

Abstract
Streptococcus parauberis, which is the main causative agent of streptococcosis among olive flounder (Paralichthys olivaceus) in northeast Asia, can be distinctly divided into two groups (type I and type II) by an agglutination test. Here, the whole genome sequences of two Japanese strains (KRS-02083 and KRS-02109) were determined and compared with the previously determined genome of a Korean strain (KCTC 11537). The genomes of S. parauberis are intermediate in size and have lower GC contents than those of other streptococci. We annotated 2,236 and 2,048 genes in KRS-02083 and KRS-02109, respectively. Our results revealed that the three S. parauberis strains contain different genomic insertions and deletions. In particular, the genomes of Korean and Japanese strains encode different factors for sugar utilization; the former encodes the phosphotransferase system (PTS) for sorbose, whereas the latter encodes proteins for lactose hydrolysis, respectively. And the KRS-02109 strain, specifically, was the type II strain found to be able to resist phage infection through the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system and which might contribute valuably to serologically distribution. Thus, our genome-wide association study shows that polymorphisms can affect pathogen responses, providing insight into biological/biochemical pathways and phylogenetic diversity.
Introduction
Streptococcosis of cultured fish contributes to major economic losses in the aquaculture industries of Israel [1], Italy [2], Korea [3], Japan [4], and the United States [5]. S. parauberis was first reported from turbot (Scophthalmus maximus) cultured in Spain [6]; since the turn of the present century, it has become important disease in the aquaculture industries of Northeast Asia (Korea, Japan and China), especially among olive flounder aquaculture farms. Recently, Nho et al. [7] reported that S. parauberis is the dominant etiological agent of streptococcosis characterized by clinical symptoms such as chronic wasting syndrome, hemorrhagic septicemia, exophthalmia and meningitis with abnormal swimming.
S. parauberis exhibits serological, genetic and biochemical variations within the species. Nho et al [7] classified S. parauberis into three serotypes based on specific antigenic bands seen on Western blot analysis using anti-S. parauberis chicken IgY. In contrast, Kanai et al. [8] differentiated S. parauberis into two serotypes based on the chemical composition of a surface capsular polysaccharide. These variations may reflect an evolutionary trend, or may suggest that there is a relationship between serotype and virulence.
Comparative genome sequence analysis of bacteria can detect sequence diversity among distinct yet closely related populations, which may have important implications among the strains are important for adaptation, strain specific genes are thought to represent the physiological and virulence properties of an organism, disease epidemiology and understanding evolutionary relationships [9,10]. These genetic variations can arise from gene mutations, insertions, deletions and/or genetic noise. Such analyses have been performed for various pathogenic bacteria, including Helicobacter pylori [11], Mycobacterium species [12], Escherichia coli [13], S. mutans [14] and the fish pathogen Edwardsiella tarda [15]. Moreover the recent determination of the complete genomic sequence for S. parauberis revealed important information on bacterial diversity, functional characteristics, environmental adaptation, and virulence components [16].
Bacteria utilize diverse carbohydrate sources for various biosynthetic processes, particularly those involved in maintenance and reproduction. Many catabolic operons are subject to carbon catabolite repression (CCR) by rapidly metabolizable carbon sources, especially glucose [17]. In Gram-positive bacteria with low GC contents, however a very different CCR pattern is used for PTS sugar transport by EI-P on HPr residue histidine, and HPr can also be phosphorylated at serine 46 (Ser-P) by a specific ATP-dependent protein kinase [18]. Then, HPr (Ser-P) interacts with CcpA, which binds to a conserved sequence in the promoter region called the catabolite-responsive element, thereby negatively controlling the expression of several carbohydrate catabolism related enzymes [19,20].
Bacteria have evolved various mechanisms to defend themselves against viral predation. One such strategy involves the CRISPR/Cas systems, which are small RNA-based defense systems that provide adaptive, heritable immunity against viruses, plasmids and other mobile elements in archaea and bacteria [21]. The RNA and protein components of these immune systems arise from the CRISPR locus and the cas genes. The six cas genes (cas1-6) are present in a wide array of bacteria [21–24]. Most bacteria have some but not all of the cas genes; however, cas1 and cas2 appear to be universal and can therefore be used as genomic markers for the CRISPR/Cas system.
In this context, we determined the whole genome sequences of two strains of S. parauberis (KRS-02083 and KRS-02109, representing serotypes I and II, respectively), that were isolated from Japan in 2002 and classified using a rabbit anti-serum based agglutination test [8]. We compared the genome sequences, genomic structures, and gene variations of these strains with those of a reference strain of serotype I (KCTC11537) isolated from South Korea in 2006. Our results show differentiation for carbon source utilization and phage resistance according geographical and serological strains which provide useful information on evolutionary events in the three S. parauberis strains and offer new insights into streptococcal species-specific survival and potential prophylactic strategies.
Materials and Methods
Bacterial strains
S. parauberis strains KRS-02083 and KRS-02109 were isolated from olive flounder at commercial aquaculture sites in Western Japan and grown in Todd Hewitt (TH; Oxoid Ltd., Cambridge, UK) broth (THB) or agar (THA) [8], at 25°C for 24 h. For long-term storage, bacteria were kept in THB containing 10% (v/v) glycerol at -70°C. The control, S. parauberis KCTC 11537 (GenBank accession no. NC_015558), was as previously described [16].
Preparation of genomic DNA
The two S. parauberis strains were cultured in THB at 25°C for 24 h, and genomic DNA was extracted using the Qiagen Genomic-tip 500/G kit (Qiagen, Hilden, Germany) and a genomic DNA buffer set (Qiagen), according to the manufacturer’s instructions.
Whole-genome sequencing
The draft genomes of S. parauberis KRS-02083 and KRS-02109 were sequenced by the National Research Institute of Fisheries Science (Yokohama, Japan) using a Roche/454 GS-FLXTM system [25], and Takara Bio, Inc. (Otsu, Japan) using Illumina Genome Analyzer (GA) [26]. Large contigs were assembled using the Newbler de novo assembler package in the 454 GS-FLXTM system, and pair-end reads were assembled with respect to the reference genome using BWA-SAMtools [27] and Edena [28] in Illumina GA.
Annotation and comparative multiple alignment analyses
The KRS-02083 and KRS-02109 genome sequences were annotated using the automated prokaryotic annotation server, Rapid Annotations using Subsystems Technology (RAST; rast.nmpdr.org) [29], and identified by manual NCBI BLAST searches. We evaluated the annotation accuracy by comparing the RAST results from our large scaffolds and pair-end reads to the coding sequences (CDSs) published reference genome (KCTC11537). Comparative circular maps of the genomes were constructed using the BRIG (BLAST Ring Image Generator) [30]. Homologies in the gene contents of the two genomes were identified using the comparative tools of the RAST algorithm, to allow for potential genome-to-genome annotation. CRISPR elements were identified and CRISPR-associated genes were annotated using CRISPRFinder [31] and our CDS results. Multiple alignments were performed using CLUSTALW [32], and various other comparisons and investigations were performed using the in silico molecular cloning tool, Genomic Edition Version 4.2.21 (In Silico Biology, Yokohama, Japan).
PCR-based assay
The presence of each lactose-, sorbose- and glucose-associated gene was determined by PCR using specific primer sets (Table S1); the tested genes included lacC, lacE and lacG for lactose, sorC and sorE for sorbose, and GCK and ptsG for glucose utilization. PCR detection of the cas1 gene was also performed for identification of CRISPR/Cas. The 24 stored S. parauberis strains utilized in this PCR-based assay had been isolated from diseased olive flounder in Korea (six strains each of type I and type II) between 2006 and 2009, and in Japan (six strains each of type I and type II) between 2002 and 2004. The isolates were cultured in TSB (for the Korean strains) and THB (for the Japanese strains) at 25°C for 24 h, and DNA was extracted using an AccuPrep® Genomic DNA Extraction Kit (Bioneer, Korea) according to the manufacturer’s instructions. PCR was performed in 20 μL reaction mixtures containing 1 μL template DNA, 0.05 μM of each primer (Bioneer), and the AccuPower PCR® premix (Bioneer). Amplification was performed using a C-1000TM thermo cycler (Bio-Rad). The PCR conditions included an initial denaturation cycle of 94°C for 3 minutes; 30 cycles of denaturation step at 94°C for 30 seconds, annealing at 54°C for 30 seconds, and extension at 72°C for 30 seconds; and a final extension step at 72°C for 5 minutes. The resulting PCR products were analyzed on a 1.5% (w/v) agarose gel and stained 1% (w/v) ethidium bromide. DNA bands were visualized using a Gel Dock system (ATTO E-Graph, AE-9000; Takara, Japan) equipped with a CS analyzer program.
Analysis of growth curves and sugar utilization
KRS-02083, KRS-02109 and KCTC11537 were maintained as previously described [8,16]. Bacterial cells were cultivated in TYE broth (tryptone, 10 g; yeast extract, 5 g; and K2HPO4, 2 g per liter) [33] supplemented with 20 mM glucose and 25 mM lactose and L-sorbose for the two Japanese strains and the Korean strain. The growth phenotypes of various strains were monitored using an xMark Microplate Absorbance Spectrophotometer (Bio-Rad, Hercules, CA, USA) at 37°C, with the optical density at 600 nm recorded every hour.
Nucleotide sequence accession numbers
The whole genome sequences of KRS-02083 and KRS-02109 have been deposited in the GenBank database under accession numbers ALYM00000000 and ALWR00000000, respectively.
Results and Discussion
General genomic features
De novo assemblies of the KRS-02083 and KRS-02109 genomes were generated using two next generation sequencing technologies: the Illumina GA and GS-FLX pyrosequencing. The BRIG program [30] was used to compare the genomes of the S. parauberis strains via genomic alignment (Figure 1).
Figure 1. Circular maps from NGS comparing the genomes of S. parauberis strains KRS-02083 and KRS-02109 with that of reference strain KCTC11537.

Beginning with the outermost ring, ring 1 shows the CRISPR/Cas and Tn916 regions encoded in the KRS-20083 genome. Rings 2 and 3 show large contigs from 454 GS-FLX and pair-end reads from the Illumina GA alignment of KRS-02083. Rings 4 and 5 show large contigs from 454 GS-FLX and pair-end reads from Illumina GA alignment in KRS-02109. Ring 6 shows four phage-associated regions in the KRS-02083 genome. Rings 7 and 8 show the GC content and GC skew, respectively, with respect to reference strain KCTC11537. The inner circle shows the scale (bp).
The 650 ~750 bp and 580 ~660 bp reads were found to be the dominant lengths in the GS-FLX results for KRS-02083 and KRS-02109, respectively. The Illumina GA results comprised more than 10,000 contigs, including numerous small contigs (≤ 500 bp) and various contigs of different sizes (≥ 500 bp, ≥ 1kb, ≥ 2kb and ≥ 4kb) in both KRS-02083 and KRS-02109. The assembled sequence data for KRS-02083 and KRS-02109 yield coverages of 65.5 and 71.6 times that of the paired-end mapped reads from GS-FLX and, 500 and 600 times that of the single reads produced using the Illunina GA, respectively. In terms of the raw data, the KRS-02083 and KRS-02109 genome sequences consisted of 55 and 31 contigs, respectively, generated by GS-FLX sequencing, and 265 and 192 contigs, respectively, generated using Illumina GA. The KRS-02083 genome was estimated to be 2,126,607 bp in length with 88.61% nucleotide similarity to the reference genome and a GC content of 35.6%, while the KRS-02109 was estimated to be 2,084,517 bp in length with 87.35% nucleotide similarity to the reference genome and a GC content of 35.5%. 2,236; 2,048 CDSs were identified from the KRS-02083 and KRS-02109 genomes, respectively, by RAST annotation. The KRS-02083 and KRS-02109 genome sequences had 13 and 6 gaps, respectively, ranging in lengths from 500 bp to 40 kb (Table 1).
Table 1. Overall features of the S. parauberis KRS-02083 and KRS-02109 genomes compared with that of strain KCTC11537.
|
Parameter
|
KCTC11537 (ref) | KRS-02083 | KRS-02109 | |
|---|---|---|---|---|
| Illumina GA | Total no. of bases (b) | - | 1,043,833,490 | 1,290,529,310 |
| No. of assembled reads | - | 29,823,814 | 36,872,266 | |
| Length of assembled reads (b) | - | 71 | 71 | |
| No. of total contigs | - | 265 | 192 | |
| No. of unmapped contigs | - | 138 | 63 | |
| 454 GS-FLX | Total no. of bases (b) | 75,170,197 | 130,986,679 | 143,264,028 |
| Length average (b) | 523 | 586.97 | 563.39 | |
| No. of total contigs | 181 | 55 | 31 | |
| RAST annotation (CDSs) | 2204 | 2,236 | 2,048 | |
| RAST annotation (RNAs) | 69 | 68 | 78 | |
| No. of gaps | 0 | 13 | 6 | |
| GC contents (%) | 35.6 | 35.55 | 35.48 | |
| Length (b) | 2,143,887 | 2,126,607 | 2,084,517 | |
Approximately 70.7% and 72.3% of the S. parauberis KRS-02083 and KRS-02109 proteins, respectively, were grouped into 27 functional groups, and the remaining 29.3% and 27.7%, respectively, were assigned to the “unknown function” group, which contained the highest proportion of annotated genes. An overview of the functional annotation in the two S. pararuberis strains is shown in Table 2. The category of carbohydrate related proteins contain the highest percentage of known genes from the RAST database, at 14.8% and 15.6% for KRS-02083 and KRS-02109, respectively. These proteins are important to the ability of the bacteria to survive in their host’s complex environmental niche. The annotated genes of KRS-02083 and KRS-02109 also included genes involved in RNA metabolism (3.6% and 3.8%), protein metabolism (6.7% and 6.3%), DNA metabolism (5.2% and 4.8%) for carbohydrate metabolism and processing. These findings may reflect that the organism is well adapted to aquatic ecosystems containing a wealth of carbohydrates and nucleic acid. We also observed relatively high levels of genes involved in regulation and cell signaling, perhaps reflecting the capacity of these strains to cope with various growth conditions and stresses [34]. In contrast, we did not identify genes involved in the photosynthesis, iron acquisition and metabolism, secondary metabolism or nitrogen metabolism.
Table 2. Functional annotation of CDSs found in the KRS-02083 and KRS-02109 genomes, according to the RAST annotated categories.
| Functional category | KRS-02083 | KRS-02109 |
|---|---|---|
| Cofactors, vitamins, prosthetic group, pigments | 101 (4.5%) | 102 (5.0%) |
| Cell wall and capsule | 123 (5.5%) | 117 (5.7%) |
| Virulence, disease and defense | 36 (1.6%) | 37 (1.8%) |
| Potassium metabolism | 3 (0.1%) | 3 (0.1%) |
| Photosynthesis | 0 (0%) | 0 (0%) |
| Miscellaneous | 82 (3.7%) | 84 (4.1%) |
| Phages, prophages, transposable elements, plasmids | 30 (1.3%) | 0 (0%) |
| Membrane transport | 39 (1.7%) | 39 (1.9%) |
| Iron acquisition and metabolism | 0 (0%) | 0 (0%) |
| RNA metabolism | 80 (3.6%) | 77 (3.8%) |
| Nucleosides and nucleotides | 87 (3.9%) | 83 (4.1%) |
| Protein metabolism | 150 (6.7%) | 129 (6.3%) |
| Cell division and cell cycle | 22 (1.0%) | 22 (1.1%) |
| Motility and chemotaxis | 2 (0.1%) | 1 (0.0%) |
| Regulation and cell signaling | 22 (1.0%) | 21 (1.0%) |
| Secondary metabolism | 0 (0%) | 0 (0%) |
| DNA metabolism | 117 (5.2%) | 99 (4.8%) |
| Fatty acids, lipids and isoprenoids | 75 (3.4%) | 76 (3.7%) |
| Nitrogen metabolism | 0 (0%) | 0 (0%) |
| Dormancy and sporulation | 1 (0.0%) | 1 (0.0%) |
| Respiration | 38 (1.7%) | 37 (1.8%) |
| Stress response | 67 (3.0%) | 63 (3.1%) |
| Metabolism of aromatic compounds | 3 (0.1%) | 3 (0.1%) |
| Amino acids and derivatives | 124 (5.5%) | 123 (6.0%) |
| Sulfur metabolism | 8 (0.4%) | 5 (0.2%) |
| Phosphorous metabolism | 39 (1.7%) | 38 (1.9%) |
| Carbohydrates | 331 (14.8%) | 320 (15.6%) |
| Unknown function | 656 (29.3%) | 568 (27.7%) |
| Total | 2236 (100%) | 2048 (100%) |
Conformation of SNPs and INDEL genes
A major goal for sequencing is to identify large and small changes in the genome that could explain the divergence of a strain or have implications for gene function. Therefore, we conducted a detailed analysis of single nucleotide polymorphisms (SNPs), variations in repeat numbers, and gene-scale gains or losses. Analysis of Illumina GA data allowed us to identify 5,360 and 4,241 single mutations in the genomic sequence of KRS-02083 and KRS-02109, respectively, compared with the reference genome.
Gene insertion-deletion (INDEL) events were abundant in the KRS-02083 and KRS-02109 genomes, which contained 346 and 232 were newly annotated genes and lacked 336 and 357 genes, respectively, with respect to the KCTC 11537 genome. Most of INDEL genes were hypothetical genes or genes of unknown function (Figure 2). INDEL genes contribute to species diversity and might encode supplementary biochemical pathways and functions that are not essential for bacterial growth but may confer selective advantages, such as adaptation to different niches and/or antimicrobial resistance. Large INDELs of gene regions can also create structural variations of the chromosome; these may be used as genotype-specific markers for epidemiological studies, and their study can offer a novel approach to understanding genetic diversity [35]. As such, our comparative analysis of the three S. parauberis genomes may provide important new insights into the evolutionary history of microbial pathogens and heterogenetic specificity in similar species.
Figure 2. Comparison of gene contents and numbers with reference strain KCTC11537.

As assessed by RAST functional categories grouped among insertion/deletion genes, between the KRS-02083 and KRS-02109 S. parauberis genomes.
Lactose (lac) operon
The KRS-02083 and KRS-02109 genomes contained lac operons homologous to those in S. bovis (NZ_AEEL01000000) and S. pyogenes (NC_002737.1) (Table 3). The KRS-02083 genome encoded lacRABCDTGF and E in scaffold 11 while KRS-02109 genome encoded lacRABCDFE, and G in scaffold 1 (Figure 3). The lacR determinant extended to the first ORF of the lac operon in both genomes which contained a DeoR-like helix-turn-helix domain and DeoR terminal sensor domain that detects diverse sugar derivatives in multiple genera, including tagatose phosphate in S. mutans [36]. In this regard, galactose-6-phosphate and tagatose-6-phosphate have been suggested to bind to the lacR cistron and facilitate its dissociation from the binding site, AGGAG. The adjacent genes in order, are galactose-6-phosphate isomerase A/B subunits, tagatose-6-phosphate kinsase and tagatose-1,6-diphosphate aldoase; designated as lacABC and D. The lacA/B enzymes convert galactose-6-phosphate to tagatose-6-phosphate, has a broad specificity for various sugars, and can be used to produce rare sugars. Previously, Zeng et al. [36] showed that a mutant strain of S. mutans lacking lacA/B failed to grow on lactose supplemented medium, indicating that they are essential for the intracellular catabolism of lactose. However, further research is required to clarify the underlying mechanism. LacC and lacD are preceded by a non-translated region containing the promoter along numerous direct and inverted repeats that are involved in regulating the lac-PTS operon.
Table 3. List of lactose-associated genes encoded in the three S. parauberis genomes (KRS-02083, KRS-02109 and KCTC11537).
| Gene function | KCTC11537 | KRS-02083 | KRS-02109 | |
|---|---|---|---|---|
| Lactose utilization (API20strep) | Negative | Positive | Positive | |
| 1 | Lactose phosphotransferase system repressor (lacR) | STP_0241 (S. equi 73%) | SPJ1_2215 (S. pyogenes 99%) | SPJ2_0543 (S. pyogenes 99%) |
| 2 | PTS, galactose-specific IIA component (gatA) | STP_0242 (S. uberis 65%) | - | - |
| 3 | PTS, galactose-specific IIB component (gatB) | STP_0243 (S. uberis 91%) | - | - |
| 4 | PTS, galactose-specific IIC component (gatC) | STP_0244 (S. uberis 94%) | - | - |
| 5 | Galactose-6-phosphate isomerase A (lacA) | STP_0245 (S. equi 84%) | SPJ1_2214 (S. pyogenes 98%) | SPJ2_0544 (S. pyogenes 98%) |
| 6 | Galactose-6-phosphate isomerase B (lacB) | STP_0246 (S. equi 92%) | SPJ1_2213 (S. pyogenes 98%) | SPJ2_0545 (S. pyogenes 98%) |
| 7 | Tagatose-6-phosphate kinase (lacC) | STP_0247 (S. gallolyticus 65%) | SPJ1_2212 (S. pyogenes 95%) | SPJ2_0546 (S. pyogenes 95%) |
| 8 | Tagatose-1,6-bisphosphate aldolase (lacD) | STP_0248 (S. gallolyticus 84%) | SPJ1_2211 (S. pyogenes 96%) | SPJ2_0547 (S. pyogenes 96%) |
| 9 | Transcriptional antiterminator (lacT) | - | SPJ1_2210 (S. bovis 86%) | - |
| 10 | PTS, lactose-specific IIB component (lacE) | - | SPJ1_2207 (S. bovis 92%) | SPJ2_0549 (S. pyogenes 98%) |
| 11 | PTS, lactose-specific IIA component (lacF) | - | SPJ1_2208 (S. bovis 100%) | SPJ2_0548 (S. pyogenes 96%) |
| 12 | 6-Phospho-beta-galactosidase (lacG) | - | SPJ1_2209 (S. bovis 99%) | SPJ2_0550 (S. pyogenes 99%) |
Symbols: parentheses indicate the sequence homology in NCBI; ‘- ’ indicates non-coding.
Figure 3. Structure variation of lactose operon among three S. parauberis strains.
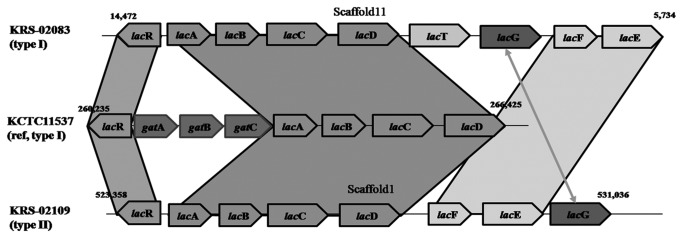
Comparison of S. parauberis KCTC11537, KRS-02083 and KRS-02109 reveals that lactose operons are sequence reservoirs for genetic diversity among strains. Schematic representation of the genetic characterization of lactose operons found in the three S. parauberis genomic sequences.
The next sets of genes in the lac-PTS operons of the two tested strains were lacTGFE in KRS-02083, and lacFEG in KRS-02109, which are similar in structure to that in Lactobacillus rhamnosus [37], and S. mutans [36], respectively. The lacF and lacE genes were clearly related to the hydrophilic PTS IIA and PTS IIB components. LacE in particular was found to have the predominantly hydrophobic N-terminal region typical of integral membrane proteins, followed by a hydrophilic domain that resembled the cytoplasmically oriented PTS lactose-specific IICB component [38]. The enzyme II components of the lactose PTS in the studied Japanese S. parauberis strains respond to intracellular inducers: IICB (lacE) and free cytosolic IIA (lacF), these domains may be part of a single polypeptide or may exist as separate interacting proteins [39]. The two Japanese S. parauberis genomes also contain 6-phospho-beta-galactosidase (lacG), an enzyme essential for the catabolism of lactose phosphate into glucose and galactose-6-phosphate, important for lactose hydrolysis (Figure S1). It has been suggested that in some cases, lactose 6-phosphate can be hydrolyzed by beta-glycosidases that are specific for beta-glucoside sugars. The absence of lacG in KCTC11537 explains the previous finding that it is unable to grow on lactose [36].
Thus, in the two Japanese S. parauberis genomes, the lacABCD genes comprise the tagatose-6-phosphate pathway and are co-transcribed with genes lacFE and G, which specify proteins for the transport and cleavage of lactose. Our findings support the previous reports that KRS-02083 and KRS-02109 showed positive reactions to lactose acidification in biochemical tests, whereas KCTC11537 did not [7,8].
Sorbose operon
Sorbose is a known carbon energy source in E. coli [40]. KCTC11537 showed a positive reaction to sorbose acidification test [7,8], and encodes L-sorbose utilization factors (sorEDCBF and R) with a 5.7-kb DNA fragment (Figure 4A). Intracellularly, L-sorbose-1-phosphate is reduced to D-sorbitol-6-phosphate by an L-sorbose-1-phosphate reductase (sorE). Then, in an NAD+-dependent step, sobitol-6-phosphate dehydrogenase (sorF) catalyzes the oxidation of D-sorbitol-6-phosphate to D-fructose-6-phosphate, which is then further catabolized by glycolytic enzymes (Figure S1). Sequence analysis revealed the presence of six sor-utilization ORFs, similar to the sor operon of Yersinia enterocolitica [41] except for the absence of sorF in the KCTC11537 (Figure 4B). The predicted gene products also showed homologies to various PTS proteins, including the IIB, IIC and IID components of the sorbose-specific PTS, and the catalytic enzyme L-sorbose 1-phosphate reductase from Y. enterocolitica (39%, 71%, 71% and 66%, respectively). The last gene, sorR, showed 43% homology with the corresponding gene of Y. enterocolitica.
Figure 4. Structure variation of L-sorbose operon among three S. parauberis strains.

(A) Comparison of S. parauberis KCTC11537, KRS-02083 and KRS-02109 reveals that sorbose operons are sequence reservoirs for genetic diversity among strains. (B) Simplified restriction map and genetic organization of the sorbose operon from S. parauberis KCTC11537.
In KCTC11537, the L-sorbose PTS EII consists of two membrane-bound proteins, IIC (sorC) and IID (sorD), and two soluble components IIA/B (sorB); BLAST searches suggested that these are fused into a single polypeptide chain, as in E. coli [42]. When the predicted amino acid sequence of sorR was used in a BLAST search, we found that its closest homologs included a transcriptional regulator from Y. enterocolitica [43], a DeoR family transcriptional regulator from Sebaldella termitidis [44] and a sor operon regulator from E. coli [45]. These results suggest that the helix-turn-helix is a major structural motif capable of binding DNA; composed of two alpha helices joined by a short strand of amino acids, this motif is found in many proteins that regulate sor gene expression [46]. KCTC11537 encodes two sorbitol 6-phosphate 2-degydrogenase genes, sorF and srlD, which were found to be 78% and 85% similar to those in S. equi [35]. This suggests that cross-talk occurs between the sorF and srlD regulatory metabolic pathways and the redox state of the cell, and further indicates that the genes involved in L-sorbose and D-sorbitol catabolism interact through common effectors to reduce sorbitol-6-phosphate by converting it to fructose-6-phosphate.
PCR-based survey of carbon utilization genes
The utilization genes for glucose, GCK and ptsG, were detected as 480- and 875- bp bands, respectively (Table 4). Our results revealed that the major pathways of carbohydrate uptake in 24 S. parauberis isolates all involved the PTS. The nucleotide sequences of the GCK and ptsG genes in the three genomes examined herein were highly homologous (over 99%) further supporting this contention. Glucose is the preferred carbon source in many bacteria, and the presence of glucose often prevents the use of other, secondary carbon sources (e.g., sorbose, lactose and many others) [47]. The lactose-utilizing genes, lacC, lacE and lacG, were amplified from 10 of the 24 tested isolates, including the Korean strain, J27, and the Japanese strains, KRS-02083, KRS-02030, KRS-02067, KRS-02109, KRS-02068, KRS-02090, KRS-02087, KRS-02091 and KRS-02102. Eleven of the 24 isolates were found to have genes for sorbose utilization, such as sorC and sorE; these isolates included the Korean strains, KCTC11537, KCTC11538, J19, J20, J24, J25 and J21, and the Japanese strains, KRS-02032, KRS-04024, KRS-02067 and KRS-04037. We failed to detect any sorbose- or lactose- associated genes in Korean strains J22, J23, J28 and J30 (Table 4). The KCTC11537 genome was found to include a sorbose-utilization, locus that contained the genes for the PTS EII domains that are involved in the transport of extracellular sorbose and the phosphorylation of sorbitol. The KRS-02083 and KRS-02109 genomes, in contrast, both encoded lac operons, but showed different gene sequences.
Table 4. PCR amplification.
| Name | Year | Serotype | lacC | lacE (J1) | lacG (J1) | lacE (J2) | lacG (J2) | sorC | sorE | GCK | ptsG | cas1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Korean strains | KCTC11537 (ref) | 2006 | I | - | - | - | - | - | + | + | + | + | - |
| KCTC11538 | 2008 | I | - | - | - | - | - | + | + | + | + | - | |
| J19 | 2009 | I | - | - | - | - | - | + | + | + | + | - | |
| J20 | 2009 | I | - | - | - | - | - | + | + | + | + | - | |
| J24 | 2009 | I | - | - | - | - | - | + | + | + | + | - | |
| J25 | 2009 | I | - | - | - | - | - | + | + | + | + | - | |
| J21 | 2009 | II | - | - | - | - | - | + | + | + | + | + | |
| J22 | 2009 | II | - | - | - | - | - | - | - | + | + | + | |
| J23 | 2009 | II | - | - | - | - | - | - | - | + | + | + | |
| J27 | 2009 | II | + | - | - | + | + | - | - | + | + | + | |
| J28 | 2009 | II | - | - | - | - | - | - | - | + | + | + | |
| J30 | 2009 | II | - | - | - | - | - | - | - | + | + | + | |
| Japanese strains | KRS-02083 | 2002 | I | + | + | + | - | - | - | - | + | + | - |
| KRS-03032 | 2003 | I | - | - | - | - | - | + | + | + | + | - | |
| KRS-02030 | 2002 | I | + | + | + | - | - | - | - | + | + | - | |
| KRS-04024 | 2004 | I | - | - | - | - | - | + | + | + | + | - | |
| KRS-02067 | 2002 | I | + | + | + | - | - | + | + | + | + | - | |
| KRS-04037 | 2004 | I | - | - | - | - | - | + | + | + | + | - | |
| KRS-02109 | 2002 | II | + | - | - | + | + | - | - | + | + | + | |
| KRS-02068 | 2002 | II | + | - | - | + | + | - | - | + | + | + | |
| KRS-02090 | 2002 | II | + | - | - | + | + | - | - | + | + | + | |
| KRS-02087 | 2002 | II | + | - | - | + | + | - | - | + | + | + | |
| KRS-02091 | 2002 | II | + | - | - | + | + | - | - | + | + | + | |
| KRS-02102 | 2002 | II | + | - | - | + | + | - | - | + | + | + |
Lactose-, sorbose- and glucose-related genes, along with gene markers of the CRISPR/Cas system, were PCR amplified from 24 stored S. parauberis strains that had been isolated from diseased olive flounder in Korean (six strains each of type I and type II) between 2006 and 2009, and Japan (six strains each of type I and type II) between 2002 and 2004.
Symbols: ‘-’ indicates no amplification; ‘+ ’ indicates amplification.
Sugar utilization
The difference in the encoded PTS components was confirmed when the bacterial strains were grown in TYE supplemented with D-glucose and with either lactose or L-sorbose. The two Japanese strains encoding the PTS components and tagatose associated enzymes involved in lactose metabolism grew in TYE supplemented with D-glucose and lactose. In contrast, the Korean strain encoding the PTS components and enzymes involved in sorbose metabolism grew well in TYE supplemented with D-glucose and L-sorbose; all strains show similar growth patterns on TYE broth containing non-carbon source (Figure 5). These results indicate that the PTS elements mediate the expression of the lac and sor operons within the cellular membrane.
Figure 5. Growth curve abservation.

Growth of KCTC11537 (A), KRS-02083 (B) and KRS-02109 (C) on TYE supplemented with glucose (▲), lactose (◆), sorbose (■), or a non-carbon source (●).
In many bacteria the amount of carbohydrates flowing through glycolysis is the primary trigger for binding of CcpA which is a CCR element that is crucial for sporulation [48,49], antibiotic resistance [50], and expression of virulence genes for pathogenicity [51]. It is important to keep in mind that the aim of pathogenic bacteria is to gain access to nutrients rather than to cause damage to the host. A number of papers have described that the carbon sources have effects on virulence particularly in terms capsule production to avoid phagocytosis and efficient attachment to host cells or tissues [52,53]. Differential nutrient availability within diverse host niches impacts upon the ability of S. parauberis cells to counteract local stresses and resist pharmacological intervention. To better understand how the catabolism of lactose and sorbose affect the virulence mechanisms in S. parauberis, future studies should examine the properties of various mutant strains lacking certain PTS-components, regulatory enzymes and other constituents of the catabolic pathways.
CRISPR/Cas systems
In S. pyogenes, phage integration, as shown by genomic rearrangement of the prophage regions, is an important source for new virulence factors [54]. Mechanisms of phage resistance have not been previously described in S. parauberis, but genomic analyses have suggested that the CRISPR/Cas system helps provide adaptive immunity against foreign genetic elements in phages. Of the genomes examined herein, only KRS-02109 was found to possess characteristic CRISPR/Cas system; it contains genes highly similar to cas1, cas2, and two csn family genes S. pseudoporcinus (Figure 6). The cas1 gene encodes a metal-dependent DNA-specific endonuclease that may play a role in the recognition, cleavage, and/or integration of foreign nucleic acids into CRISPRs [55].
Figure 6. Genetic structure of the chromosomal region where the CRISPR/Cas system is integrated in KRS-02109.

The biological significance of the CRISPR/Cas system against phage infection has been examined in S. thermophilus [54], where the presence of a CRISPR spacer identical to a phage sequence adjacent to cas provides resistance against phages containing that particular sequence (direct sequence consensus: GTT TTG GAA TCA TTC AAA ATA ACA TAG CTC TAA AAC). The spacer sequences found in the CRISPR loci of KRS-02109 showed sequence similarity to a lytic phage suggesting that S. parauberis KRS-02109 might resist phage infection via its CRISPR/Cas system. KCTC11537 and KRS-02083 each encode four phage associated loci in their genomes (Figure 1), suggesting that these four regions of the KCTC11537 and KRS-02083 genomes may be involved in the acquisition of foreign genes via natural transformation or bacteriophage activity. In contrast, no such phage-associated gene was found in the KRS-02109 genome, suggesting that this strain may resist phage infection through its CRISPR/Cas system. Interestingly, the cas1 gene was amplified from type II strains obtained from both Korea and Japan (Table 4), further suggesting that the CRISPR/Cas system might contribute to serology.
In the genus streptococcus, specific species may have evolved particular functions by acquiring foreign genes via natural transformation or bacteriophages. However, the acquisition of new foreign genes via phage infection may not be have always favored their lifestyles. In the future, elucidation of the mechanism by which S. parauberis acquires new genes could help clarify the species-specific evolutionary strategies among the streptococci.
Conclusion
Our genomic comparison of three S. parauberis strains revealed numerous biological, virulence, and pathogenetic factors, reflecting the organism’s adaptation as an obligate and versatile fish pathogen. The genomic features of the two Japanese strains (KRS-02083 and KRS-02109) were overall similar to those of the reference genome (KCTC11537). The S. parauberis genome appears to encode a number of carbohydrate related proteins (comprising the highest percentage of the annotated genes) and seems to confer the ability to synthesize all of the amino acids and regulatory factors required to survive in the host’s complex environmental niche.
The genomes of the two Japanese strains each encoded a lac operon, which was found to be homologous in both type I and type II strains, and the loci were structurally similar to those of L. rhamnosus and S. mutans. In contrast, KCTC11537 exhibited a positive reaction to sorbose and was found to encode L-sorbose utilization factors. Its genome includes a 5.7-kb chromosomal DNA fragment that harbors six sor ORFs, which encode intracellular inducers and proteins capable of responding to external sorbose. Of the three examined strains, only KRS-02019 appears to preferentially use a CRISPR/Cas system to defend against phage integration. The cas1 gene, however, was amplified from all type II strains, regardless of geographic distribution.
Our genome sequence analyses showed that sequence diversities can exist among closely related, but distinct populations. We observed distinct variations, most notably in carbohydrate utilization, between similar species from different geographic regions, suggesting that the bacteria may have adapted to using certain carbohydrate sources that are abundant in particular area and/or utilizing secondary carbohydrate sources in the absence or lack of a primary source. Our findings may also be useful for the development of new prophylactic and therapeutic strategies to counter fish streptococcal infection.
Supporting Information
PCR primers for this study.
(DOCX)
Sugar utilization. Metabolic pathway in three S. parauberis for the conversion of lactose (two Japanese strains) and sorbose (A Korean strain) to glycosysis.
(DOCX)
Funding Statement
This work was supported by a grant from the World Class University Program (R32-10253), funded by the Korean Ministry of Education, Science, and Technology (Republic of Korea). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Eldar A, Frelier PF, Assenta L, Varner PW, Lawhon S et al. (1995) Streptococcus shiloi, the Name for an Agent Causing Septicemic Infection in Fish, Is a Junior Synonym of Streptococcus iniae . Int J Syst Bacteriol 45: 840-842. doi: 10.1099/00207713-45-4-840. [DOI] [PubMed] [Google Scholar]
- 2. Ghittino C, Prearo M (1992) Report of streptococcosis in rainbow trout (Oncorhynchus mykiss) in Italy: preliminary note. Soc ital pathol ittica bull 8:4-11. [Google Scholar]
- 3. Shin GW, Palaksha KJ, Yang HH, Shin YS, Kim YR et al. (2006) Discrimination of streptococcosis agents in olive flounder (Paralichthys olivaceius). Bull Eur Assn Fish: 26:68-79. [Google Scholar]
- 4. Aoki T, Takami K, Kaito T (1990) Drug resistance in a non-hemolytic Streptococcus sp. isolated from cultured yellowtail Seriola quinqueradiata . Dis Aquat Organ 8(3): 171-177. [Google Scholar]
- 5. Perera RP, Johnson SK, Collins MD, Lewis DH (1994) Streptococcus iniae associated with mortality of Tilapia nilotica XT aurea hybrids. J Aquat Anim Health 6: 335-340. doi: 10.1577/1548-8667(1994)006. [DOI] [Google Scholar]
- 6. Doménech A, Fernández-Garayzábal JF, Pascual C, García JA, Cutuli MT et al. (1996) Streptococcosis in cultured turbot (Schophtalmus maximus) associated with Streptococcus parauberis . J Fish Dis 19: 33–38. doi: 10.1111/j.1365-2761.1996.tb00117.x. [DOI] [Google Scholar]
- 7. Nho SW, Shin GW, Park SB, Jang HB, Cha IS et al. (2009) Phenotypic characteristics of Streptococcus iniae and Streptococcus parauberis isolated from olive flounder (Paralichthys olivaceus). FEMS Microbiol Lett 293(1): 20-27. doi: 10.1111/j.1574-6968.2009.01491.x. PubMed: 19278524. [DOI] [PubMed] [Google Scholar]
- 8. Kanai K, Yamada M, Meng F, Takahashi I, Nagano T et al. (2009) Serological differentiation of Streptococcus parauberis strains isolated from cultured Japanese flounder in Japan. Fish Pathol 44: 33–39. doi: 10.3147/jsfp.44.33. [DOI] [Google Scholar]
- 9. Dobrindt U, Hacker J (2001) Whole genome plasticity in pathogenic bacteria. Curr Opin Microbiol 4(5): 550-557. Review. doi: 10.1016/S1369-5274(00)00250-2. PubMed: 11587932. [DOI] [PubMed] [Google Scholar]
- 10. Wren BW (2000) Microbial genome analysis: insights into virulence, host adaptation and evolution. Nat Rev Genet 1(1): 30-39. Review. doi: 10.1038/35049551. PubMed: 11262871. [DOI] [PubMed] [Google Scholar]
- 11. Garcia-Vallvé S, Janssen PJ, Ouzounis CA (2002) Genetic variation between Helicobacter pylori strains: gene acquisition or loss? Trends Microbiol 10(10): 445-447. doi: 10.1016/S0966-842X(02)02446-0. PubMed: 12377549. [DOI] [PubMed] [Google Scholar]
- 12. Brosch R, Gordon SV, Pym A, Eiglmeier K, Garnier T et al. (2000) Comparative genomics of the mycobacteria. Int J Med Microbiol 290(2): 143-152. Review. doi: 10.1016/S1438-4221(00)80083-1. PubMed: 11045919. [DOI] [PubMed] [Google Scholar]
- 13. Ogura Y, Ooka T, Asadulghani, Terajima J, Nougayrède JP et al. (2007) Extensive genomic diversity and selective conservation of virulence-determinants in enterohemorrhagic Escherichia coli strains of O157 and non-O157 serotypes. Genome Biol 8(7): R138. doi: 10.1186/gb-2007-8-7-r138. PubMed: 17711596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maruyama F, Kobata M, Kurokawa K, Nishida K, Sakurai A et al. (2009) Comparative genomic analyses of Streptococcus mutans provide insights into chromosomal shuffling and species-specific content. BMC Genomics 10: 358. doi: 10.1186/1471-2164-10-358. PubMed: 19656368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang M, Lv Y, Xiao J, Wu H, Zheng H et al. (2012) Edwardsiella comparative phylogenomics reveal the new intra/inter-species taxonomic relationships, virulence evolution and niche adaptation mechanisms. PLOS ONE 7(5): e36987. doi: 10.1371/journal.pone.0036987. PubMed: 22590641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nho SW, Hikima J, Cha IS, Park SB, Jang HB et al. (2011) Complete genome sequence and immunoproteomic analyses of the bacterial fish pathogen Streptococcus parauberis . J Bacteriol 193(13): 3356-3366. doi: 10.1128/JB.00182-11. PubMed: 21531805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saier MH Jr, Chauvaux S, Cook GM, Deutscher J, Paulsen IT et al. (1996) Catabolite repression and inducer control in Gram-positive bacteria. Microbiology 142 ( 2): 217-230. Review. doi: 10.1099/13500872-142-2-217. PubMed: 8932696. [DOI] [PubMed] [Google Scholar]
- 18. Deutscher J, Saier MH Jr. (1983) ATP-dependent protein kinase-catalyzed phosphorylation of a seryl residue in HPr, a phosphate carrier protein of the phosphotransferase system in Streptococcus pyogenes . Proc Natl Acad Sci U S A 80(22): 6790-6794. doi: 10.1073/pnas.80.22.6790. PubMed: 6359157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deutscher J, Küster E, Bergstedt U, Charrier V, Hillen W (1995) Protein kinase-dependent HPr/CcpA interaction links glycolytic activity to carbon catabolite repression in Gram-positive bacteria. Mol Microbiol 15(6): 1049-1053. doi: 10.1111/j.1365-2958.1995.tb02280.x. PubMed: 7623661. [DOI] [PubMed] [Google Scholar]
- 20. Hueck CJ, Hillen W (1995) Catabolite repression in Bacillus subtilis: a global regulatory mechanism for the gram-positive bacteria? Mol Microbiol 15(3): 395-401. Review. doi: 10.1111/j.1365-2958.1995.tb02252.x. PubMed: 7540244. [DOI] [PubMed] [Google Scholar]
- 21. Jansen R, Embden JD, Gaastra W, Schouls LM (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43(6): 1565-1575. doi: 10.1046/j.1365-2958.2002.02839.x. PubMed: 11952905. [DOI] [PubMed] [Google Scholar]
- 22. Haft DH, Selengut J, Mongodin EF, Nelson KE (2005) A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLOS Comput Biol 1: e60. doi: 10.1371/journal.pcbi.0010060. PubMed: 16292354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV (2006) A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1: 1-7. doi: 10.1186/1745-6150-1-1. PubMed: 16542032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E et al. (2011) Evolution and classification of the CRISPR/Cas systems. Nat Rev Microbiol 9: 467-477. doi: 10.1038/nrmicro2577. PubMed: 21552286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS et al. (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437(7057): 376-380. PubMed: 16056220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bennett S (2004) Solexa Ltd. Pharmacogenomics 5(4): 433-438. doi: 10.1517/14622416.5.4.433. PubMed: 15165179. [DOI] [PubMed] [Google Scholar]
- 27. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14): 1754-1760. doi: 10.1093/bioinformatics/btp324. PubMed: 19451168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hernandez D, François P, Farinelli L, Osterås M, Schrenzel J (2008) De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res 18: 802–809. doi: 10.1101/gr.072033.107. PubMed: 18332092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T et al. (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9: 75. doi: 10.1186/1471-2164-9-75. PubMed: 18261238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA (2011) BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12: 402. doi: 10.1186/1471-2164-12-402. PubMed: 21824423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grissa I, Vergnaud G, Pourcel C (2007) The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8: 172. doi: 10.1186/1471-2105-8-172. PubMed: 17521438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ et al. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31(13): 3497-3500. doi: 10.1093/nar/gkg500. PubMed: 12824352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hamilton IR, Lo GC (1978) Co-induction of beta-galactosidase and the lactose-P-enolpyruvate phosphotransferase system in Streptococcus salivarius and Streptococcus mutans . J Bacteriol 136(3): 900-908. PubMed: 214423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ward PN, Holden MT, Leigh JA, Lennard N, Bignell A et al. (2009) Evidence for niche adaptation in the genome of the bovine pathogen Streptococcus uberis . BMC Genomics 28: 10:54. PubMed: 19175920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holden MT, Heather Z, Paillot R, Steward KF, Webb K et al. (2009) Genomic evidence for the evolution of Streptococcus equi: host restriction, increased virulence, and genetic exchange with human pathogens. PLOS Pathog 5(3):e1000346 PubMed: 19325880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zeng L, Das S, Burne RA (2010) Utilization of lactose and galactose by Streptococcus mutans: transport, toxicity, and carbon catabolite repression. J Bacteriol 192(9): 2434-2444. doi: 10.1128/JB.01624-09. PubMed: 20190045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsai YK, Lin TH (2006) Sequence, organization, transcription and regulation of lactose and galactose operons in Lactobacillus rhamnosus TCELL-1. J Appl Microbiol 100(3): 446-459. doi: 10.1111/j.1365-2672.2005.02790.x. PubMed: 16478484. [DOI] [PubMed] [Google Scholar]
- 38. Yu Y, Tangney M, Aass HC, Mitchell WJ (2007) Analysis of the mechanism and regulation of lactose transport and metabolism in Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 73(6): 1842-1850. doi: 10.1128/AEM.02082-06. PubMed: 17209069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. de Vos WM, Boerrigter I, van Rooyen RJ, Reiche B, Hengstenberg W (1990) Characterization of the lactose-specific enzymes of the phosphotransferase system in Lactococcus lactis . J Biol Chem 265(36): 22554-22560. PubMed: 2125052. [PubMed] [Google Scholar]
- 40. Crichton PB, Old DC (1982) A biotyping scheme for the subspecific discrimination of Escherichia coli . J Med Microbiol 15(2): 233-242. doi: 10.1099/00222615-15-2-233. PubMed: 6754944. [DOI] [PubMed] [Google Scholar]
- 41. Garzetti D, Bouabe H, Heesemann J, Rakin A (2012) Tracing genomic variations in two highly virulent Yersinia enterocolitica strains with unequal ability to compete for host colonization. BMC Genomics 13: 467. doi: 10.1186/1471-2164-13-467. PubMed: 22963272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wehmeier UF, Nobelmann B, Lengeler JW (1992) Cloning of the Escherichia coli sor genes for L-sorbose transport and metabolism and physical mapping of the genes near metH and iclR. J Bacteriol 174(23): 7784-7790. PubMed: 1447144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thomson NR, Howard S, Wren BW, Holden MT, Crossman L et al. (2006) The complete genome sequence and comparative genome analysis of the high pathogenicity Yersinia enterocolitica strain 8081. PLOS Genet 2(12): e206. doi: 10.1371/journal.pgen.0020206. PubMed: 17173484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harmon-Smith M, Celia L, Chertkov O, Lapidus A, Copeland A et al. (2010) Complete genome sequence of Sebaldella termitidis type strain (NCTC 11300). Stand Genomics Sci 2(2): 220-227. doi: 10.4056/sigs.811799. PubMed: 21304705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Iguchi A, Ooka T, Ogura Y, Asadulghani, Nakayama K et al. (2008) Genomic comparison of the O-antigen biosynthesis gene clusters of Escherichia coli O55 strains belonging to three distinct lineages. Microbiology 154(2): 559-570. doi: 10.1099/mic.0.2007/013334-0. PubMed: 18227260. [DOI] [PubMed] [Google Scholar]
- 46. Brennan RG, Matthews BW (1989) The helix-turn-helix DNA binding motif. J Biol Chem 264(4): 1903-1906. Review. PubMed: 2644244. [PubMed] [Google Scholar]
- 47. Görke B, Stülke J (2008) Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6(8): 613-624. doi: 10.1038/nrmicro1932. PubMed: 18628769. [DOI] [PubMed] [Google Scholar]
- 48. Iyer R, Baliga NS, Camilli A (2005) Catabolite control protein A (CcpA) contributes to virulence and regulation of sugar metabolism in Streptococcus pneumoniae . J Bacteriol 187(24): 8340-8349. doi: 10.1128/JB.187.24.8340-8349.2005. PubMed: 16321938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Abranches J, Nascimento MM, Zeng L, Browngardt CM, Wen ZT et al. (2008) CcpA regulates central metabolism and virulence gene expression in Streptococcus mutans . J Bacteriol 190(7): 2340-2349. doi: 10.1128/JB.01237-07. PubMed: 18223086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Seidl K, Stucki M, Ruegg M, Goerke C, Wolz C et al. (2006) Staphylococcus aureus CcpA affects virulence determinant production and antibiotic resistance. Antimicrob Agents Chemother 50(4): 1183-1194. doi: 10.1128/AAC.50.4.1183-1194.2006. PubMed: 16569828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Browngardt CM, Wen ZT, Burne RA (2004) RegM is required for optimal fructosyltransferase and glucosyltransferase gene expression in Streptococcus mutans . FEMS Microbiol Lett 240(1): 75-79. doi: 10.1016/j.femsle.2004.09.012. PubMed: 15500982. [DOI] [PubMed] [Google Scholar]
- 52. Deutscher J, Francke C, Postma PW (2006) How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev 70(4): 939-1031. doi: 10.1128/MMBR.00024-06. PubMed: 17158705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Marion C, Stewart JM, Tazi MF, Burnaugh AM, Linke CM et al. (2012) Streptococcus pneumoniae can utilize multiple sources of hyaluronic acid for growth. Infect Immun 80(4): 1390-1398. doi: 10.1128/IAI.05756-11. PubMed: 22311922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bolotin A, Quinquis B, Sorokin A, Ehrlich SD (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151(8): 2551–2561. doi: 10.1099/mic.0.28048-0. PubMed: 16079334. [DOI] [PubMed] [Google Scholar]
- 55. Wiedenheft B, Zhou K, Jinek M, Coyle SM, Ma W et al. (2009) Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure 17(6): 904-912. doi: 10.1016/j.str.2009.03.019. PubMed: 19523907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PCR primers for this study.
(DOCX)
Sugar utilization. Metabolic pathway in three S. parauberis for the conversion of lactose (two Japanese strains) and sorbose (A Korean strain) to glycosysis.
(DOCX)