

Abstract
Peroxiredoxins (Prxs) make up a ubiquitous class (proposed EC 1.11.1.15) of cysteine-dependent peroxidases with roles in oxidant protection and signal transduction. An intriguing biophysical property of typical 2-Cys Prxs is the redox-dependent modulation of their oligomeric state between decamers and dimers at physiological concentrations. The functional consequences of this linkage are unknown, but on the basis of structural considerations, we hypothesized that decamer-building (dimer–dimer) interactions serve to stabilize a loop that forms the peroxidatic active site. Here, we address this important issue by studying mutations of Thr77 at the decamer-building interface of AhpC from Salmonella typhimurium. Ultracentrifugation studies revealed that two of the substitutions (T77I and T77D) successfully disrupted the decamer, while the third (T77V) actually enhanced decamer stability. Crystal structures of the decameric forms of all three mutant proteins provide a rationale for their properties. A new assay allowed the first ever measurement of the true kcat and Km values of wild-type AhpC with H2O2, placing the catalytic efficiency at 4 × 107 M−1 s−1. T77V had slightly higher activity than wild-type enzyme, and both T77I and T77D exhibited ca. 100-fold lower catalytic efficiency, indicating that the decameric structure is quite important for, but not essential to, activity. The interplay between decamer formation and active site loop dynamics is emphasized by a decreased susceptibility of T77I and T77D to peroxide-mediated inactivation, and by an increase in the crystallographic B-factors in the active site loop, rather than at the site of the mutation, in the T77D variant.
Peroxiredoxins (Prxs) make up a ubiquitous family (proposed EC 1.11.1.15) of cysteine-based peroxidases that not only protect cells against oxidative damage by peroxides but also play an important role in regulating H2O2-mediated cell signaling events in eukaryotes (2, 3). Hydrogen peroxide, alkyl hydroperoxides, and peroxynitrite are all detoxified by Prx action through a catalytic cycle which depends on an essential peroxidatic Cys (CP).1 In the first step of the reaction, CP is oxidized by the hydroperoxide substrate to a Cys sulfenic acid (abbreviated CP-SOH). In the case of 2-Cys Prxs, the CP-SOH condenses with a second redox-active cysteine called the “resolving” cysteine (CR), forming a disulfide bond (4). Typical 2-Cys Prxs are obligate homodimers, with the resolving cysteine located in a different subunit than the peroxidatic cysteine. Recycling of the oxidized enzyme requires a “disulfide reductase” which can consist of a single protein, as with the bacterial flavoprotein AhpF, or can involve two or more flavin and/or CXXC-containing proteins (5). In other classes of Prxs, the CR is located in the same subunit as the CP (the “atypical 2-Cys” Prxs) or missing altogether (the “1-Cys” Prxs), and recycling by reductase systems likely follows mechanisms similar to that for the typical 2-Cys Prxs, although details of this step are not clear for all the family members.
X-ray crystal structures for a number of typical 2-Cys Prxs have shown them to exist either as α2 homodimers or as doughnut-shaped (α2)5 decamers (4). Analyses of AhpC using analytical ultracentrifugation showed that the oligomeric state was linked to the catalytic cycle, with the reduced protein forming a strong decamer and the oxidized protein tending to dissociate into dimers (6). The crystal structures of various 2-Cys Prxs allowed a detailed understanding of the redox-linked conformational changes that lead to changes in oligomerization (Figure 1). Briefly, in its thiol(ate) form, the CP is present in the first turn of an α-helix protruding into a highly conserved active site pocket, interacting with conserved Arg and Thr residues that likely serve to enhance its nucleophilicity. In this conformation, the CR in the C-terminal end of the partner subunit is remote, ~13 Å from CP (3). For disulfide bond formation, the α-helix containing CP and the β-strand containing CR must both undergo a local unfolding. If this unfolding is unfavorable, then the enzyme is highly sensitive to inactivation via further oxidation of CP by a second molecule of peroxide and formation of a sulfinic acid (CP-SO2H) (inactivation shunt in Figure 1); this inactivation pathway appears to be important for peroxide signaling in eukaryotes (3, 7). Disulfide bond formation results in a loss in stability for the decamer, because the active site loop in the fully folded conformation acts to buttress the decamer-building interface (Figure 1) (6). Given that the fully folded active site loop stabilizes the decamer through packing interactions, it must also be true that the decamer contributes to the stability of the folded conformation of the active site. If the folded conformation of the active site in the decamer is significantly more stable than in the dimer, this could create a situation in which the decameric form is effectively required for peroxidatic activity.
Figure 1.

Structurally detailed model of the 2-Cys peroxiredoxin catalytic cycle and redox-sensitive oligomerization. Each panel represents a different structure of the proposed mechanism. Hatched surfaces at the top right represent the interfacial region (residues 73–84) contributed by an adjacent dimer in the decameric enzyme; panels lacking it indicate dissociation of the decamer to dimers. Thr77 in this interfacial region was targeted for mutagenesis in this study to disfavor decamer formation and potentially affect the catalytic properties of the enzyme. Boxed structures represent determined crystal structures identified by name in the top right corner [TryP (1E2Y), HBP-23 (PrxI, 1QQ2), TPx-B (PrxII, 1QMV), and AhpC (1KYG)]. The dashed box represents an alternate conformation present in the TryP structure. Unboxed structures are proposed intermediates. The active site loop (residues 45–49) containing the peroxidatic cysteine thiol(ate) (SPH), cysteine sulfenic acid (SPOH), or cysteine sulfinic acid (SPO2H) is represented in either its fully folded conformation (hashed cylinder) or its locally unfolded conformation (thick line). The dynamic equilibrium between folded and unfolded states is represented by KLU. Loop residues 40–44 leading up to the active site loop are represented as well-ordered (curved line), loosely packed (dotted line), or restructured (distorted curved line with hydrogen bonds represented by small dashed lines, species 5 and 6). The C-terminus containing the resolving cysteine (SRH or SR) is depicted as a thick line. Redox steps are represented by one-way arrows, and equilibrium steps are denoted with two-way arrows, with the length relative to the proposed direction of the reaction. The catalytic cycle of AhpC is identified by a circular arrow. See the original reference for further discussion (6).
To address this important issue, whether the decameric structure of the reduced 2-Cys Prxs contributes significantly to catalysis through stabilization of the fully folded active site, we set out to disrupt the decamer through specific mutations and to determine the effect of these disruptions on catalysis. Focusing on the high-resolution structures, we noted that the AhpC decamer-building interface contains a local 2-fold symmetry axis close to Thr77 of each chain (Figure 2). The fully buried, well-packed nature of Thr77 led us to believe that its mutation would affect the stability of the decamer. We therefore constructed and studied mutants of Thr77 substituted with Val, Ile, and Asp, with the expectation that Val would be a minimally disruptive control, Ile would be more disruptive, and Asp would be the most disruptive due to the introduction of two (one from each chain contributing to the interface) uncompensated, buried negative charges. We report herein hydrodynamic and structural studies on these decamer disruption mutants of AhpC and the effect of the perturbation of the oligomeric state on both catalysis and oxidative inactivation.
Figure 2.

Three views emphasizing the location of proximal Thr77 residues at the AhpC decamer-building interface. The dimer–dimer interface (panels a and b) in decameric AhpC (also known as the “alternate” or “A” interface) includes Thr residues in the opposing subunits (Thr77 and Thr77′) which are near one another, suggesting a potentially useful target for mutagenesis to destabilize decamers. In panel c, the close-up of this interface region shows that Oγ of Thr77 is proximal to Cγ of Thr77′ (3.8 Å) and to Oδ of Asp73′ (3.7 Å), across the dimer–dimer interface. Chain C (gray carbons) and chain B (green carbons) are colored according to atom type (red oxygens and blue nitrogens). Dashed lines represent hydrogen bonds (2.4–3.2 Å), and the striped lines represent the van der Waals interaction between Cγ2 of Thr77 and C∊1 of Phe42 (3.8 Å). An approximate 2-fold axis relates the two molecules at this interface and also relates the preferred hydration sites (water 2131/3131, and water 2097/3097, 2xxx and 3xxx designation for subunits b and c, respectively).
EXPERIMENTAL PROCEDURES
Materials
Sodium dodecyl sulfate (SDS), ultrapure glycine, ethylenediaminetetraacetic acid (EDTA) disodium salt, dithiothreitol (DTT), ammonium sulfate, Tris base, and other buffer reagents were purchased from Research Organics (Cleveland, OH). Bacteriological medium components were from Difco Laboratories (Detroit, MI). Ethanol was obtained from Warner Graham Co. (Cockeysville, MD). Isopropyl β-d-thiogalactopyranoside (IPTG) was from Inalc (Milan, Italy). Restriction enzymes, T4 DNA ligase, and restriction buffers were obtained from Promega (Madison, WI). Agarose Medium EEO (electrophoresis grade), acetic acid, sodium chloride, and H2O2 were from Fisher (Fairlawn, NJ). An acrylamide/bisacrylamide (40%) solution was purchased from Bio-Rad (Hercules, CA). Ampicillin powder, streptomycin sulfate, and dimethyl sulfoxide were from Sigma (St. Louis, MO). NADPH and NADH were from Roche Molecular Biochemicals (Mannheim, Germany). Stratagene (LaJolla, CA) supplied the PfuTurbo polymerase, DpnI, and Escherichia coli XL-1 Blue. All ultrafiltration was carried out using Centricon CM-30 concentrators (Millipore, Bedford, MA).
Methods
Site-Directed Mutagenesis and Protein Preparation
The three mutant AhpC enzymes, T77V, T77I, and T77D, were generated from an overexpression plasmid for Salmonella typhimurium AhpC, pPROK-ahpC. This expression vector was engineered from pAC1 (8) by ligation of the excised 677 bp HpaI–XmaI fragment from pAC1 into the EcoRI (blunted) and XmaI sites of the pPROK-1 expression vector from Clontech (Palo Alto, CA). Mutagenesis using this template employed the protocol outlined in the QuikChange site-directed mutagenesis kit (Stratagene) and the following primers: 5′-CAACCGATACTCACTTCGTGCACAAAG-CATGGCACAG-3′ for T77V, 5′-CAACCGATACTCACT-TCATCCACAAAGCATGGCACAGC-3′ for T77I, and 5′-GTCTCAACCGATACTCACTTCGATCACAAAGCA-TGGCACAGC-3′ for T77D (mismatches introducing the mutations shown in bold). The exact reverse complementary oligonucleotides were also synthesized as reverse primers for the mutagenesis procedures. Reaction mixtures (50 μL) contained 10–50 ng of pPROK-ahpC, 12.5 pmol of each primer, 200 μM dNTPs, and 2.5 units of PfuTurbo polymerase.
To introduce a fluorescent probe for the redox state, Trp was substituted for Ser at position 128 adjacent to the active site CXXC sequence (analogous to the WCXXC redox center of thioredoxins) of the N-terminal domain (NTD) of AhpF, also termed F[1–202]. The previously generated NTD expression construct, pAF[1–202] (1), was used as template for mutagenesis. The mutant was created as described above using the following primers: 5′-CGAAACTTATTAT-TCACTCTGGTGCCATAACTGCCCGGACG-3′ (forward) and 5′-CGTCCGGGCAGTTATGGCACCAGAGTGAA-TAATAAGTTTCG-3′ (reverse, with the mismatches introducing the mutation shown in bold).
Products from mutagenesis were transformed into competent XL1-Blue cells. Single colonies were selected on Luria-Bertani (LB) plates containing ampicillin (100 μg/mL) for AhpC expression constructs or chloramphenicol (30 μg/mL) for the NTD construct; AhpC mutated protein (mutein) expression was evaluated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) after induction with 0.4 mM IPTG. Isolated plasmid DNA for the final constructs was sequenced throughout the coding region by automated DNA sequencing at the Comprehensive Cancer Center of Wake Forest University. The NTD S128W expression construct was retransformed into JM109(DE3) and checked for T7-dependent expression. Bacterial stocks containing each plasmid with the subcloned gene were prepared from a single colony and stored at −80 °C in LB broth containing 15% (v/v) glycerol. Culture procedures were generally the same as those reported previously (25).
Purifications of AhpC mutants and the S128W NTD fragment were carried out as described previously for the wild-type proteins (1, 8). S. typhimurium AhpF (8) and E. coli thioredoxin reductase (TrxR) and thioredoxin (Trx) (1, 9) were also expressed and purified as described previously.
Due to our expectation that the introduction of a third Trp into the NTD would significantly affect its absorbance properties, the extinction coefficient for S128W NTD was determined experimentally by comparisons of 280 nm absorbance and the protein content as measured by the microbiuret assay, using bovine serum albumin as a standard (1). The new value of 21 250 M−1 cm−1 for S128W NTD at 280 nm is indeed higher than that for wild-type NTD, at 15 100 M−1 cm−1 (1).
Enzyme Assays
For peroxide-dependent assays, a new procedure for monitoring the loss of fluorescence from S128W NTD as it is oxidized in the presence of AhpC (wild-type or mutants) and hydrogen peroxide was developed. Briefly, S128W NTD was prereduced with a 100-fold molar excess of DTT for 1 h at room temperature. Excess DTT was removed by passing the mixture through a small Sephadex G-25 column equilibrated in reaction buffer [50 mM potassium phosphate (pH 7.0) containing 0.5 mM EDTA and 100 mM ammonium sulfate]. Prereduced S128W NTD and wild-type or mutant AhpC in one syringe were then mixed with reaction buffer containing H2O2 in the second syringe of an Applied Photophysics SX.18MV stopped-flow spectrophotometer. Fluorescence changes were observed, with excitation at 280 nm and emission at >320 nm (using an emission filter). The temperature was maintained at 25 °C. All the rates reported here are averages of at least six rate measurements. The fluorescence changes were calibrated by measuring the total change in fluorescence upon oxidation of a known concentration of reduced S128W NTD. Initial rate data were plotted and analyzed using the Hanes–Woolf representation of the Michaelis–Menten equation for a two-substrate, substituted reaction once this was determined to be appropriate. True values for the Km for each of the substrates, for kcat, and for kcat/Km for hydrogen peroxide were obtained, along with the standard errors for the fit, directly from global fits of all the data for a single protein using the multiple-function nonlinear regression capability of Sigma-Plot (Jandel Scientific, San Rafael, CA).
To assess peroxide-dependent inactivation rates of the various AhpC mutants, assays were conducted in the presence of TrxR and Trx and millimolar concentrations of hydrogen peroxide as described previously (3).
Analytical Ultracentrifugation
To determine the oligomeric state of the mutant AhpC proteins, samples at different concentrations were analyzed by sedimentation velocity at various speeds on an Optima XL-A analytical ultracentrifuge (Beckman Instruments, Palo Alto, CA) outfitted with absorbance optics. AhpC, with or without a 1 h pretreatment with 20[notdef] DTT and subsequent removal of the reductant by gel filtration on a Sephadex G-25 column, was concentrated and exchanged into 25 mM potassium phosphate buffer with 1 mM EDTA and 0.15 M NaCl at pH 7 using CM-30 Centricon ultrafiltration units (Millipore). Samples (at 2.5, 5, 10, and 100 μM) of oxidized and reduced AhpC mutants were loaded into double-sectored cells and equilibrated to 20 °C. Sedimentation data at 230 or 280 nm were collected every 3 or 4 min at rotor speeds of 35 000 or 42 000 rpm and a radial step size of 0.003 cm. From these data, sedimentation and diffusion coefficient values at each concentration of reduced AhpC were calculated using the Fujita–MacCosham function based on the Lamm equation and the SVEDBERG software (version 6.39) from J. S. Philo (Thousand Oaks, CA). DCDTplus (version 1.16, also from J. S. Philo) was also used to prepare the plots of the data. These parameters were standardized to pure water (correcting for density and viscosity) and extrapolated to zero concentration (10). The molecular weight was then calculated from the Svedberg equation (11) using this s20,w0 value and the corrected translational diffusion coefficient (D20,w0). Partial specific volumes of 0.7370, 0.7378, 0.7381, and 0.7364 cm3/g were calculated on the basis of the amino acid compositions of wild-type, T77V, T77I, and T77D proteins, respectively (10). The buffer density of 1.00773 g/cm3 was determined using a DA-310M precision density meter (Mettler Toledo, Hightstown, NJ) at 20 °C.
X-ray Crystallography
All crystals were grown at 4 °C in hanging drops using a 0.4 mL reservoir solution, and a drop containing a 1:1 ratio of protein [at 10 mg/mL in 25 mM potassium phosphate (pH 7) and 1 mM EDTA] to reservoir solution. Conditions were optimized on the basis of leads generated from commercial screening kits (Hampton Research). For T77V, a 20% PEG 3350, 0.1 M citrate, pH 5.5 reservoir resulted in 0.4 mm × 0.4 mm × 0.4 mm parallelopipeds. For T77I, a 1.6 M (NH4)2SO4, 0.1 M MES, pH 6.5, 10% (v/v) dioxane reservoir (authentic Hampton Screen II #23 gave the best results) yielded 0.4 mm × 0.4 mm [notdef] 0.4 mm triangular prism-shaped (Toblerone) crystals. For T77D, a 1.6 M (NH4)2SO4, 0.1 M MES, pH 6.0 reservoir resulted in 0.3 mm × 0.4 mm × 0.2 mm crystals which were also triangular prism-shaped.
For data collection, crystals were mounted in loops and flash-frozen in liquid nitrogen. T77V crystals were harvested in 30% PEG 3350 and 0.1 M citrate (pH 5.5) and then placed in the same buffer with successive 5, 15, and 17.5% concentrations of glycerol, each for 1–2 min, before being frozen. T77I and T77D crystals were frozen after 25 μL of Hampton Screen II #23 with 17.5% glycerol was placed directly on the drop for 3–5 min.
Data were collected at the Advanced Light Source (Lawrence Berkeley National Laboratory, Berkeley, CA). T77V data were collected at beam line 5.0.2 (λ = 1.0 Å, Δϕ = 1.0°, 162 15 s images). The crystals belong to orthorhombic space group P21212 with two half-decamers in the asymmetric unit and the following unit cell dimensions: a = 136.35 Å, b = 169.96 Å, and c = 117.65 Å. T77I and T77D data were collected at beam line 8.2.1. Both crystals belonged to space group C2221 with the following unit cell dimensions: a = 127.45 Å, b = 170.87 Å, and c = 135.90 Å for T77I and a = 127.47 Å, b = 171.61 Å, and c = 135.81 Å for T77D. These crystals are isomorphous with wild-type AhpC (6) and contain one half-decamer in the asymmetric unit. T77I data were merged from a low-resolution (λ = 1.0 Å, Δϕ = 1.0°, 100 3 s images) and a high-resolution (λ = 1.0 Å, Δϕ = 0.8°, 100 10 s images) pass. T77D data were similarly collected (low-resolution pass, λ = 1.0 Å, Δϕ = 1.0°, 90 3 s images; high-resolution pass, λ = 1.0 Å, Δϕ = 0.7°, 120 10 s images). The T77V, T77I, and T77D data extended to 2.6, 2.5, and 2.3 Å resolution, respectively (Table 1).
Table 1.
Data Collection and Refinement Statistics for Each Chain in the Asymmetric Unita
| wild-type | T77V | T77I | T77D | |
|---|---|---|---|---|
| data collection | ||||
| resolution (Å) | 50–2.5 (2.6–2.5) | 100–2.6 (2.7–2.6) | 100–2.5 (2.6–2.5) | 100–2.3 (2.4–2.3) |
| no. of unique observations | 51663 | 83807 | 50126 | 129628 |
| multiplicity | 7.2 (6.7) | 5.3 (4.7) | 6.2 (3.3) | 5.4 (3.1) |
| completeness (%) | 97.4 (92.2) | 99.2 (97.8) | 99.4 (100.0) | 99.3 (99.0) |
| 〈I/σ〉 | 16.0 (4.5) | 16.8 (1.9) | 16.5 (2.6) | 9.3 (1.9) |
| Rmeasb (%) | 8.0 (52) | 6.3 (51) | 6.3 (30) | 8.5 (40) |
| Rmrgd-Fb (%) | 7.1 (36) | 8.7 (47) | 6.4 (33) | 11.0 (49) |
| refinement | ||||
| no. of residues | 827 | 1650 | 826 | 826 |
| no. of waters | 311 | 826 | 431 | 425 |
| no. of SO42− ions | 9 | 0 | 10 | 8 |
| total no. of atoms | 6871 | 13829 | 6992 | 6976 |
| 〈B〉 for protein (Å2) | 54 | 42 | 52 | 53 |
| 〈B〉 for waters (Å2) | 63 | 44 | 59 | 60 |
| Rcryst (%) | 17.8 | 18.1 | 17.4 | 18.9 |
| Rfree (%) | 22.7 | 23.7 | 22.8 | 23.3 |
| rmsd for bond lengths (Å) | 0.008 | 0.010 | 0.007 | 0.010 |
| rmsd for bond angles (deg) | 1.4 | 1.5 | 1.3 | 1.4 |
Numbers in parentheses correspond to values in the highest-resolution bin.
Rmeas is the multiplicity-weighted merging R-factor, and Rmrgd-F is an indicator of the quality of reduced data as defined by Diederichs and Karplus (26).
The structure of T77V was determined by molecular replacement using CNS version 1.1 (12). To produce the search model for T77V from wild-type AhpC (PDB entry 1KYG), Thr77 was changed to a Gly, and the waters and ions were removed. Ten percent of the data were randomly picked for cross validation. The T77I and T77D refinement also began with the wild-type AhpC structure after Thr77 was changed to a Gly and the ions (but not waters) were removed. The cross validation set to 2.5 Å resolution was taken from the wild-type data (so that it would be unbiased), and a random 10% of the reflections were selected for T77D from 2.5 to 2.3 Å to extend the test set for both T77I and T77D. All three structures underwent rounds of automated positional and B-factor refinement followed by manual rebuilding in O (13). A bulk solvent correction was applied so all low-resolution data could be used. Waters were initially added to T77V using the water pick routine in CNS. During the final stages of all refinements, waters were placed manually into Fo − Fc map peaks of ≥3ρrms, if the density in the 2Fo − Fc map was ≥1ρrms and if a reasonable hydrogen bonding environment was present. The Fo − Fc maps were calculated using a low-resolution cutoff of 6 Å. Refinements were stopped when the largest difference density peaks were not clearly interpretable and refinement statistics (Table 1) had converged. For consistency, the wild-type AhpC structure was retrieved from the Protein Data Bank (PDB) and subjected to further refinement using the protocols described above. Changes made include a few different side chain conformations and placement of 16 additional waters, including water 131 near Thr77. Using structural overlays, we determined which waters were in equivalent positions (within 1 Å) in the different protein chains. Waters were then numbered according to their peak electron density (averaged over noncrystallographically equivalent occurrences and with lower numbers having stronger density). Common waters between the various structures were given the same names. Unique water sites are numbered 1–334, and the digit in the thousands position indicates the chain with which it is associated (1000–5000 for chains A–E, respectively; for chains F–J in the T77V structure, water numbers are incremented by 500, i.e., Wat1001 of chain A is equivalent to Wat1501 in chain F). All chains have some residues missing at their C-termini because of a lack of density. Residues not modeled in the wild-type, T77V, T77I, and T77D are A168–188 (also 167 for the wild-type and T77V), B166–188, C164–188, D167–188, and E166–188. The second half-decamer of T77V has the same residues missing (i.e., F167–188, G166–188, H164–188, I167–188, and J166–188). The geometry of the final model was analyzed with PROCHECK (14). The refined coordinates have been deposited in the PDB as entries 1YF0 for T77I, 1YF1 for T77V, 1YEX for T77D, and 1YEP for the further refined wild-type coordinates (replacing entry 1KYG).
RESULTS AND DISCUSSION
Oligomeric States of AhpC Mutants in Solution
Sedimentation velocity studies with reduced and disulfide forms of the AhpC mutants reveal that our strategy to influence oligomerization state through the introduction of mutations at Thr77 was successful (Figure 3). As reported by Wood et al. (6), the reduced (CP-SH) form of wild-type AhpC was decameric at all concentrations (≥2.5 μM), whereas the disulfide-containing form was dimeric at 2.5 μM, a mixture of oligomers at higher concentrations, and decameric at the highest concentration that was analyzed (485 μM). Thus, for the Thr77 muteins, stabilization of the decamer would be indicated by increased amounts of higher-order oligomeric species for the disulfide form at 2.5 μM, and destabilization of the decamer would be indicated by the presence of species smaller than a decamer for the reduced form of the enzyme at 2.5 μM. For T77V, sedimentation velocity analyses showed that the mutation is stabilizing; the disulfide form of the protein is primarily decameric at 2.5 μM, and fully decameric at 10 μM (Figure 3). Reduced T77V sediments as a decamer at all concentrations that were studied. In contrast, the disulfide forms of both the T77I and T77D muteins remain dimeric even at concentrations as high as 485 μM, indicating that these mutations are very destabilizing to the decamer building interface. In a similar pattern of destabilization, the reduced forms of these muteins are also fully dimeric at 2.5 μM (not shown).
Figure 3.
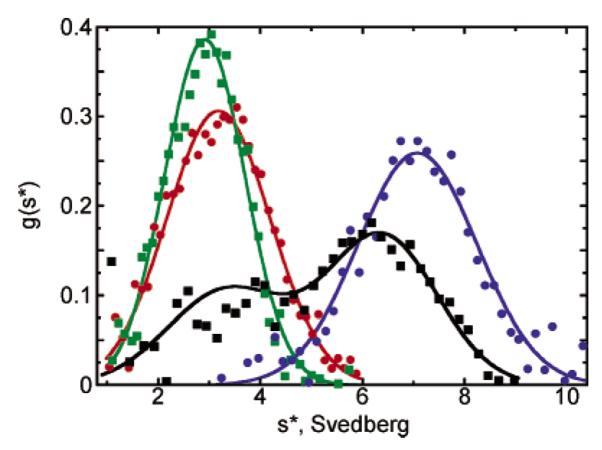
Sedimentation velocity studies of oxidized wild-type and Thr77 mutant AhpC proteins. Analytical ultracentrifugation studies of AhpC proteins were carried out with centrifugation at 42 000 rpm, 20 °C, and neutral pH. Eight to 16 consecutive 280 nm data sets for each run, where the boundaries had moved to approximately the middle of the cells, were included in the analyses by DCDT+ software to give the g*(s) distributions that are shown. Only data for the oxidized proteins (40 of at least 240 data points used in the fit) are shown. Curves fit to a single-species model (except for wild-type AhpC, which was fit to a two-species model) represent data for wild-type AhpC at 10 μM (black), T77V at 10 μM (blue), T77D at 100 μM (red), and T77I at 100 μM (green). All plots were normalized by area to ease comparisons. Results show that the T77V mutation actually promotes decamerization of the oxidized enzyme, whereas the T77I and T77D mutations destabilize the decameric forms of both the oxidized and reduced proteins (latter not shown).
Crystal Structures of AhpC Mutants
To elucidate the details of the structural perturbations imparted by the Thr77 mutations, the disulfide forms of the mutants were crystallized, their structures were determined, and the final models were compared with that of wild-type AhpC. All three muteins crystallized as (α2)5 decamers, presumably due to the high protein and precipitant concentration during crystallization. In each case, residue 77 had clear density and its conformation could be unambiguously modeled (Figure 4). The next three paragraphs describe the mutant structures, using chain C as the representative molecule with chain B being the molecule with which it interacts across the local 2-fold symmetry axis at the decamer-building interface (Figure 2). All changes that are presented are representative of the entire decamer.
Figure 4.

Structures at the mutation site are well-defined. Each mutant model (chain C) is displayed along with its 2Fo – Fc electron density. Atom coloring is as in Figure 2c. As a reference, the wild-type structure is shown as a semitransparent model. Because of their relevance, AspB73 and water 131 are also shown. Hydrogen bonds are represented as dashed lines: (a) ValC77 contoured at 1.2ρrms, (b) IleC77 contoured at 2.5ρrms, and (c) AspC77 contoured at 2.1ρrms.
In the T77V model, Val adopts the same conformation as Thr in wild-type AhpC. The only significant difference between these two structures is a shift in water 131 away from Cγ1 of Val (Figure 4). In the wild-type, this water is hydrogen bonded to Oγ1 of Thr77 (Figure 2), and its movement places it a reasonable 3.5 Å from the Val methyl. AspB73 shifts slightly to maintain its hydrogen bonding with the moved water. As a consequence of the mutation, very small shifts in packing (≤0.2 Å) are seen in the decamer-building interface, consistent with the need to accommodate the slightly larger radius of Cγ1 of Val. The increased stability of the T77V decamer implies that the energy associated with the burial of an extra methyl group more than makes up for the slight changes in packing.
The Ile side chain in the T77I model also adopts the same χ1 rotamer as the wild-type, with Cδ angled toward PheB42 (χ2 = −47°). This Phe rotates slightly to provide room for the new Cδ atom. As for the Val mutant, small shifts in water 131 and AspB73 at this interface are also observed. Compared to the T77V mutant, it must be the collisions of Cδ of Ile77 with Phe72 (twice due to the pseudo-2-fold) that induce the overall instability in the decamer that we observe in solution.
The T77D model contains the largest differences from the wild-type structure (Figures 4 and 5). The introduced Asp adopts a χ1 of −61°, placing Cγ into the same space as Oγ1 of Thr77 in wild-type AhpC (Figure 2). To fill the void created by the absence of Cγ2 of Thr77, the entire helix (residues 75–82) shifts (ca. 0.3 Å) toward PheC42, and the Phe side chain also swivels (Δχ1 = 7°) closer to Cβ of Asp. Despite the rearrangements, a void is formed between Cβ of Asp and C∊1 of Phe (atom–atom separation of 4.7 Å), contributing to the destabilization of the interface. With regard to the Asp77 carboxylate, one of the oxygens (Oδ2) is within 1.3 Å of the wild-type position of water 131, replacing it, and making hydrogen bonds with both N of ThrC74 (3.2 Å) and Oδ2 of AspC73 (2.6 Å). The latter is a surprising interaction because the crystals were grown at pH 6.0, but the geometry strongly suggests that Asp77 is protonated. At neutral pH, the pKa shift required to support this interaction would be expected to further destabilize the interface. One other interesting observation provides indirect structural evidence that oligomerization influences the mobility of the active site loop. Even though the locally unfolded conformation of the active site loop is unchanged, a comparison of the B-factors reveals that among the mutants, T77D has a unique increase in the mobility of residues 43–55, whereas B-factors in the region surrounding the mutation at Thr77 do not change significantly (Figure 6). Thus, while the interface itself is fully intact, something related to the mutation (it cannot be a crystal packing interaction since the crystals are isomorphous) has allowed the active site loop to have more freedom. We propose that the previously mentioned void near Phe42 has resulted in a looser packing of this residue, which served as an anchor for the loop. The weaker packing of the anchor allows a wider range of movement for each successive residue in the loop until the point halfway to where it is anchored at the other end. This behavior is not seen in the T77I structure, because it does not have a packing void near Phe42. That the small increase in the degree of freedom of the Phe42 side chain in T77D leads to such an increase in the mobility of the active site loop implies that the large increase in the degree of freedom of Phe42 upon dissociation of the decamer (wild-type or mutant) will have an even greater effect on active site loop mobility.
Figure 5.
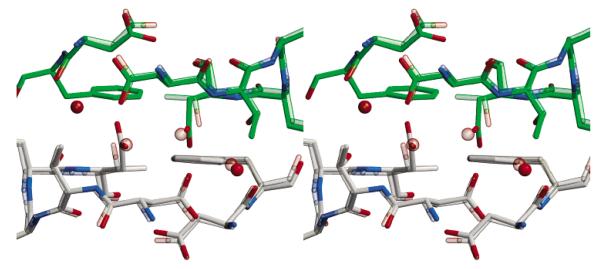
Structural differences between T77D and wild-type AhpC. T77D and wild-type AhpC are superimposed in this stereoview. All colors are as in Figure 2c with the wild-type structure shown as a semitransparent image. The view is as in Figure 2c.
Figure 6.
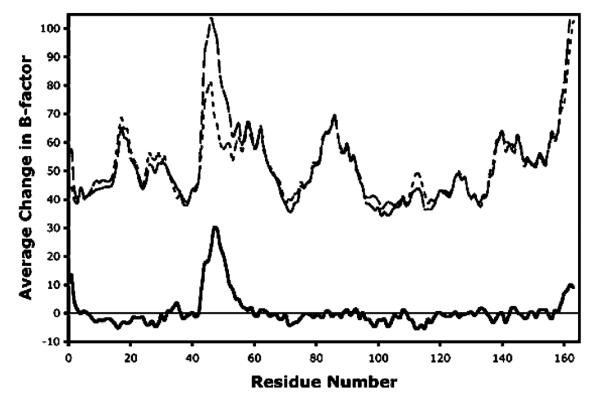
Active site loop becomes much less ordered in T77D. For each residue, the average T77D main chain B-factor values are averaged over all five chains (dashed line). The wild-type main chain B-factor values are also averaged over all five chains (dotted line). The difference between these two (solid line) displays a sharp peak from residue 43 to 55. Values greater than zero are for residues for which T77D is on average more mobile than the wild-type. T77I shows no such difference in B-factors.
A New Fluorescence-Based AhpC Assay
Previous measurements of S. typhimurium AhpC peroxidase activity, even in the presence of limiting amounts of AhpC, showed little or no dependence of the rate on H2O2 concentration (8, 15). These results and others suggested that the rates were not limited by AhpC peroxidase activity, but by the slower reduction of AhpC by its native disulfide reductase, AhpF, or by Trx (15). We have since confirmed that this was a limitation of those assays (data not shown). To reliably assess the effects of the Thr77 mutations on catalysis, it was imperative that we develop an assay that could be used to measure accurate kcat and Km(H2O2) values for AhpC. We therefore designed a new fluorescence-based assay that uses as the reducing substrate a truncated form of AhpF consisting of only the NTD, the direct electron donor to AhpC (1, 9, 16, 17). Whereas the reoxidation of Trx can be monitored by following the change in the fluorescence of a Trp residue in the proximity of the active site disulfide/dithiol (18), there are no equivalent fluorescence changes for the AhpF NTD. On the basis of the Trx assay, we mutated the NTD (1) to introduce a Trp residue adjacent to the redox-active disulfide, resembling the active site of Trx (S128W; Chart 1). Initial experiments indicated that oxidation of the reduced S128W NTD led to an 11% decrease in fluorescence (excitation at 280 nm and emission peak at 343 nm). To assess if the S128W mutation impaired the ability of the NTD to reduce AhpC, it was assayed with AhpC-FAM (carboxyfluorescein attached via a linker and disulfide bond to Cys165 of the C46S mutant of AhpC) (1). The mixed disulfide of AhpC-FAM mimics the intermolecular disulfide bond of wild-type AhpC, and the large and rapid increase in fluorescence upon reaction to release the carboxyfluorescein moiety allows direct measurement of the reaction with reduced NTD. Using this benchmark, the S128W mutant was found to be as reactive toward AhpC-FAM as wild-type NTD.
Chart 1.

Catalytic Parameters of Wild-Type and Mutant AhpCs
Using our new assay, low levels (40–100 nM) of wild-type or mutant AhpC proteins were mixed with the two substrates, reduced S128W NTD and hydrogen peroxide, and the loss of NTD fluorescence was monitored using a stopped-flow spectrofluorometer. Wild-type AhpC and the three Thr77 muteins display intercepts on the y-axis in primary Hanes plots ([S]/v vs [S]), indicating that they follow a substituted (ping-pong) enzyme mechanism (not shown). The kinetic parameters (Table 2) show that the kcat values for all four proteins are within an ~3-fold range, but the Km values for peroxide for T77D and T77I are roughly 50-fold higher than those for the wild-type and T77V. The catalytic efficiency [kcat/Km(H2O2)] of these mutants is therefore decreased by ~2 orders of magnitude with respect to that of the wild-type enzyme in the peroxide reaction. On the other hand, all mutants exhibit little change in Km for S128W NTD.
Table 2.
Kinetic Constants for Wild-Type and Thr77 Mutant AhpC Proteins
| enzyme | kcat(s−1) | Km(S128W) (μM) | Km(H2O2) (μM) | kcat/Km(H2O2)(M−1 s−1) |
|---|---|---|---|---|
| wild-type | 55.1 ± 0.8 | 5.4 ± 0.2 | 1.4 ± 0.2 | (3.9 ± 0.5) × 107 |
| T77V | 75.8 ± 1.6 | 3.6 ± 0.4 | 1.6 ± 0.2 | (4.7 ± 0.4) × 107 |
| T77I | 25.0 ± 1.0 | 12.0 ± 1.0 | 93.4 ± 8.8 | (2.7 ± 0.2) × 105 |
| T77D | 31.5 ± 1.9 | 9.5 ± 0.9 | 62.0 ± 9.5 | (5.1 ± 0.6) × 105 |
The first important point about these results from our new assay and previous results from another bacterial Prx, E. coli thiol peroxidase (Tpx) (19), as well as a recent study of a 2-Cys Prx from Toxoplasma gondii (20), is that they challenge the popular belief that Prxs are “comparatively poor” enzymes when compared with heme peroxidases and selenocysteine-containing peroxidases (2). With catalytic efficiencies with peroxides of 0.8–5 × 107 M−1 s−1, at least some Prxs are very efficient peroxidases when compared to the heme peroxidases/catalases (kcat/Km ~ 107 M−1 s−1 and the selenium-containing glutathione peroxidases (kcat/Km ~ 108 M−1 s−1) (2). With their naturally high abundance in many cell types (0.1–1% of total soluble protein) (4), the antioxidant role of Prxs cannot be assumed to be inferior relative to these other, better known cellular peroxidases.
With regard to the role of the decamer in catalysis, the above peroxidase assays are highly informative. Given that AhpC concentrations in the assays were more than 20-fold lower than the lowest concentrations used in the analytical ultracentrifugation experiments, it is very likely that both T77D and T77I are exclusively in the dimeric form throughout the catalytic cycle. On the other hand, the reduced forms of wild-type AhpC and the T77V mutant are expected to be decameric. The high activity of T77V allows us to conclude that the loss in activity of T77I and T77D is correlated with the loss of the decamer rather than the loss of the Thr77 side chain itself, and that the decamer is very important for the integrity of the fully folded peroxidatic active site. As mentioned above, we attribute this difference in reactivity to the added stabilization of the loop leading up to the active site Cys imparted by the buttressing effect of the adjacent dimer in the decameric protein, which would be missing in the dimeric T77I and T77D mutants. That the loss in activity is more associated with Km than kcat is consistent with the expectation that the binding pocket for peroxide is less well formed in the dimeric enzymes, but that in the presence of high substrate concentrations it can be driven to adopt the normal fully folded active site configuration associated with catalysis by wild-type AhpC. Val in place of Thr77 does not interfere with, and may actually enhance, the activity of the enzyme; this result is consistent with it having a more stable decamer (Table 2). Interestingly, Val is naturally found at this position in Helicobacter pylori AhpC (21).
Decreased Sensitivity of Thr77 Mutants of AhpC toward Turnover-Induced Inactivation by Peroxide
If the loss in activity of T77I and T77D muteins is due to destabilization of the fully folded form of the active site, then these muteins should have more facile disulfide bond formation and thus a decreased tendency to succumb to oxidative inactivation by peroxide (Figure 1). To test this hypothesis, we examined the rates of inactivation of wild-type AhpC and the Thr77 muteins as a function of hydrogen peroxide concentration. The activity with high H2O2 concentrations can be compared with the rate observed with the low 0.1 mM H2O2 concentration (not shown), which is fully consumed before any inactivation of the enzyme is seen. The wild-type and the stabilized T77V enzyme both displayed a time-dependent decrease in activity when turning over in the presence of relatively high peroxide concentrations (Figure 7a for the wild-type and similar, but not shown, for T77V), as reported earlier for the peroxide-dependent inactivation of the wild-type enzyme (3). In contrast, the destabilized T77D and T77I mutants show only the expected decrease in rate when the peroxide concentration approaches the Km value (Figure 7b for T77D and not shown for T77I; Table 2). With the limited sensitivity of these bacterial enzymes toward peroxide-mediated inactivation (3), it is difficult to obtain a precise quantitative evaluation of the further decreased sensitivity of the T77D and T77I mutants relative to the wild-type and T77V AhpC, but the data suggest an ~4–5-fold decrease in the fraction of inactivation per turnover for the more robust T77D and T77I mutants. The reduced sensitivity of the T77I and T77D, but not the T77V, AhpC muteins to inactivation provides another line of evidence that the mutations increase the propensity of the active site to unfold, facilitating disulfide bond formation.
Figure 7.
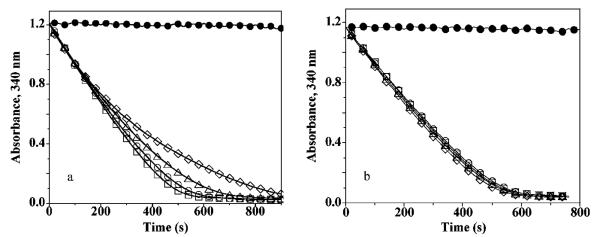
Wild-type and T77V AhpC show a susceptibility to peroxide inactivation that T77I and T77D mutants do not. NADPH oxidation was assessed at 25 °C in the presence of 50 mM HEPES-NaOH (pH 7.0) with 1 mM EDTA and 0.1 M ammonium sulfate, and with 80 nM thioredoxin reductase, 2.5 μM thioredoxin, 6 μM AhpC [wild-type (a) or T77D mutant (b)] and H2O2 at 0 (●), 1 (◻), 5 (◯), 10 (▵), and 30 mM (◇). T77V results were very similar to those from wild-type AhpC (a), while T77I results were essentially identical to those with T77D (b).
Implications of the Linkage between Oligomeric State and Activity
These studies have clarified the connection between decamer formation and active site loop dynamics and the impact of their disruption on both the catalytic activity and the susceptibility of Prxs to inactivation during turnover. We would predict, then, that any modification or other condition that would promote decamer dissociation would also result in decreased catalytic activity but more resistance to inactivation by peroxide. In this regard, the speculation by Wood et al. (4) that phosphorylation of Prxs decreases catalytic activity via decamer dissociation is a viable theory. The site of this cell cycle-dependent phosphorylation in mammalian Prxs (Thr90 in PrxI and Thr89 in PrxII) (22) is quite close to the site of these Thr77 mutations in AhpC, and phosphorylation of adjacent dimers would likely have effects similar to those of the T77D mutation in AhpC. This concept of oligomeric state-linked activity control may also apply more generally to enzymes capable of changing oligomeric state in a way that impinges on their biological function (23).
Finally, we consider the extent to which the activity–oligomerization linkage observed here for AhpC applies to other Prxs. The high level of sequence and structural similarity of the typical 2-Cys Prxs (2, 3, 24) leads us to confidently predict that they will all have a similar linkage. Beyond this, we note that available evidence indicates that all Prxs (both 2-Cys and 1-Cys) require a local unfolding of the peroxidatic active site for resolution of the sulfenic acid form, and this implies that for all Prxs the relative stabilities of the fully folded and locally unfolded conformations of the Cp loop will be delicately balanced. Given such a balance, and the nearly universal presence among Prxs of the decamer-building interface [described as the A-type interface by Sarma et al. (25)], we hypothesize that some level of linkage between activity and quaternary structure is common to all Prxs with the A-type interface. If true, this would support the suggestion by Sarma et al. that the A-type interface preceded the B-type interface in evolution, and would provide a rationale for its strong conservation in the family.
Footnotes
Abbreviations: Trx, thioredoxin; TrxR, thioredoxin reductase; AhpC, alkyl hydroperoxide reductase C component (peroxidase); AhpF, alkyl hydroperoxide reductase F component (flavoprotein reductase); NTD, N-terminal domain of AhpF (including residues 1–202); DTT, 1,4-dithiothreitol; EDTA, ethylenediaminetetraacetic acid; IPTG, isopropyl α-d-thiogalactopyranoside; HEPES, N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid; PDB, Protein Data Bank; CP, peroxidatic cysteine (Cys46 in AhpC); CR, resolving cysteine (Cys165 in AhpC); AhpC-FAM, carboxyfluorescein attached through a linker and a disulfide bond to Cys165 of the C46S mutant of AhpC, as previously described (1); ρrms, root-mean-squared density of map.
This study was supported by a grant from the National Institutes of Health (NIH) to L.B.P. with a subcontract to P.A.K. (RO1 GM50389) and an Established Investigatorship from the American Heart Association to L.B.P. (0140175N). D.S.Y. was partially supported by an undergraduate research fellowship funded by Howard Hughes Medical Institute Grant 57003741 awarded to Oregon State University (OSU) and an OSU Undergraduate Research, Innovation, Scholarship, Creativity award. This work was also made possible in part by Grant P30 ES00210 from the National Institute of Environmental Health Sciences, NIH.
The coordinates and structure factors have been deposited in the Protein Data Bank as entries 1YF0, 1YF1, and 1YEX for T77I, T77V, and T77D, respectively, and 1YEP for the further refined wild-type AhpC coordinates (replacing entry 1KYG).
REFERENCES
- 1.Poole LB, Godzik A, Nayeem A, Schmitt JD. AhpF can be dissected into two functional units: Tandem repeats of two thioredoxin-like folds in the N-terminus mediate electron transfer from the thioredoxin reductase-like C-terminus to AhpC. Biochemistry. 2000;39:6602–6615. doi: 10.1021/bi000405w. [DOI] [PubMed] [Google Scholar]
- 2.Hofmann B, Hecht H-J, Flohé L. Peroxiredoxins. Biol. Chem. 2002;383:347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 3.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 4.Wood ZA, Schroder E, Harris JR, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 5.Poole LB, Reynolds CM, Wood ZA, Karplus PA, Ellis HR, Li Calzi M. AhpF and other NADH: peroxiredoxin oxidoreductases, homologues of low Mr thioredoxin reductase. Eur. J. Biochem. 2000;267:6126–6133. doi: 10.1046/j.1432-1327.2000.01704.x. [DOI] [PubMed] [Google Scholar]
- 6.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: Redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 7.Georgiou G, Masip L. An overoxidation journey with a return ticket. Science. 2003;300:592–594. doi: 10.1126/science.1084976. [DOI] [PubMed] [Google Scholar]
- 8.Poole LB, Ellis HR. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 1. Purification and enzymatic activities of overexpressed AhpF and AhpC proteins. Biochemistry. 1996;35:56–64. doi: 10.1021/bi951887s. [DOI] [PubMed] [Google Scholar]
- 9.Reynolds CM, Poole LB. Attachment of the N-terminal domain of Salmonella typhimurium AhpF to Escherichia coli thioredoxin reductase confers AhpC reductase activity but does not affect thioredoxin reductase activity. Biochemistry. 2000;39:8859–8869. doi: 10.1021/bi000826d. [DOI] [PubMed] [Google Scholar]
- 10.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. In: Computer-aided interpretation of analytical sedimentation data for proteins, in Analytical ultracentrifugation in biochemistry and polymer science. Harding SE, Rowe AJ, Horton JC, editors. The Royal Society of Chemistry; Cambridge, U.K.: 1992. pp. 90–125. [Google Scholar]
- 11.Van Holde KE. Physical Biochemistry. Prentice-Hall, Inc.; Englewood Cliffs, NJ: 1971. pp. 110–136. [Google Scholar]
- 12.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54(Part 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 13.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47(Part 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 14.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 15.Poole LB, Higuchi M, Shimada M, Calzi ML, Kamio Y. Streptococcus mutans H2O2-forming NADH oxidase is an alkyl hydroperoxide reductase protein. Free Radical Biol. Med. 2000;28:108–120. doi: 10.1016/s0891-5849(99)00218-x. [DOI] [PubMed] [Google Scholar]
- 16.Li Calzi M, Poole LB. Requirement for the two AhpF cystine disulfide centers in catalysis of peroxide reduction by alkyl hydroperoxide reductase. Biochemistry. 1997;36:13357–13364. doi: 10.1021/bi9713660. [DOI] [PubMed] [Google Scholar]
- 17.Poole LB, Ghisla S, Kroneck P, Mach-eroux P, Sund H. Flavin-linked redox components required for AhpC reduction in alkyl hydroperoxide reductase systems, in Flavins and Flavoproteins 1999. Agency for Scientific Publications; Berlin: 1999. pp. 691–694. [Google Scholar]
- 18.Holmgren A. Tryptophan fluorescence study of conformational transitions of the oxidized and reduced form of thioredoxin. J. Biol. Chem. 1972;247:1992–1998. [PubMed] [Google Scholar]
- 19.Baker LM, Poole LB. Catalytic mechanism of thiol peroxidase from Escherichia coli. Sulfenic acid formation and overoxidation of essential CYS61. J. Biol. Chem. 2003;278:9203–9211. doi: 10.1074/jbc.M209888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akerman SE, Müller S. Peroxiredoxin-linked detoxification of hydroperoxides in Toxoplasma gondii. J. Biol. Chem. 2005;280:564–570. doi: 10.1074/jbc.M406367200. [DOI] [PubMed] [Google Scholar]
- 21.Baker LM, Raudonikiene A, Hoffman PS, Poole LB. Essential thioredoxin-dependent peroxiredoxin system from Helicobacter pylori: Genetic and kinetic characterization. J. Bacteriol. 2001;183:1961–1973. doi: 10.1128/JB.183.6.1961-1973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang TS, Jeong W, Choi SY, Yu S, Kang SW, Rhee SG. Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J. Biol. Chem. 2002;277:25370–25376. doi: 10.1074/jbc.M110432200. [DOI] [PubMed] [Google Scholar]
- 23.Breinig S, Kervinen J, Stith L, Wasson AS, Fairman R, Wlodawer A, Zdanov A, Jaffe EK. Control of tetrapyrrole biosynthesis by alternate quaternary forms of porphobilinogen synthase. Nat. Struct. Biol. 2003;10:757–763. doi: 10.1038/nsb963. [DOI] [PubMed] [Google Scholar]
- 24.Copley SD, Novak WR, Babbitt PC. Divergence of function in the thioredoxin fold suprafamily: Evidence for evolution of peroxiredoxins from a thioredoxin-like ancestor. Biochemistry. 2004;43:13981–13995. doi: 10.1021/bi048947r. [DOI] [PubMed] [Google Scholar]
- 25.Sarma GN, Nickel C, Rahlfs S, Fischer M, Becker K, Karplus PA. Crystal structure of a novel Plasmodium falciparum 1-Cys peroxiredoxin. J. Mol. Biol. 2005;346:1021–1034. doi: 10.1016/j.jmb.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 26.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]