of metastasize to lung was generated from wide-type MDA-MB-231 by in vivo screening method; mainly, at 28 days after inoculation, pulmonary metastasis was occurred in 100% mice orthotopic inoculated with the cells [10]. Stable BRMS1 expressing MDA-MB-231HM cells were generated by transfection of a pcDNA3.1/BRMS1 expression vector containing the human BRMS1 cDNA. Transfection was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Briefly, 1×105 cells in 1 ml of DMEM containing 10% FBS were seeded in 60-mm dishes and grew to 80%-85% confluence. pcDNA3.1/BRMS1 (8 μg) and Lipofectamine 2000 were added respectively in tubes each containing 500 μl of DMEM, mixed well and allowed to stand for 5 min at room temperature. These two mixtures were then combined together and incubated for 20 min at room temperature. One milliliter of medium containing Lipofectamine packaged plasmid DNA was added to the 60-mm dish and mixed gently by rocking the plate back and forth. The medium was refreshed after 6 h of transfection and cells were incubated at 37°C with 5% CO2 in a humidified atmosphere for 24 h. Then cells were passaged at 1:10 into fresh DMEM supplemented with 10% FBS. Stable clones were obtained by G418 selection (800 μg/ml) (Invitrogen, Carlsbad, CA) for 2-3 weeks. Individual clones were isolated and cultured further in the presence of 400 μg/ml G418. For the control, empty vector pcDNA3.1 was transfected and cells were harvested after selection.
Cell migration assay
A three-dimensional cell migration assay was performed with the Transwell system, which allows cells to migrate through an 8-μm pore size polycarbonate membrane (Costar, Cambridge, USA). Briefly, the lower compartment was filled with 600 μl DMEM medium with 10% fetal bovine serum. Cells were harvested by trypsinization and resuspended in DMEM medium without fetal calf serum (1×106 cells/ml). A total of 100 ul of the cell suspension, 1×106 cells/ml, treated with pertussis toxin (PTX) (100 ng/ml) or untreated, was added to the upper compartment and then the chambers were incubated at 37°C in 5% CO2 for 18 h. Cells on the upper side of the filter were removed mechanically. Cells which had migrated to the lower side were fixed on the filter with methanol, stained with hematoxylin-eosin and 5 random fields were counted at 200-fold magnification. Migration was evaluated by the number of cells penetrating through the membrane per field.
Measurement of cAMP
Determination of intracellular cAMP was preformed according to the instructions of the manufacture by a cAMP RIA kit (China Institute of Atomic Energy). Briefly, the cells were grown to confluence in 60-mm-diameter cell culture dishes. Assay buffer was 50-mM sodium acetate buffer with 0.1% sodium azide (pH 4.75). All samples and standards were acetylated with 10 μl of a 2:1 solution of triethylamine: acetic anhydride. Each assay tube contained 5 μl of each sample, 95 μl of buffer, 100 μl of anti-cAMP antibody and 100 μl of 125I-cAMP, total assay volume was 310 μl. Tubes were vortexed and stored at 4°C overnight. Antibody-bound radioactivity was then separated by the addition of 25 μl of goat anti-rabbit IgG, followed by vortexing and further incubation at 4°C for 1 h. To these samples, 1 ml of 12% polyethylene glycol, 50 mM sodium acetate buffer (pH 6.75) was added, and all tubes were centrifuged at 1700 g for 15 min. Supernatants were aspirated, and radioactivity in the resulting pellet was determined using a gamma counter. cAMP content was normalized to protein content of each cell, which was determined by the Bradford assay with BSA as a standard.
Adenylate cyclase (AC) assay
Membrane preparations isolated from cells were tested for AC activity. Plasma membrane preparations were isolated by differential centrifugation. Briefly, cells were homogenized with a Dounce homogenizer in 20 mM HEPES, 0.25 M sucrose, 1 mM EDTA, 5 mM benzamidine, and a protease inhibitor cocktail (1:500). Membranes were collected by two steps of differential centrifugation (1,000x g for 5 min at 4°C and 40,000x g for 20 min at 4°C). The protein concentrations in the samples were determined. For AC assay, 30 μgof membrane protein was added to reaction buffer [50 mM Tris-HCl (pH 7.4), 5.0 mM MgCl2, 0.5 mM EDTA, 1 mM ATP, 0.1 μM GTP, 0.2 IU pyruvate kinase, 0.1 U myokinase, and 2.5 mM phosphoenolpyruvate] and incubated for 15 min at 37 C. The converted cAMP from ATP in the supernatant of the samples was determined with an EIA assay kit.
cAMP-specific phosphodiesterase (PDE) assay
The cells were scraped into lysis buffer [20 mM HEPES (pH 7.4), 1 mM EDTA, 1 mM dithiothreitol, and 1:500 protease inhibitor cocktail], Dounce homogenized, and then sonicated for 30 sec on ice. Cells lysates were centrifuged (14,000x g for 5 min at 4°C), and 30 μg protein of cell lysates was incubated in reaction buffer [20 mM HEPES (pH 7.4), 90 mM KCl, 5 mM MgCl2, 0.75 mM CaCl2, and 100 nM cAMP] for 30 min at 30°C. The reaction was terminated by the addition of 0.1 N HCl and centrifuged. The cAMP remaining in the supernatants was measured by an EIA kit. The PDE activity was determined as the amount of cAMP hydrolyzed during the reaction time.
PKA activity
Cell extracts were prepared and lysed in three volumes of 0.25 M mannitol, 100 mM Tris-HCl buffer, pH 7.4, at 4°C, containing 50 mM NaF, 2 mM NaPPi, 1 mM EDTA, 1 mM benzamidine, 0.2 mM PMSF, 1 μg/mL soya bean trypsin inhibitor (SBTI), and 1 mM isobutyl methylxanthine (IBMX). The lysates were then centrifuged at 14,000 rpm for 10 min. For the PKA assays, supernatants were diluted 1:20 in 100 mM Tris-HCl buffer, pH 7.4, containing 50 mM NaF, 100 mM NaCl, 1 mM EDTA, 1 mM IBMX, 1 mM DTT, 1 mM benzamidine, 1 μg/mL SBTI, and 1 mg/mL BSA. Assay incubation (50 μL, 50 μg protein lysate) was carried out at 37°C in the buffer containing 0.2 mM kemptide (a peptide substrate for PKA) (Sigma), 4 mM MgCl2, and 0.2 mM 32P-ATP (specific activity 200,000-400,000 cpm/nmol phosphate). Aliquots (20 μL) were removed at 4 and 8 min of incubation and spotted onto P81 phosphocellulose paper squares that were then washed extensively in three changes of 77 mM phosphoric acid before being transferred to scintillation vials for determination of 32P incorporation into kemptide. PKA activity was taken as the 32P incorporated into kemptide.
Total RNA isolation and RT-PCR
Total RNA was extracted by using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. In brief, 106 cells were lysed with 1 ml of TRIzol, and the proteins were separated by chloroform. RNAs were precipitated with isopropanol, and cDNAs were synthesized by using a RevertAid First Strand cDNA Synthesis Kit (MBI Fermentas, Vilnius, Lithuania) according to the manufacturer’s protocol. Reverse transcription was carried out at 42°C for 30 min following incubation at 95°C for 5 min.
A conventional PCR using GeneAmp PCR System from Perkin Elmer (Norwalk, CT, USA) was based on standard amplification conditions: 30-35 cycles of denaturation at 94°C (45 s), annealing at 58°C (30 s), and extension at 72°C (45 s) followed by 5 min of final extension at 72°C. All data were normalized relative to the expression of GAPDH mRNA in respective samples.
The quantitative real-time PCR reaction mixtures were amplified with SYBR Green PCR Master Mix (Takara Shuzo, Japan) in the presence of 0.2 μmol/L each of the sense and antisense primers, and 100 ng sample, by the following thermal cycling programs: stage 1, 95°C for 10 seconds; stage 2, 40 cycles of 94°C for 5 seconds, 60°C for 20 seconds and 72°C for 15 seconds; stage 3, 72°C for 5minutes. Fluorescence intensity was measured in real-time during extension steps for a SYBR Green assay by using the Mini Opticon Real-Time PCR System (Bio-Rad). Relative expression levels of the target gene was normalized using GAPDH as an endogenous reference using the comparative CT (threshold cycle) method. The amount of gene expression that was normalized to endogenous reference is given by: 2-ΔΔCT. The information of PCR primers were showed in Table 1.
Table 1.
Information of PCR primers
| Gene | Primer | Length of product | |
|---|---|---|---|
| BRMS1 | sense | cagcctccaagcaaagaca | 160bp |
| antisense | ggcgtcgctcatagtcctcat | ||
| Cx26 | sense | gctgcaagaacgtgtgctac | 196bp |
| antisense | tgggttttgatctcctcgat | ||
| Cx32 | sense | accaattcttccccatctcc | 250bp |
| antisense | aagacggcctcaaacaacag | ||
| Cx43 | sense | aggagttcaatcacttggcg | 168bp |
| antisense | gcaggattcggaaaatgaaa | ||
| GAPDH | sense | agaaggctggggctcatttg | 258bp |
| antisense | aggggccatccacagtcttc |
Western blot
Cells were washed twice with ice-cold PBS and scarped into 1 ml of ice-cold NP40 lysis buffer (10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% (v/v) NP40, 1 mM EDTA (ethylene diaminetetraacetic acid, edetic acid), 50 mM NaF, 5 mM NaPPI, 1 mM phenylmethylsulfonyl fluoride, 1 mg/ml leupeptin and 1 mg/ml pepstatin A).Cells were then sonicated for 5 s at 5 W. Insoluble debris was removed by centrifugation at 1000 g for 15 min. Total proteins (50 microgram) were analyzed by 10% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis). Western blot, using mouse anti-human monoclonal antibody against to BRMS1, rabbit anti-human polyclonal antibody against to Cx26 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was performed according to standard protocols. Normalization of protein loading was done with GAPDH. Blot quantification was done with a Molecular Dynamics Laser Densitometer (Model PSD) and the Image Quant Version 1 software.
Statistical analysis
All data are expressed as mean ± SEM. Differences between groups were analyzed by Student’s t test or one-factor analysis of variance, as required. Differences were considered significant if P values were less than 0.05.
Results
Stable transfection of BRMS1cDNA in MDA-MB-231HM cells
In order to investigate the effect of BRMS1 on cell migration, we transfected BRMS1 expression vector into MDA-MB-231HM cells and generated stable transfectant. BRMS1 expression in this clone was confirmed by reverse transcription PCR and western blot (Figure 1A, 1B). Transfectant clones from MDA-MB-231HM cells with high expression levels of BRMS1 protein were named MDA-MB-231HM-BRMS1. The levels of BRMS1 mRNA and protein expression in MDA-MB-231HM-BRMS1 were significantly increased compared with MDA-MB-231HM and MDA-MB-231HM-vector (Figure 1C).
Figure 1.
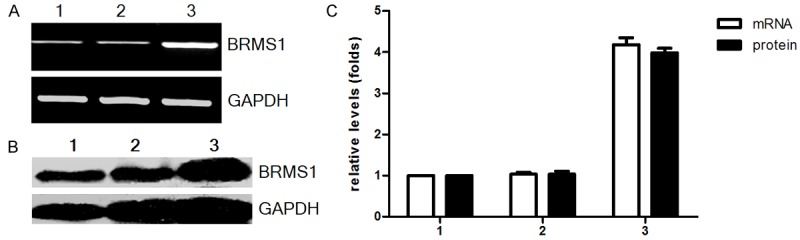
Expression of BRMS1 in MDA-MB-231HM after transfection was detected by RT-PCR (A) and Western blot (B). The relative expression of BRMS1 mRNA and protein was shown in (C). Representative quantitation of three independent experiments. The error bars represent the SEM. For (A-C): lane 1, MDA-MB-231 HM; lane 2, MDA-MB-231 HM-vector; lane 3, MDA-MB-231HM-BRMS1.
BRMS1 inhibited the migration of breast cancer cell in vitro
We examined the migration activities of BRMS1 stable transfectant and the parental cells using the Boyden chamber. Migration was significantly inhibited in BRMS1 stable transfectant (Figure 2A, 2B).
Figure 2.
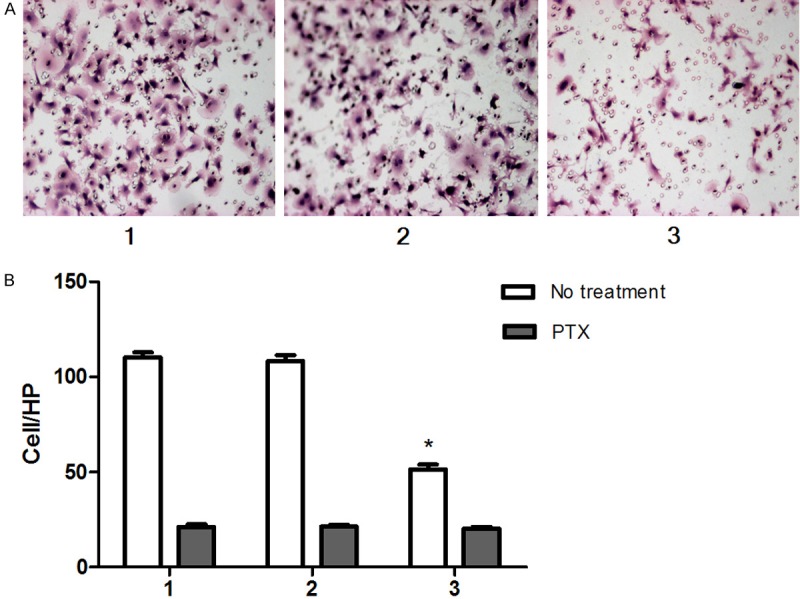
BRMS1 inhibited the migration of MDA-MB-231HM in vitro (A). Quantification of migration assay for the BRMS1-transfected cells and control cells. PTX eliminated the effect of BRMS1 on cell migration (B). Cells were counted in triplicate wells and in three identical experiments. *, P < 0.05. Columns, mean of three independent experiments; bars, SEM. HP, high-power objective. For (A, B): lane 1, MDA-MB-231 HM; lane 2, MDA-MB-231 HM-vector; lane 3, MDA-MB-231HM-BRMS1.
PTX eliminated the effect of BRMS1 on cell migration
We speculated that cAMP signaling pathway may involve in BRMS1 related cell migration. To investigate this possibility, we have use PTX to treat breast cancer cells before migration assay. The results showed that PTX inhibit the migration of BRMS1-transfected MDA-MB-231HM cells, mock-transfected and wild-type cells. But, there is no significant difference among three groups after pretreating with PTX (Figure 2B).
BRMS1 activated cAMP signaling pathway
To future support for the idea that cAMP signaling pathway involves in BRMS1 related cell migration. Firstly, we examined the effect of BRMS1 on the intracellular cAMP concentration and PKA activity in breast cancer cells. The results demonstrated that BRMS1-transfected MDA-MB-231HM cells increased intracellular cAMP and PKA activity than mock-transfected and wild-type cells (Figure 3A, 3B).
Figure 3.
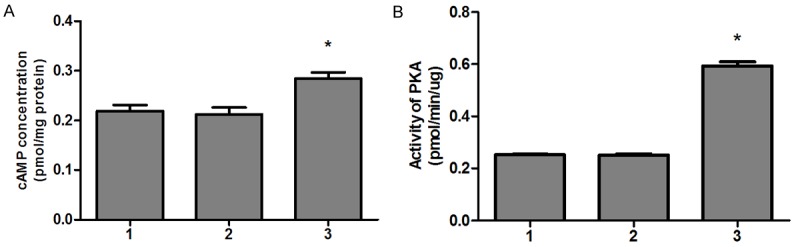
BRMS1 increased MDA-MB-231HM cells intracellular cAMP (A) and PKA activity (B). The experiment was repeated three times in triplicate, and data were expressed as means ± SEM. *, P < 0.05. For (A, B): lane 1, MDA-MB-231 HM; lane 2, MDA-MB-231 HM-vector; lane 3, MDA-MB-231HM-BRMS1.
It is well known that cAMP is raised by increased synthesis via activation of AC and/or decreased degradation via inhibition of PDEs. Then, we investigated the activities of AC and PDE. We observed an increased activity of AC in BRMS1-transfected MDA-MB-231HM cells compared to mock-transfected and wild-type cells (Figure 4A). But there were no significant difference in activity of PDE among BRMS1-transfected MDA-MB-231HM cells, mock-transfected and wild-type cells (Figure 4B).
Figure 4.
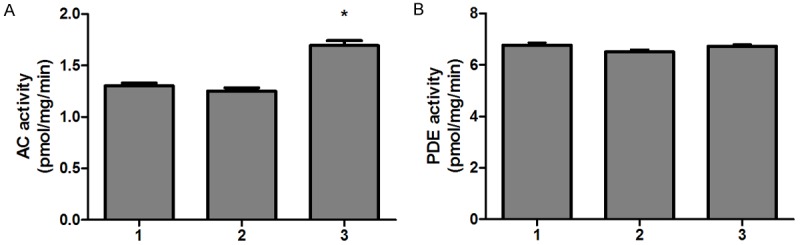
BRMS1 stimulated AC activity (A) and had no effect on PDE activity (B) in MDA-MB-231HM cells. The experiment was repeated three times and data were expressed as means ± SEM. *, P < 0.05. For (A, B): lane 1, MDA-MB-231 HM; lane 2, MDA-MB-231 HM-vector; lane 3, MDA-MB-231HM-BRMS1.
These data thus indicate that BRMS1 activated G-protein-coupled cAMP signaling pathway through activation of AC.
BRMS1 up-regulated Cx26 expression
In order to investigate whether BRMS1 overexpression could modulate the expression of connexins, Cx26, Cx32, Cx43 expression of cells were examined by RT-PCR and western blot. We observed that BRMS1-transfected MDA-MB-231HM cells expressed higher levels of Cx26 mRNA and protein than mock-transfected and wild-type cells (Figure 5). We did not find any significant difference in Cx32, Cx43 mRNA expression among BRMS1-transfected MDA-MB-231HM cells compared with control groups (Figure 5A).
Figure 5.
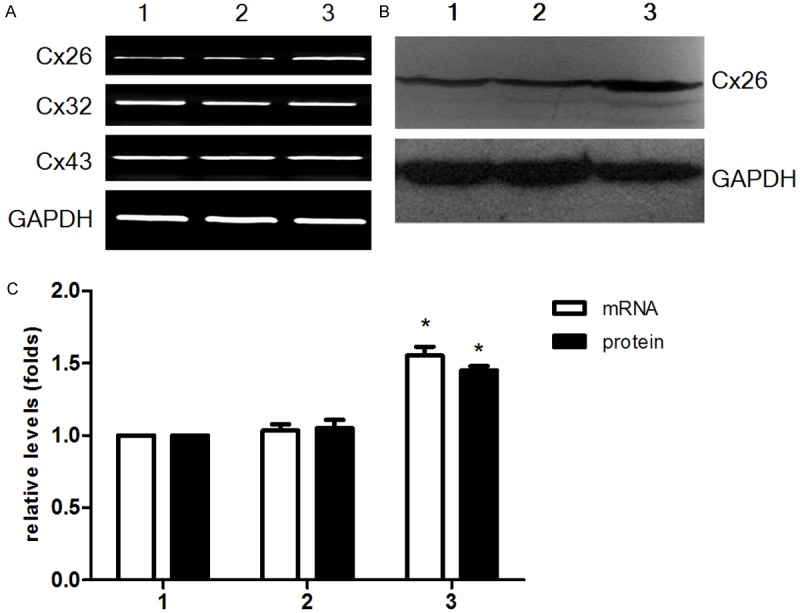
Expression of connexins in MDA-MB-231HM after transfection. Cx26 was detected by RT-PCR (A) and Western blot (B). The relative expression of Cx26 mRNA and protein was shown in (C). They are the means ± SEM of three separate experiments. For (A-C): lane 1, MDA-MB-231 HM; lane 2, MDA-MB-231 HM-vector; lane 3, MDA-MB-231HM-BRMS1. *, P < 0.05.
Discussion
In the present study, we observed that overexpression of the BRMS1 reduced MDA-MB-231HM cell migration in vitro. The result is consistent with prior study [3,7]. Migration, invasion and avoidance of cell death or apoptosis are three separate steps required for cancer metastasis [11]. Migration is one of the key functional activities of cancer cells directly associated with their metastatic potential. Previous study have indicated that signaling through phosphoinositide in the regulation of breast cancer metastasis that can be suppressed by BRMS1 [12]. Vaidya KS. et al. reported BRMS1 expression reduced epidermal growth factor receptor (EGFR) expression and downstream (Akt) signaling; but signaling through another receptor tyrosine kinase, hepatocyte growth factor receptor (c-Met) remains unchanged [13]. However, participation of other signaling molecules, including cAMP remains not to be explored.
In our experiment, migration of MDA-MB-231HM-BRMS1 was markedly reduced comparing with MDA-MB-231HM, whereas cells migration were all decreased after pretreating with PTX. That indicated the inhibitory effect of BRMS1 on MDA-MB-231HM-BRMS1 cell migration could be abolished by PTX pretreatment. PTX specifically inactivates Gαi protein mediated signaling pathways. The results indicated that PTX-sensitive G-protein-coupled signaling may be involved in BRMS1 related cell migration. Further analysis showed that the intracellular cAMP concentration of MDA-MB-231HM-BRMS1 cells significantly increased. The activity of PKA which is a serine kinase activated by cAMP also increased significantly, compared with MDA-MB-231HM cells. Intracellular cAMP concentration was determined by activities of AC and cAMP dependent PDE. The study showed that BRMS1 overexpression stimulated AC activity, but had no effect on PDE activity. Those results definitely suggested that BRMS1 related cell migration involved in activated G protein coupled cAMP signaling pathway. To the best of our knowledge, our findings provide the first indication that the G-protein-coupled cAMP signaling pathway is involved in BRMS1 related cell migration. The effects of cAMP signaling on BRMS1 expression, cell migration and metastasis are worth to be researched further.
In the present study, we also found that overexpression of BRMS1 significantly increase Cx26 expression. Recently, increasing evidences have suggested that Cx26, Cx32, Cx43 could act as a suppressor gene against metastasis of cancer cell lines [14-18]. In almost all tumors, the function of the gap junction is down-regulated through one or more of a variety of mechanisms, including no or reduced expression, aberrant localization, and aberrant phosphorylation or dephosphorylation of connexin protein. A earlier report showed that exogenous expression of BRMS1 in MDA-MB-435 cells can lead to up regulation of Cx43, downregulation of Cx32 and restoration of GJIC [19]. Interestingly, in our experiment we have demonstrated that BRMS1 can up regulate of Cx26, but not change Cx32 and Cx43 in MDA-MB-231HM cells. The disagreement with prior result may be due to that BRMS1 can regulate different connexins transcription in different cancer cell types.
Connexins have multifaceted functions in both normal mammary gland development and homeostasis, as well as in cancer metastasis [20]. However connexins have GJIC-dependent cellular effects through GJ channel activity, by the exchange of small molecules or current between cells on cancer cells metastasis including migration. However, extensive studies suggested that connexins also exert their functions in a GJIC-independent manner. The notion was supported by the study on the role of Cx26, Cx43 in breast [21-23]. Take together with earlier study, our results imply that BRMS1 might change connexins expression profile to affect metastasis in breast cancer cells.
Conclusion
Our results indicated that BRMS1 plays a negative regulatory role as a metastasis suppressor gene in human breast cancer. G-protein-coupled cAMP signaling pathway is involved in BRMS1 related breast cancer cells migration. Furthermore, overexpression of BRMS1 can result in change of connexins expression profile in breast cancer cells. There findings may provide new sights into the BRMS1 mechanisms of action.
Acknowledgements
This work was supported by the Medical Science Research Foundation of Zhejiang Province of China (NO. 2011KYA108) and Wenzhou City Science and Technology Bureau grant (NO. Y20110065).
Disclosure of conflict of interest
None.
References
- 1.Steeg PS. Perspectives on classic article: metastasis suppressor genes. J Natl Cancer Inst. 2004;96:E4. doi: 10.1093/jnci/djh107. [DOI] [PubMed] [Google Scholar]
- 2.Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000;60:2764–2769. [PubMed] [Google Scholar]
- 3.Shevde LA, Samant RS, Goldberg SF, Sikaneta T, Alessandrini A, Donahue HJ, Mauger DT, Welch DR. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp Cell Res. 2002;273:229–239. doi: 10.1006/excr.2001.5452. [DOI] [PubMed] [Google Scholar]
- 4.Yang J, Zhang B, Lin Y, Yang Y, Liu X, Lu F. Breast cancer metastasis suppressor 1 inhibits SDF-1alpha-induced migration of non-small cell lung cancer by decreasing CXCR4 expression. Cancer Lett. 2008;269:46–56. doi: 10.1016/j.canlet.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Zhang S, Lin QD, Di W. Suppression of human ovarian carcinoma metastasis by the metastasis-suppressor gene, BRMS1. Int J Gynecol Cancer. 2006;16:522–531. doi: 10.1111/j.1525-1438.2006.00547.x. [DOI] [PubMed] [Google Scholar]
- 6.Hedley BD, Vaidya KS, Phadke P, MacKenzie L, Dales DW, Postenka CO, MacDonald IC, Chambers AF. BRMS1 suppresses breast cancer metastasis in multiple experimental models of metastasis by reducing solitary cell survival and inhibiting growth initiation. Clin Exp Metastasis. 2008;25:727–740. doi: 10.1007/s10585-008-9184-0. [DOI] [PubMed] [Google Scholar]
- 7.Samant RS, Seraj MJ, Saunders MM, Sakamaki TS, Shevde LA, Harms JF, Leonard TO, Goldberg SF, Budgeon L, Meehan WJ, Winter CR, Christensen ND, Verderame MF, Donahue HJ, Welch DR. Analysis of mechanisms underlying BRMS1 suppression of metastasis. Clin Exp Metastasis. 2000;18:683–693. doi: 10.1023/a:1013124725690. [DOI] [PubMed] [Google Scholar]
- 8.Trosko JE, Ruch RJ. Cell-cell communication in carcinogenesis. Front Biosci. 1998;3:d208–236. doi: 10.2741/a275. [DOI] [PubMed] [Google Scholar]
- 9.Omori Y, Zaidan Dagli ML, Yamakage K, Yamasaki H. Involvement of gap junctions in tumor suppression: analysis of genetically-manipulated mice. Mutat Res. 2001;477:191–196. doi: 10.1016/s0027-5107(01)00120-8. [DOI] [PubMed] [Google Scholar]
- 10.Chang XZ, Li DQ, Hou YF, Wu J, Lu JS, Di GH, Jin W, Ou ZL, Shen ZZ, Shao ZM. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007;9:R76. doi: 10.1186/bcr1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fidler IJ. Critical factors in the biology of human cancer metastasis. Am Surg. 1995;61:1065–1066. [PubMed] [Google Scholar]
- 12.DeWald DB, Torabinejad J, Samant RS, Johnston D, Erin N, Shope JC, Xie Y, Welch DR. Metastasis suppression by breast cancer metastasis suppressor 1 involves reduction of phosphoinositide signaling in MDA-MB-435 breast carcinoma cells. Cancer Res. 2005;65:713–717. [PubMed] [Google Scholar]
- 13.Vaidya KS, Harihar S, Phadke PA, Stafford LJ, Hurst DR, Hicks DG, Casey G, DeWald DB, Welch DR. Breast cancer metastasis suppressor-1 differentially modulates growth factor signaling. J Biol Chem. 2008;283:28354–28360. doi: 10.1074/jbc.M710068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirschi KK, Xu CE, Tsukamoto T, Sager R. Gap junction genes Cx26 and Cx43 individually suppress the cancer phenotype of human mammary carcinoma cells and restore differentiation potential. Cell Growth Differ. 1996;7:861–870. [PubMed] [Google Scholar]
- 15.Li Z, Zhou Z, Donahue HJ. Alterations in Cx43 and OB-cadherin affect breast cancer cell metastatic potential. Clin Exp Metastasis. 2008;25:265–272. doi: 10.1007/s10585-007-9140-4. [DOI] [PubMed] [Google Scholar]
- 16.Momiyama M, Omori Y, Ishizaki Y, Nishikawa Y, Tokairin T, Ogawa J, Enomoto K. Connexin26-mediated gap junctional communication reverses the malignant phenotype of MCF-7 breast cancer cells. Cancer Sci. 2003;94:501–507. doi: 10.1111/j.1349-7006.2003.tb01473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao Q, Wang H, McLachlan E, Veitch GI, Laird DW. Down-regulation of Cx43 by retroviral delivery of small interfering RNA promotes an aggressive breast cancer cell phenotype. Cancer Res. 2005;65:2705–2711. doi: 10.1158/0008-5472.CAN-04-2367. [DOI] [PubMed] [Google Scholar]
- 18.Yano T, Fujimoto E, Hagiwara H, Sato H, Yamasaki H, Negishi E, Ueno K. Connexin 32 as an anti-invasive and anti-metastatic gene in renal cell carcinoma. Biol Pharm Bull. 2006;29:1991–1994. doi: 10.1248/bpb.29.1991. [DOI] [PubMed] [Google Scholar]
- 19.Saunders MM, Seraj MJ, Li Z, Zhou Z, Winter CR, Welch DR, Donahue HJ. Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res. 2001;61:1765–1767. [PubMed] [Google Scholar]
- 20.McLachlan E, Shao Q, Laird DW. Connexins and gap junctions in mammary gland development and breast cancer progression. J Membr Biol. 2007;218:107–121. doi: 10.1007/s00232-007-9052-x. [DOI] [PubMed] [Google Scholar]
- 21.Kalra J, Shao Q, Qin H, Thomas T, Alaoui-Jamali MA, Laird DW. Cx26 inhibits breast MDA-MB-435 cell tumorigenic properties by a gap junctional intercellular communication-independent mechanism. Carcinogenesis. 2006;27:2528–2537. doi: 10.1093/carcin/bgl110. [DOI] [PubMed] [Google Scholar]
- 22.McLachlan E, Shao Q, Wang HL, Langlois S, Laird DW. Connexins act as tumor suppressors in three-dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res. 2006;66:9886–9894. doi: 10.1158/0008-5472.CAN-05-4302. [DOI] [PubMed] [Google Scholar]
- 23.Qin H, Shao Q, Curtis H, Galipeau J, Belliveau DJ, Wang T, Alaoui-Jamali MA, Laird DW. Retroviral delivery of connexin genes to human breast tumor cells inhibits in vivo tumor growth by a mechanism that is independent of significant gap junctional intercellular communication. J Biol Chem. 2002;277:29132–29138. doi: 10.1074/jbc.M200797200. [DOI] [PubMed] [Google Scholar]