

Abstract
Alzheimer's disease (AD, OMIM: 104300), a progressive neurodegenerative disorder with no cure to date, is caused by the generation of amyloid-beta-42 (Aβ42) aggregates that trigger neuronal cell death by unknown mechanism(s). We have developed a transgenic Drosophila eye model where misexpression of human Aβ42 results in AD-like neuropathology in the neural retina. We have identified an apical-basal polarity gene crumbs (crb) as a genetic modifier of Aβ42-mediated-neuropathology. Misexpression of Aβ42 caused upregulation of Crb expression, whereas downregulation of Crb either by RNAi or null allele approach rescued the Aβ42-mediated-neurodegeneration. Co-expression of full length Crb with Aβ42 increased severity of Aβ42-mediated-neurodegeneration, due to three fold induction of cell death in comparison to the wild type. Higher Crb levels affect axonal targeting from the retina to the brain. The structure function analysis identified intracellular domain of Crb to be required for Aβ42-mediated-neurodegeneration. We demonstrate a novel neuroprotective role of Crb in Aβ42-mediated-neurodegeneration.
Background
Alzheimer's disease (AD) is a progressive neurodegenerative disorder with no effective cure to date. AD is characterized by the progressive loss of neurons in the hippocampus and cortex causing decline in cognitive and behavioral functions eventually leading to the death of the patient [1], [2]. AD neuropathology is associated with two types of abnormal protein deposition in the human brain viz.: (1) neurofibrillary tangles (NFTs) containing hyperphosphorylated forms of a microtubule associated protein Tau, and (2) the accumulation of the amyloid-beta (Aβ42) peptide [1]–[8]. Aβ42 is generated by improper (β- and γ-) cleavage of the transmembrane receptor amyloid precursor protein (APP), as well as by mutations linked to familial AD that affect APP processing [9]. The abnormal cleavage of APP causes the protein to be 42 amino acids long (Aβ42), whereas, the normal length of the protein is 40 amino acids long (Aβ40) [1], [2], [7], [8]. The amyloid hypothesis suggests that Aβ42 forms protofibrils and fibrils. Accumulation of Aβ42 impairs basic cellular processes due to oxidative stress, misregulation of intracellular calcium, ER stress [10], and aberrant signaling through interaction with several receptors [3], [5], [6], [8], which results in the death of neurons [7]. Therefore, it is important to understand the mechanism underlying Aβ42 mediated cell death and neurotoxicity.
Since the genetic machinery and basic cell biological pathways are conserved from insects to humans, several animal models have been employed to model AD. Despite the immense amount of information available from modeling AD in animal models such as the mouse [2], [7] and the fruit fly [7], [11]–[15], the exact mechanism(s) mediating Aβ42-dependent cell death are yet to be determined. The fruit fly has been a model organism for human diseases for many years since nearly 70% of human disease genes are conserved in flies [16]. We have used a Drosophila melanogaster eye model to express the human Aβ42 peptide [3].
The Drosophila eye model has been extensively employed to investigate patterning, growth, and cell biological processes [7], [14]–[16]. The adult Drosophila compound eye develops from an epithelial bi-layer structure housed inside the larva called the eye-antennal imaginal disc, which gives rise to an eye, antenna and head cuticle of the adult fly [17]. A synchronous differentiation event in the developing third instar larval eye imaginal disc differentiates retinal precursor cells to photoreceptor neurons [18]. The eye imaginal disc metamorphose to a pupal retina which develops into the adult eye comprising of about 800 units called ommatidia [18]. Each ommatidium contains eight photoreceptors, pigment cells and several support cells. In the pupal retina, the extra undifferentiated cells are eliminated by programmed cell death (PCD) [19]. PCD is not observed during earlier stages of larval eye development, however, abnormal extracellular signaling due to inappropriate levels of morphogens may trigger cell death in the developing larval eye imaginal disc [20]. We have found that Aβ42 dependent cell death is mediated, in part, through activation of the JNK signaling pathway [3]. However, blocking the JNK signaling pathway does not completely rescue the Aβ42-dependent cell death [3]. Therefore, there may be other genetic components that remain to be identified.
Using the Gal4/UAS system [21], we have developed an AD model with transgenic flies [3] where high levels of Aβ42 are misexpressed in the differentiating photoreceptor neurons of the fly retina using a Glass Multiple Repeat driver [22] (GMR-Gal4>UAS-Aβ42, hereafter GMR>Aβ42). These GMR>Aβ42 transgenic flies exhibit progressive neurodegenerative pathology in the developing retina, which is similar to that observed in AD [3]. Moreover, the misexpression of Aβ42 in the differentiating retina (GMR>Aβ42) exhibits a stronger neurodegenerative phenotype at 29°C [3]. The expression of the cell fate marker like disc large (dlg, a membrane specific marker) in the developing eye imaginal disc was studied. In comparison to the wild type adult eye (Figure 1A) and the larval eye imaginal disc (Figure 1B), misexpression of Aβ42 (GMR>Aβ42) in the Drosophila eye imaginal disc resulted in a reduced eye size with disorganized photoreceptors on the posterior margin as evident from the expression of pan neural marker, Elav (DSHB), in the photoreceptor neurons (Figure 1G), and a highly reduced adult eye which did not show any wild type ommatidium within the compound eye (Figure 1F) [3].
Figure 1. Levels of apical basal polarity gene crb modulates Aβ42 mediated neurodegeneration.

Wild type (A) adult compound eye, a highly organized structure comprising of 750–800 ommatidia [18], which develops from (B, C) eye-imaginal disc. Third-instar eye imaginal disc stained with membrane specific marker, Disc large (Dlg; white), a pan neural marker Elav (red, marks photoreceptors), and (C) Crb protein expression. The Crb expression is localized on the apical surface of epithelial cells and accumulates at the apical membrane's outer margin [51]. (D–F) Misexpression of Aβ42 using GMR-Gal4 driver [22] in the differentiating photoreceptor neurons results in the induction of neurodegeneration as seen in (D) the highly reduced adult eye with a glazed surface and (E, F) eye imaginal disc. Note that in GMR>Aβ42 eye imaginal discs (E) pan neural marker, Elav, exhibits clumping of the photoreceptor neurons and holes in the developing retina, and (F) strong enrichment of Crb expression in the GMR domain. (G–I) Misexpression of Crb full length [41] in GMR>Aβ42 background (GMR>Aβ42+Crb FL) strongly enhances the neurodegeneration phenotype which results in (G) pupal lethality (adults failed to form due to early pupal lethality and as a result lacked the adult eye structure) and (H, I) severe neurodegeneration observed in the eye imaginal disc as evident from (H) fusion of Elav positive photoreceptor neurons, and (I) several fold increase in Crb protein levels. (J–O) Reducing Crb protein levels by using (J–L) crb11A22 allele [33] (GMR>Aβ42+crb11A22) or (M–O) crb RNAi (Vienna Drosophila RNAi Center) (GMR>Aβ42+RNAi) result in the significant rescue of GMR>Aβ42 mediated neurodegeneration as seen in (J, M) the adult eye and (K, L, N, O) the eye imaginal disc. Note that (L, O) the Crb levels are reduced in these backgrounds.
Our earlier studies showed that in the GMR>Aβ42 retina, the ommatidia delaminated from the retinal layers possibly due to loss of polarity and/or cell adhesion [3]. We tested various components of the apical-basal polarity gene pathway in a forward gain of function genetic screen by individually co-expressing the apical basal polarity genes with Aβ42 (GMR>Aβ42+apical basal polarity genes) in the differentiating photoreceptor neurons. From this screen, we identified a transmembrane protein Crumbs (Crb), as a strong genetic modifier of the Aβ42 mediated neurodegenerative phenotype. Crb is highly conserved and has three homologs CRB1, CRB2 and CRB3 in humans. An apical basal polarity gene crb encodes Crb protein, which is localized to the apical domain of the epithelial cells, where it is involved in setting up the apico-basal axis of the cell [23]. Furthermore, Crb is required for organizing apico-basal polarity specification, adherens junctions (AJ) and remodeling in epithelial cells [23], [24]. Crb works by forming a complex with Stardust (Sdt/Pals1) [25]. Sdt, in turn, binds to the intracellular domain of Crb and recruits Pals associated tight junction protein (Patj) [26] and Lin7 [27]. To date, Crb has not been reported to play any role in Aβ42 mediated neurodegeneration.
Materials and Methods
Fly stocks
All fly stocks used in this study are described in Flybase (http://flybase.bio.indiana.edu). The fly stocks used in this study were GMRGal4>UAS-Aβ42 (GMR>Aβ42) [3], UAS-crb Full Length (II), , UAS- crbintra, UAS-crbintraΔPBM, UAS- crbintraΔJM, UAS-crbintraΔJMΔPBM [28], V39177, V39178 crumbs RNAi (Vienna Drosophila RNAi Center) and FRT82B crb 11A22/TM6B [23], GMR Gal4 [22].
We have employed Gal4/UAS system for targeted misexpression studies [21]. All Gal4/UAS crosses were maintained at 18°C, 25°C and 29°C, unless specified, to sample different induction levels. The adult fly cultures were maintained at 25°C, while the egg laying (progeny) were transferred to 29°C. Misexpression of Aβ42 in the differentiating retina (GMRGal4>UAS-Aβ42, GMR>Aβ42) exhibits a stronger neurodegenerative phenotype at 29°C [3]. All the targeted misexpression experiments were conducted using the Glass Multiple Repeat driver line (GMR-Gal4), which directs expression of transgenes in the differentiating retinal precursor cells of the developing eye imaginal disc and pupal retina [22].
Immunohistochemistry
Eye-antennal imaginal discs were dissected from third-instar larvae and stained following standard protocol [29]. Antibodies used were rat anti-Elav (1∶100), rat anti Chaoptin {24B10 (1∶100)}, mouse anti-crumbs (1∶10) (Developmental Studies Hybridoma Bank), rabbit anti-Dlg (1∶200; a gift from K. Cho). Secondary antibodies (Jackson Laboratory) used were goat anti-rat IgG conjugated with Cy5 (1∶200), donkey anti-rabbit IgG conjugated to Cy3 (1∶250), donkey anti-mouse IgG conjugated to Cy3 (1∶200). The tissues were mounted in Vectashield (Vector Laboratories) and immunofluorescent images were captured using the Olympus Fluoview 1000 Confocal Microscope. A modified protocol was used for Crb staining in the eye imaginal disc [30].
Detection of cell death
Cell death was detected using TUNEL assays from Roche Diagnostics [3], [31]. TUNEL assays were used to identify the cells undergoing cell death where the cleavage of double and single stranded DNA is labeled by a Fluorescein. The fluorescently labeled nucleotides are added to 3′ OH ends in a template-independent manner by Terminal Deoxynucleotidyl transferase (TdT). The fluorescent label tagged fragmented DNA within a dying cell can be detected by fluorescence microscopy. Eye antennal discs after secondary antibody staining [32] were blocked in 10% normal donkey serum in phosphate buffered saline with 0.2% Triton X-100 (PBT) and labeled for TUNEL assays using a cell death detection kit from Roche Diagnostics.
The TUNEL positive cells were counted from five sets of imaginal discs and were used for statistical analysis using Microsoft Excel 2010. The P-values were calculated using one-tailed t-test and the error bars represent Standard Deviation from Mean [3].
Adult eye imaging
Adult eye images were taken on the Axioimager.Z1 Zeiss Apotome. Adult flies were mounted onto a needle and the image was completed by using extended depth of focus function of the Axiovision software version 4.6.3 by compiling the individual stacks from the Z-sectioning approach. The final images and figures were prepared using Adobe Photoshop CS4 software.
Results
We tested Crb protein levels using Crb antibody (Cq4, DSHB) [23] in the GMR>Aβ42 eye imaginal disc using a modified protocol [30]. The Crb protein is localized to the apical domain of the epithelial cells. We observed higher levels of Crb protein in the GMR>Aβ42 background (Figure 1F) as compared to the wild type eye imaginal disc (Figure 1C). Misexpression of Aβ42 peptide with full length Crb [28] using GMR-Gal4 driver (GMR>Aβ42+Crb (FL), as evident from Crb antibody staining (Figure 1I), resulted in increased neurodegeneration as shown by highly disorganized morphology due to clumping of photoreceptor neurons (Red channel, marked by Elav) of neighboring ommatidia of the eye imaginal disc (Figure 1H). Large gaps were observed among the photoreceptors of the ommatidia where the cells begin to die or clump together. The adults failed to form due to early pupal lethality (Figure 1G). These animals died in the early pupal stages; as a result we could not observe any pupal retina like structures (data not shown). Downregulating Crb levels by using a heterozygous combination of FRT82B crb11A22 allele [33] (Figure 1L) or crb RNAi (Figure 1 O) resulted in the rescue of the GMR>Aβ42 mediated neurodegeneration as seen in the eye imaginal disc (Figure 1K, N) as well as in the adult eye (Figure 1J, M). We found significant rescue although complete restoration to the wild type eye was not seen. These results suggested that higher levels of crbs are associated with the retina undergoing neurodegeneration due to misexpression of Aβ42. Furthermore, Aβ42 mediated neurodegeneration can be rescued by downregulating crb function.
We employed TUNEL staining to discern the mechanism of neurodegeneration due to misexpression of Crb in the developing retina. The TUNEL staining marks the nuclei of the dying cells, where the cleavage of double and single stranded DNA is labeled by Fluorescein [31]. Here we utilized TUNEL staining to quantitate the effects of Crb protein levels on neurodegeneration in the GMR>Aβ42 background (Figure 2A–F). The TUNEL positive cells were counted from five sets of imaginal discs and were used for statistical analysis using Microsoft Excel 2010. The P-values were calculated using one-tailed t-test and the error bars represent Standard Deviation from the Mean [3]. It is known that a few cells undergo cell death in the wild-type eye imaginal disc (Figure 2A) which does not affect the final morphology of the adult compound eye (Figure 1A). The number of TUNEL positive nuclei of the dying cells in the GMR>Aβ42 flies (Figure 2B) was almost three times as high when compared to the wild-type eye imaginal disc (p = 1.943×10−6; Figure 2F). We investigated the levels of Crb with reference to the induction of cell death and found that when Crb levels were increased in a GMR>Aβ42 background (GMR>Aβ42+Crb FL), the TUNEL positive cell number increased (Figure 2C) and was almost seven times higher than the wild type eye imaginal disc (p = 9.536×10−8; Figure 2F) and nearly two times higher than the GMR>Aβ42 eye imaginal disc (Figure 2F). Reducing levels of crb by using crb11A22 allele [33] (Figure 2D) or crb RNAi (Figure 2E) reduced cell death as evident from reduction in the number of TUNEL positive cells to almost two fold with respect to the GMR>Aβ42 eye imaginal disc (for crb11A22 p = 8.386×10−5, for crb RNAi p = 8.030×10−5; Figure 2F).
Figure 2. Downregulation of crb can block neurodegeneration in the Aβ42 background.

TUNEL assays are commonly employed to mark the cells undergoing cell death where the cleavage of double and single stranded DNA is labeled by a Fluorescein [31]. (A) Wild type eye imaginal disc showing a few TUNEL positive nuclei. (B) Misexpression of Aβ42 using GMR-Gal4 driver [22] in the differentiating photoreceptor neurons results in induction of neurodegeneration (B) as seen by a three-fold induction of cell death as evident from number of TUNEL positive nuclei of the dying cells in comparison to (A) wild type eye imaginal disc. Misexpression of Crb full length (FL) in GMR>Aβ42 background (GMR>Aβ42+Crb FL) strongly enhances (C) the neurodegeneration phenotype which results in nearly seven fold increase in number of TUNEL positive nuclei of dying cells in comparison to wild type eye imaginal disc. (D, E) Reducing Crb levels by using (D) crb11A22 mutant allele [33] (GMR>Aβ42+crb11A22) or (E) crb RNAi (VDRC) (GMR>Aβ42+RNAi) result in the rescue of GMR>Aβ42 mediated neurodegeneration as evident from reduction in numbers of TUNEL positive nuclei of the dying cells. (F) Quantitatively, the number of TUNEL cells have been counted and recorded with all five constructs shown. These phenotypes of enhancement of neurodegenerative phenotype and rescue, based on the number of TUNEL positive cells, are significant as seen by the calculation of P-values based on the one-tailed t-test using Microsoft Excel 2010.
Next, we investigated the effects of modulating levels of Crb on retinal axon targeting from the retina to the brain using chaoptin (24B10, a marker for photoreceptor cells and their axons [34], DSHB) staining. Disruption of axonal transport mechanisms that leads to axonal vesicle stalling has been shown to contribute to the neurodegenerative phenotypes in the AD fly model [35]. During Drosophila visual system development, stereotypical targeting of the axons from the retinal neurons to the special layers of the optic ganglion, medulla and lamina of the brain occurs. The axons of the eight photoreceptor neurons from each ommatidium [36] fasciculate together and project as a single bundle towards the optic lobes of the brain. The Drosophila photoreceptors (R cells) seek specific targets to connect in distinct layers of the optic lobes of the brain, viz., R1–R6 axons project to the lamina; R7 and R8 axons project to the separate layers of the medulla [37]. In comparison to the wild-type eye disc where retinal neurons innervate different layers (medulla and lamina) in the brain (Figure 3A), the GMR>Aβ42 eye disc shows complete loss of axonal targeting (Figure 3B). Additional upregulation of full length Crb levels in GMR>Aβ42 (GMR>Aβ42+Crb FL) strongly affected the retinal axon targeting from the retina to the brain (Figure 3C) as compared to the wild type (Figure 3A) and the GMR>Aβ42 alone (Figure 3B). The axonal targeting was restored when crb levels were reduced in the GMR>Aβ42 background by using either FRT82B crb11A22 allele (Figure 3D) or crb RNAi (Figure 3E). These results further validated our hypothesis that higher levels of Crb enhanced the neurodegenerative phenotype of Aβ42 aggregate accumulation.
Figure 3. Modulating crb levels in the Aβ42 background leads to defects in axonal targeting from retina to the brain.
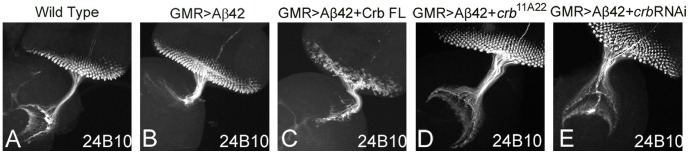
(A) Wild Type eye disc stained with sensory neuron marker, Chaoptin (24B10) [34], which marks only photoreceptor neurons and their axons. The photoreceptor neurons extends through the optic stalk and innervate the medulla and lamina of the larval brain. Note that misexpression of Aβ42 (GMR>Aβ42) in the eye imaginal discs, (B) there is mislocalization of 24B10 expression showing aberrant axonal targeting from retina to brain. The retinal axons fail to innervate the two layers of the brain and end abruptly. (C) Misexpression of Crb full length (FL) in the GMR>Aβ42 background (GMR>Aβ42+Crb FL) strongly enhances the neurodegeneration phenotype which results in (C) lack of axonal targeting from retina to brain. Reducing Crb levels by using (D) crb11A22 allele [33] (GMR>Aβ42+crb11A22) or (E) crb RNAi (VDRC) (GMR>Aβ42+RNAi) result in the significant rescue of GMR>Aβ42 mediated neurodegeneration as evident from the (D, E) restoration of retinal axon targeting.
In order to discern how different domains of Crb protein (Figure 4A) are involved in preventing GMR>Aβ42 mediated neurodegeneration, we used the structure function analysis approach. The full length Crb, a type I transmembrane protein, has 28 EGF domains and four Laminin- AG like repeats in its large extracellular domain (ECD), a transmembrane domain (TM), and a short intracellular domain (ICD) (Figure 4A). The Crb protein's TM domain consists of 37 amino acids spanning the region of the membrane [38]. The ICD contains two motifs, juxtamembrane FERM-binding motif (FBM or JM) domain and C-terminal PDZ (Postsynaptic density/Discs large/ZO-1) binding motif (PBM) domain (Figure 4A). Through its PBM domain, Crb forms a complex with PDZ domain proteins, Stardust and PatJ [25]. It is important to note that the ICD of Crb protein interacts with a variety of conserved proteins including apical basal polarity genes Par6 and aPKC [39], [40]. Prior structure-function studies using the different Crb domains, for example, in the gastrulating embryo, showed that the ubiquitous expression of a membrane-bound cytoplasmic ICD, suppressed the crb mutant phenotype to the same extent as full length crb [23], [28]. Thus, the different domains of Crb carry out different downstream signaling interactions of the protein, so it is important to investigate which domains are involved in the rescue or enhancement of the neurodegeneration caused by Aβ42.
Figure 4. Intracellular domain (ICD) of Crb is required for Aβ42 mediated neurodegeneration.

(A) A cartoon depicting full length type I transmembrane Crb protein and various truncated constructs used in this study. The full length Crb protein consists of an extracellular domain (ECD), transmembrane domain (TM), and a short cytoplasmic intracellular domain (ICD), which consists of the juxtamembrane Ferm-binding motif (JM) and PDZ-binding motif (PBM) domains [28]. GMR-Gal4 driver was used for the misexpression studies in the differentiating photoreceptor neurons [22]. (B–D) Adult eyes of (B) Wild-Type, (C) GMR>Aβ42 (GMR enhancer driving overexpression of human Aβ42 in the developing neural retina), and (D) GMR>Crb (FL) are shown as controls. (A, E–F) Misexpression of (E) Crbintra alone, comprising of fully intact ICD, shows a severe phenotype with a small scab on the head cuticle in the adult eye, which is similar to the (F) GMR>Aβ42+Crbintra adult eye. (G) In the GMR>Aβ42+Crbintra eye disc big gaps and holes between photoreceptors of the ommatidia are seen, Dlg (white) marks the membrane and provide an outline of the imaginal disc and pan neural marker Elav [52] marks the photoreceptors. (A, H–P) In the three other Crb constructs, one of the two domains (JM and PBM) of the ICD is either missing or both of them are missing. (H–P) When Crb is missing either (H–J) PBM, or (K–M) JM, or (N–P) both the PBM and JM domain of the ICD, the GMR>Aβ42 neurodegenerative phenotype is restored significantly with the adult eye having a larger size, higher number of ommatidia, and interommatidial bristles. Furthermore, the Elav staining in the eye-imaginal discs shows more organized photoreceptors in comparison to the GMR>Aβ42 eye imaginal disc. (H, K, N) The controls (H) GMR>Crbintra ΔPBM, (K) GMR>Crbintra ΔJM, and (N) GMR>Crbintra ΔPBM ΔJM showed adult eye phenotypes that are significantly closer to the wild-type. (I, J) When the PBM domain (GMR>Aβ42+Crbintra ΔPBM) is missing, (I) the adult eye and (J) the eye imaginal disc showed significant rescue in comparison to the GMR>Aβ42 phenotype. (L, M) When the JM domain (GMR>Aβ42+Crbintra ΔJM) is missing, (L) the adult eye and (M) the eye-imaginal disc showed significant rescue in comparison to the GMR>Aβ42 phenotype. (O, P) Finally, when both PBM and JM domains of the ICD are missing (GMR>Aβ42+Crbintra ΔPBM ΔJM), a significant rescue was seen in (O) the adult eye and (P) the eye imaginal disc in comparison to the GMR>Aβ42 phenotype.
We employed targeted misexpression [21] of Aβ42 and various domains of Crb protein using the GMR-Gal4 driver [22] for a structure function analysis. The rationale of these studies was to determine which domain of Crb protein is required for its function in Aβ42 mediated neurodegeneration (Figure 1D, E). As discussed previously, in comparison to the wild type eye (Figure 4B), GMR>Aβ42 exhibited strong reduction in size due to neurodegeneration as seen in the adult eye (Figure 4C), whereas GMR>Crb [41] resulted in an increase of the adult eye size with minimal necrosis on the margin (Figure 4D) [42]. Targeted misexpression of Crb ICD (the Crb ICD construct used has been referred to as Crbintra [28]; Figure 4A) in a GMR>Aβ42 background (GMR>Aβ42+Crbintra) resulted in strong enhancement of the neurodegenerative phenotype of GMR>Aβ42 alone as seen in the eye imaginal disc (Figure 4G) as well as in the adult eye (Figure 4F). The GMR>Aβ42+Crbintra adult eye showed strong neurodegeneration as evident from the dark necrotic patch in place of the adult eye (Figure 4F). However, the control GMR>Crbintra also showed some neurodegeneration (Figure 4E), which was not as strong as GMR>Aβ42+Crbintra (Figure 4F). Since both the control (Figure 4E) as well as GMR>Aβ42+Crbintra (Figure 4F, G) showed a neurodegenerative phenotype, it raised the possibility of an additive effect. Further experimentation using the truncated constructs of Crbintra domains disproved this additive effect hypothesis. Targeted misexpression of GMR>Aβ42 with Crbintra ΔPBM [28] or Crbintra ΔJM [28] in developing retina resulted in the rescue of the GMR>Aβ42 neurodegenerative phenotype as seen in the eye imaginal disc (Figure 4J, M) as well as the adult eye (Figure 4I, L). The controls GMR>Crbintra ΔPBM (Figure 4H) and GMR>Crbintra ΔJM (Figure 4K) exhibit a slightly reduced adult eye. The Crbintra construct lacking both the JM and PBM domains (GMR>Crbintra ΔJM ΔPBM (Figure 4A)) resulted in a near normal adult eye (Figure 4N). Targeted misexpression of GMR>Aβ42 with Crbintra ΔJM ΔPBM resulted in the rescue of the GMR>Aβ42 neurodegenerative phenotype as seen in the eye imaginal disc (Fig. 4P), and the adult eye (Fig. 4O). All these results clearly demonstrated that like the full length Crb (Crb FL), the entire ICD (Crbintra) is also responsible for the enhancement of the neurodegenerative phenotype of GMR>Aβ42. It suggests that Crb ICD is sufficient enough to carry out the Crb FL function in Aβ42 mediated neurodegeneration. When we remove either one or both of the JM and PBM domains from the ICD of Crb, the GMR>Aβ42 phenotype is rescued and the ommatidia are restored to near wild-type. This data strongly indicates that both the JM and PBM domains in Crb are essential to suppress the Aβ42 effects. There might be a correlative interaction between the JM and PBM domains of Crb in the Aβ42 mediated neurodegeneration. However, when we have an intact ICD or full length Crb, there is a severe enhancement of the GMR>Aβ42 phenotype. Also, in the loss-of-function crb flies (GMR>Aβ42+crb11A22 and GMR>Aβ42+crb RNAi) where we see reduced Crb level expression (Figure 1L, O) as compared to the wild-type (Figure 1C), there is a rescue of Aβ42 mediated neurodegeneration further validating our hypothesis that Crb levels can modify the neurodegenerative phenotype of Aβ42 accumulation. Thus, Crb levels can serve as an excellent biomarker for AD.
To further verify the structure function analysis results, TUNEL assays were performed on all of the constructs. The rationale was to examine if the reduced eye phenotype seen in GMR>Aβ42+Crbintra was due to cell death or, on the other hand, if the restored eye as shown by removing either or both of the JM and PBM domains of ICD motifs (Figure 4A) is due to reduced number of TUNEL cells. As mentioned earlier, TUNEL marks the nuclei of dying cells, therefore a reduced number of TUNEL positive cells nuclei corresponds to less cells dying, which will lead to a rescue of GMR>Aβ42 neurodegenerative phenotype in the adult eye. We found that the severely reduced adult eye phenotype of GMR>Aβ42+Crbintra is in fact due to an increase in the number of TUNEL positive cells as compared to the wild-type and the GMR>Aβ42 eye disc (Figure 5A, B, I). The GMR>Aβ42+Crbintra exhibits strong neurodegenerative phenotype as evident from disorganized photoreceptor neurons (marked by Elav, green) in the ommatidia. Furthermore, the number of TUNEL positive cells nuclei are increased (Figure 5A, B; red). The TUNEL staining explains the reason for a highly reduced adult eye in GMR>Aβ42+Crbintra (Figure 2F). Additionally, when any either JM or PBM or both JM and PBM domains of the ICD motifs were removed in the GMR>Aβ42 background, the severity of neurodegenerative phenotypes was significantly reduced. In GMR>Aβ42+Crbintra ΔJM (Figure 5C, D), GMR>Aβ42+Crbintra ΔPBM (Figure 5E, F), or GMR>Aβ42+Crbintra ΔJM ΔPBM (Figure 5G, H), the number of TUNEL positive dying cells nuclei were significantly less than GMR>Aβ42 and GMR>Aβ42+Crbintra (Figure 5I). All of these results further validate the data shown in Figure 4 and conforms to the adult eye phenotypes of each of these structures.
Figure 5. Misexpression of Crb intracellular domain triggers neuronal cell death.

(A, C, E, G) The eye-antennal discs are stained with pan neural marker Elav (green), marking the photoreceptor neurons, and TUNEL (red), which marks the nuclei of dying cells. (B, D, F, H) The split channels of the TUNEL cells are shown for better depiction of the TUNEL cells alone. (A, B) In the GMR>Aβ42+crbintra eye disc, the neurodegenerative phenotype of GMR>Aβ42 is enhanced due to increased number of dying photoreceptor neurons as evident from the large number of TUNEL (red) positive cells nuclei, which are (I) calculated quantitatively for all constructs in the bar graph. (A)The dying photoreceptors are clumped and fused together. When we removed either of the PBM, JM or both of these domains within the intracellular domain (ICD) motifs, we see a rescue in the adult eye (Figure 2) and also a (I) decrease in the number of TUNEL positive cells. (C, D) GMR>Aβ42+Crbintra ΔJM (when the JM motif is removed) shows an increase in the (C) organization of the photoreceptors within the ommatidia (Elav) and (C, D, I) a decrease in the number of TUNEL positive cells nuclei as compared to the GMR>Aβ42+Crbintra. (D) The number of dying cells in GMR>Aβ42+Crbintra ΔJM is closer to that seen in the wild-type. A similar result was found (E, F, I) when PBM domain was removed from the ICD motif, GMR>Aβ42+Crbintra ΔPBM or (G, H, I) when both the JM and PBM domains were removed from the ICD motif Aβ42+Crbintra ΔJM ΔPBM. In comparison to GMR>Aβ42+Crbintra, we see a significant decrease in the number of TUNEL positive cells in (E, F, I) GMR>Aβ42+Crbintra ΔPBM and (G, H, I), Aβ42+Crbintra ΔJM ΔPBM. (E–H) The number of dying cell nuclei is closer to that seen in the wild-type. Thus, when the ICD is intact (A, B), there is a large number of TUNEL positive cells, which accounts for the adult eye phenotype observed in Figure 2B. However, when either or both of the ICD motifs are removed (C, D, E, F, G, H), there is a significant reduction in the number of TUNEL positive cells as compared to GMR>Aβ42+Crbintra and GMR>Aβ42.
For all the ICD motifs of Crb, the TUNEL positive cells were counted from five sets of imaginal discs and were used for statistical analysis using Microsoft Excel 2010. The P-values were calculated using one-tailed t-test and the error bars represent Standard Deviation from the Mean [3]. All the p-values showed the TUNEL count to be significantly different from GMR>Aβ42 and the wild-type (Figure 5I). By studying the domains of Crb with reference to the cell death, we found that misexpression of intact Crb ICD domain in GMR>Aβ42 background (GMR>Aβ42+Crbintra), resulted in the increased number of TUNEL positive cell (Figure 5I) and was almost six times higher than the wild type eye imaginal disc (p = 1.5559×10−7) and nearly two times higher than the GMR>Aβ42 eye imaginal disc (p = 8.7869×10−8). Removing the JM motif alone (GMR>Aβ42+Crbintra ΔJM (Figure 5C, D), PBM motif alone (GMR>Aβ42+Crbintra ΔPBM (Figure 5E, F), or by removing both the ICD motifs (GMR>Aβ42+Crbintra ΔJM ΔPBM (Figure 5G, H) resulted in reduced numbers of TUNEL positive dying cells nuclei. The dying cells nuclei in these truncated constructs (Figure 5C–H) were significantly lower than GMR>Aβ42 (for Crbintra ΔJM p = 3.3329×10−5, for Crbintra ΔPBM p = 1.5028×10−5, for Crbintra ΔJM ΔPBM p = 8.9278×10−6; Figure 5I). This TUNEL assay further validated our hypothesis that the reduced eye phenotype seen in GMR>Aβ42+Crbintra (with its fully intact ICD) is primarily due to induction of cell death and the restored eye phenotypes seen when any one or both of the ICD motifs of Crb is/are removed, does indeed have reduced number of dying cells as evident from TUNEL staining.
To further test our hypothesis, we looked at the axonal targeting from the retina to the brain using 24B10 (Chaoptin) in these constructs (Figure 4A). As mentioned earlier, 24B10 shows an organized and orderly axon branching from the retina to the brain in the wild-type background (Figure 3A). However, when we observed the 24B10 staining in the GMR>Aβ42+Crbintra eye there is extreme disorganization marked by the clumping of axons, as well as Elav (red) positive photoreceptors which results in impairing of axonal targeting from retina to the brain (Figure 6A, B). This data further confirms our TUNEL data using GMR>Aβ42+Crbintra. Additionally, when we analyzed other constructs of Crb by removing either or both of the JM or PBM domains from the ICD motif, there is a rescue of the adult eye (Figure 4A, H–P), a reduction in the number of TUNEL positive (Figure 5C–I), and restoration of the organization of axons from the retina to the brain (Fig. 6 C–H) in all three constructs (GMR>Aβ42+Crbintra ΔJM (Figure 6C, D), GMR>Aβ42+Crbintra ΔPBM (Figure 6E, F), GMR>Aβ42+Crbintra ΔJM ΔPBM (Figure 6G, H). When the JM motif (GMR>Aβ42+Crbintra ΔJM (Figure 6C, D) or the PBM motif (GMR>Aβ42+Crbintra ΔPBM (Figure 6E, F) was removed, there is restoration of the axonal targeting as evident from the 24B10 staining and marking the axonal projections innervate the two layers of the brain. Furthermore, when we remove both of the ICD motifs (GMR>Aβ42+Crbintra ΔJMΔPBM (Figure 6G, H), the axonal connection to the brain is restored to near wild type axonal targeting. These data further validates that the ICD domain of Crb is sufficient enough for Crb function in Aβ42 mediated neurodegeneration.
Figure 6. Misexpression of Crb intracellular domain (ICD) can impair axonal targeting.

The eye-antennal disc is stained with Elav (red), which marks the photoreceptors, and 24B10 (chaoptin; green), which stains the axons from the retina to the brain [34]. (A, B) Misexpression of intact instar cellular domain ICD GMR>Aβ42+Crbintra results in clumping of photoreceptors (Elav; A), disorganization of axonal targeting from the retina to the brain as evident from 24B10 staining. (C, D)When the JM motif using GMR>Aβ42+Crbintra ΔJM, photoreceptor organization as well as the axonal targeting is restored to the wild type. Similarly, removing the (E, F) PBM motif of the ICD using GMR>Aβ42+Crbintra ΔPBM, or both PBM and JM domain in GMR>Aβ42+Crbintra ΔJMΔPBM, result in restoration of axonal targeting and photoreceptors. Thus, ICD domain of Crb is required for its role in neurodegeneration.
Discussion
Our studies strongly suggest that transmembrane protein Crb is involved in Aβ42 mediated neurodegeneration. During wing development, N upregulates crb transcription at the dorso-ventral (DV) boundary, and the ability of Crb to inhibit the activity of the γ-secretase complex has been proposed to help refine the N activity domain [43]. Crb functions as a negative regulator of the N signaling pathway [42]. Notch is involved in the development and organization of the dorso-ventral boundary through cell proliferation of the developing eye. Because N and Amyloid Precurssor Protein (APP) are cleaved by similar secretases [44] and Crb regulates N, the Crb effects on Aβ42 could be caused through N regulation. However, in the GMR>Aβ42 model used in our studies, the Aβ42 protein is already cleaved from of APP and does not require cleavage by β- and γ-secretase. Therefore, our data using the transgenic model suggests that Crb also acts downstream of γ-secretase mediated cleavage of APP. Furthermore, higher levels of Crb can enhance human Aβ42 mediated neurodegeneration [3]. Thus, Crb role in modulating Aβ42 mediated neurodegeneration is downstream of N signaling pathway.
In addition, Crb is an upstream regulator of the organ size growth control pathway viz., Hippo signaling pathway. Recently, it was shown that Crb interacts with its juxtamembrane FERM-binding motif (JM) with the FERM domain of Expanded (Ex) to regulate growth by affecting the Hippo pathway activity [45]–[47]. Our structure function analysis studies exhibited that ICD of Crb is sufficient for its role in Aβ42 mediated neurodegeneration suggesting that Crb may act independent of its interaction with Hippo pathway member Ex in Aβ42 mediated neurodegeneration.
Since Crb ICD is involved in its interaction with apical basal polarity gene localization, there is a strong possibility that higher level of Crb in a GMR>Aβ42 background might affect the apical basal polarity of the retinal photoreceptor neurons which result in neurodegeneration. Mutations in Crb homolog 1 (CRB1) has been shown to cause autosomal recessive retinitis pigmentosa (arRP) and autosomal Leber congenital amaurosis (arLCA) [36]. During Drosophila eye development, Crb is required in photoreceptors for stalk elongation [42], [48], and in preventing light-dependent retinal degeneration [49]. Mutations in the human Crb homolog (CRB1) result in abnormalities like thick retina and lamination problems [50]. Furthermore, mutant Crb protein is thought to be responsible for retinal degenerations [50]. However, in GMR>Aβ42 background higher levels of Crb protein were responsible for neurodegeneration. Therefore, it is a strong possibility that higher Crb levels may impair apical basal polarity leading to the Aβ42 neurodegeneration. Thus, regulating Crb levels can help prevent the onset of neurodegeneration and Crb may serve as one of the biomarker as well as the key therapeutic targets for the AD.
Acknowledgments
Authors thank Bloomington Stock Centre, Developmental Studies Hybridoma Bank (DSHB), Sangchul Nam, Pedro Fernandez-Funez, Justin Kumar, and K Cho for fly reagents and members of Singh and Kango-Singh Lab for the comments on the manuscript. AMS is a Berry Summer Research Institute Scholar and a member of the undergraduate Honors Program at UD. MT and ORP are supported by the graduate program at the University of Dayton.
Funding Statement
Funding from National Institutes of Health (NIH) grant (1R15 HD064557-01), University of Dayton, Start-up Support. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hardy J (2009) The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem 110: 1129–1134. [DOI] [PubMed] [Google Scholar]
- 2. O'Brien RJ, Wong PC (2010) Amyloid Precursor Protein Processing and Alzheimers Disease. Annu Rev Neurosci [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tare M, Modi RM, Nainaparampil JJ, Puli OR, Bedi S, et al. (2011) Activation of JNK signaling mediates amyloid-ss-dependent cell death. PLoS One 6: e24361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, et al. (2008) Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14: 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rincon-Limas DE, Jensen K, Fernandez-Funez P (2012) Drosophila Models of Proteinopathies: the Little Fly that Coulds. Curr Pharm Des [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pandey UB, Nichols CD (2011) Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev 63: 411–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirth F (2010) Drosophila melanogaster in the study of human neurodegeneration. CNS Neurol Disord Drug Targets 9: 504–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crews L, Masliah E (2010) Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum Mol Genet 19: R12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M (2004) A model for studying Alzheimer's Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci 26: 365–375. [DOI] [PubMed] [Google Scholar]
- 10. Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, et al. (2011) The ER stress factor XBP1s prevents amyloid-{beta} neurotoxicity. Hum Mol Genet 20: 2144–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moloney A, Sattelle DB, Lomas DA, Crowther DC (2010) Alzheimer's disease: insights from Drosophila melanogaster models. Trends Biochem Sci 35: 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iijima-Ando K, Iijima K (2010) Transgenic Drosophila models of Alzheimer's disease and tauopathies. Brain Struct Funct 214: 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iijima K, Iijima-Ando K (2008) Drosophila models of Alzheimer's amyloidosis: the challenge of dissecting the complex mechanisms of toxicity of amyloid-beta 42. J Alzheimers Dis 15: 523–540. [DOI] [PubMed] [Google Scholar]
- 14. Cowan CM, Shepherd D, Mudher A (2010) Insights from Drosophila models of Alzheimer's disease. Biochem Soc Trans 38: 988–992. [DOI] [PubMed] [Google Scholar]
- 15. Cao W, Song HJ, Gangi T, Kelkar A, Antani I, et al. (2008) Identification of novel genes that modify phenotypes induced by Alzheimer's beta-amyloid overexpression in Drosophila. Genetics 178: 1457–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bier E (2005) Drosophila, the golden bug, emerges as a tool for human genetics. Nat Rev Genet 6: 9–23. [DOI] [PubMed] [Google Scholar]
- 17. Kumar JP (2010) Retinal determination the beginning of eye development. Curr Top Dev Biol 93: 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ready DF, Hanson TE, Benzer S (1976) Development of the Drosophila retina, a neurocrystalline lattice. Dev Biol 53: 217–240. [DOI] [PubMed] [Google Scholar]
- 19. Brachmann CB, Cagan RL (2003) Patterning the fly eye: the role of apoptosis. Trends Genet 19: 91–96. [DOI] [PubMed] [Google Scholar]
- 20. Mehlen P, Mille F, Thibert C (2005) Morphogens and cell survival during development. J Neurobiol 64: 357–366. [DOI] [PubMed] [Google Scholar]
- 21. Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415. [DOI] [PubMed] [Google Scholar]
- 22. Moses K, Rubin GM (1991) Glass encodes a site-specific DNA-binding protein that is regulated in response to positional signals in the developing Drosophila eye. Genes Dev 5: 583–593. [DOI] [PubMed] [Google Scholar]
- 23. Tepass U, Theres C, Knust E (1990) crumbs encodes an EGF-like protein expressed on apical membranes of Drosophila epithelial cells and required for organization of epithelia. Cell 61: 787–799. [DOI] [PubMed] [Google Scholar]
- 24. Campbell K, Knust E, Skaer H (2009) Crumbs stabilises epithelial polarity during tissue remodelling. J Cell Sci 122: 2604–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bachmann A, Schneider M, Theilenberg E, Grawe F, Knust E (2001) Drosophila Stardust is a partner of Crumbs in the control of epithelial cell polarity. Nature 414: 638–643. [DOI] [PubMed] [Google Scholar]
- 26. Bhat MA, Izaddoost S, Lu Y, Cho KO, Choi KW, et al. (1999) Discs Lost, a novel multi-PDZ domain protein, establishes and maintains epithelial polarity. Cell 96: 833–845. [DOI] [PubMed] [Google Scholar]
- 27. Bachmann A, Grawe F, Johnson K, Knust E (2008) Drosophila Lin-7 is a component of the Crumbs complex in epithelia and photoreceptor cells and prevents light-induced retinal degeneration. Eur J Cell Biol 87: 123–136. [DOI] [PubMed] [Google Scholar]
- 28. Klebes A, Knust E (2000) A conserved motif in Crumbs is required for E-cadherin localisation and zonula adherens formation in Drosophila. Curr Biol 10: 76–85. [DOI] [PubMed] [Google Scholar]
- 29. Singh A, Kango-Singh M, Sun YH (2002) Eye suppression, a novel function of teashirt, requires Wingless signaling. Development 129: 4271–4280. [DOI] [PubMed] [Google Scholar]
- 30. League GP, Nam SC (2011) Role of kinesin heavy chain in Crumbs localization along the rhabdomere elongation in Drosophila photoreceptor. PLoS One 6: e21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. White K, Grether ME, Abrams JM, Young L, Farrell K, et al. (1994) Genetic control of programmed cell death in Drosophila. Science 264: 677–683. [DOI] [PubMed] [Google Scholar]
- 32. Singh A, Shi X, Choi KW (2006) Lobe and Serrate are required for cell survival during early eye development in Drosophila. Development 133: 4771–4781. [DOI] [PubMed] [Google Scholar]
- 33. Johnson K, Grawe F, Grzeschik N, Knust E (2002) Drosophila crumbs is required to inhibit light-induced photoreceptor degeneration. Curr Biol 12: 1675–1680. [DOI] [PubMed] [Google Scholar]
- 34. Zipursky SL, Venkatesh TR, Teplow DB, Benzer S (1984) Neuronal development in the Drosophila retina: monoclonal antibodies as molecular probes. Cell 36: 15–26. [DOI] [PubMed] [Google Scholar]
- 35. Gunawardena S, Goldstein LS (2001) Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 32: 389–401. [DOI] [PubMed] [Google Scholar]
- 36. Richard M, Roepman R, Aartsen WM, van Rossum AG, den Hollander AI, et al. (2006) Towards understanding CRUMBS function in retinal dystrophies. Hum Mol Genet 15 Spec No 2: R235–243. [DOI] [PubMed] [Google Scholar]
- 37. Garrity PA, Lee CH, Salecker I, Robertson HC, Desai CJ, et al. (1999) Retinal axon target selection in Drosophila is regulated by a receptor protein tyrosine phosphatase. Neuron 22: 707–717. [DOI] [PubMed] [Google Scholar]
- 38. Laprise P, Beronja S, Silva-Gagliardi NF, Pellikka M, Jensen AM, et al. (2006) The FERM protein Yurt is a negative regulatory component of the Crumbs complex that controls epithelial polarity and apical membrane size. Dev Cell 11: 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nam SC, Choi KW (2003) Interaction of Par-6 and Crumbs complexes is essential for photoreceptor morphogenesis in Drosophila. Development 130: 4363–4372. [DOI] [PubMed] [Google Scholar]
- 40. Sotillos S, Diaz-Meco MT, Caminero E, Moscat J, Campuzano S (2004) DaPKC-dependent phosphorylation of Crumbs is required for epithelial cell polarity in Drosophila. J Cell Biol 166: 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, et al. (2001) Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci 21: 7551–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Richardson EC, Pichaud F (2010) Crumbs is required to achieve proper organ size control during Drosophila head development. Development 137: 641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Herranz H, Stamataki E, Feiguin F, Milan M (2006) Self-refinement of Notch activity through the transmembrane protein Crumbs: modulation of gamma-secretase activity. EMBO Rep 7: 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kimberly WT, Esler WP, Ye W, Ostaszewski BL, Gao J, et al. (2003) Notch and the amyloid precursor protein are cleaved by similar gamma-secretase(s). Biochemistry 42: 137–144. [DOI] [PubMed] [Google Scholar]
- 45. Chen CL, Gajewski KM, Hamaratoglu F, Bossuyt W, Sansores-Garcia L, et al. (2010) The apical-basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc Natl Acad Sci U S A 107: 15810–15815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ling C, Zheng Y, Yin F, Yu J, Huang J, et al. (2010) The apical transmembrane protein Crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc Natl Acad Sci U S A 107: 10532–10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robinson BS, Huang J, Hong Y, Moberg KH (2010) Crumbs regulates Salvador/Warts/Hippo signaling in Drosophila via the FERM-domain protein Expanded. Curr Biol 20: 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Richard M, Muschalik N, Grawe F, Ozuyaman S, Knust E (2009) A role for the extracellular domain of Crumbs in morphogenesis of Drosophila photoreceptor cells. Eur J Cell Biol 88: 765–777. [DOI] [PubMed] [Google Scholar]
- 49. Bulgakova NA, Knust E (2009) The Crumbs complex: from epithelial-cell polarity to retinal degeneration. J Cell Sci 122: 2587–2596. [DOI] [PubMed] [Google Scholar]
- 50. Jacobson SG, Cideciyan AV, Aleman TS, Pianta MJ, Sumaroka A, et al. (2003) Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum Mol Genet 12: 1073–1078. [DOI] [PubMed] [Google Scholar]
- 51. Tepass U (1996) Crumbs, a component of the apical membrane, is required for zonula adherens formation in primary epithelia of Drosophila. Dev Biol 177: 217–225. [DOI] [PubMed] [Google Scholar]
- 52. Dhanasekaran DN, Reddy EP (2008) JNK signaling in apoptosis. Oncogene 27: 6245–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]