

Summary
Exosomes (EXOs) are nano-sized secreted microvesicles that can function as potent endogenous carriers of adjuvant and antigens. To examine a possible role in autoimmunity for EXOs, we studied EXO-induced immune responses in non-obese diabetic (NOD) mice, an autoimmune-prone strain with tissue-specific targeting at insulin-secreting beta cells. EXOs released by insulinoma cells can activate various antigen-presenting cells to secrete several proinflammatory cytokines and chemokines. A subset of B cells responded to EXO stimulation in culture by proliferation, and expressed surface markers representing marginal zone (MZ) B cells, which was independent of T helper cells. Importantly, splenic B cells from prediabetic NOD mice, but not diabetic-resistant mice, exhibited increased reactivity to EXOs, which was correlated with a high level of serum EXOs. We found that MyD88-mediated innate TLR signals were essential for the B-cell response; transgenic B cells expressing surface immunoglobulin specific for insulin reacted to EXO stimulation, and addition of a calcineurin inhibitor FK506 abrogated the EXO-induced B-cell response, suggesting that both innate and antigen-specific signals may be involved. Thus, EXOs may contribute to the development of autoimmunity and type 1 diabetes (T1D) in NOD mice, partially via activating autoreactive MZ-like B cells.
Keywords: Autoimmunity, autoreactive B cells, exosomes, NOD, T1D
Introduction
Tissue-specific autoimmune disease such as type 1 diabetes (T1D) can be initiated following loss of tolerance to a single tissue-specific antigen and subsequent activation of its cognate T-cell clones [1, 2]. Several islet antigens including insulin, GAD65 and IA-2 are considered candidate autoantigens in T1D due to the high frequency of autoantibodies to these molecules in patients. Intriguingly, the expression of these autoantigens is not restricted to the pancreatic islets and can be expressed in thymus and peripheral lymphoid organs [3, 4]. It remains unclear whether these autoreactive T cells are activated in the periphery by the candidate islet antigens, or by unknown endogenous molecules or exogenous antigens. One plausible explanation is that processing and presentation [5–7] or modification [8] of the islet antigens in the pancreas may be different from that in thymus or lymphoid organs and thus generate high affinity peptides or peptide-MHC complexes that can activate autoreactive T cells locally in the pancreas. But, such high affinity self peptide(s) have not been identified.
Non-obese diabetic (NOD) mice are highly genetically predisposed to T1D and serve as an excellent animal model of T1D [9]. In these mice, autoreactive T cells are activated very early in life (2–3 weeks of age) in the pancreas of almost all pups, suggesting that antigenic trigger(s) arise endogenously in the pancreas. It has been suggested that beta cell apoptosis occurs early during development and may act as the trigger of an autoimmune response [10], but the process of apoptosis commonly induces tolerance rather than a Th1-biased inflammatory response. Interestingly, type I IFN has been found upregulated only in the islets of 2 week-old NOD mouse strain [11], indicating unique endogenous inflammatory trigger(s) may be produced locally. Peri-islet Schwann-like glial cells have been suggested as the early autoimmune targets [12]; however, these cells do not express the candidate diabetes-causing autoantigens, and not all lymphocyte-infiltrated islets have peri-insulitis. The initial triggering event remains unclear. Nevertheless, this event likely leads to activation of effector T cells specific for insulin, which appears the primary autoimmune target, at least in NOD mice [2].
Exosomes (EXOs) are small (30–100 nm) extracellular microvesicles that are produced by fusion of late endosomal compartments and/or multivesicular bodies with the plasma membrane. Under normal physiological conditions or in response to stress or tissue damage, a number of cell types secrete microvesicles containing RNA and/or proteins as a form of intercellular communication. Recent evidence suggests that some extracellular microvesicles, particularly EXOs, can stimulate immune responses [13]. Tumor cells actively release proinflammatory EXOs that can induce tumor-specific immunity [14, 15]; however, such responses are frequently immunosuppressive, possibly due to the induction of myeloid immune suppressor cells [16] and/or regulatory T cells [17]. Our group has pioneered the study of the immune response to EXOs in autoimmunity, a situation where pathogenic effectors, rather than regulatory T cells, are preferentially activated. We found that insulinoma-released EXOs contain innate stimuli and candidate islet antigens, and can activate autoreactive Th1 cells in prediabetic NOD mice [18]. In this continuing study, we found that EXOs can stimulate MZ-like B cells to proliferate, which does not require T-cell help, but requires MyD88-mediated innate signals and is partially dependent on antigen-specific signals. In agreement with the notion that EXOs may trigger autoimmunity, we demonstrate that prediabetic NOD mice have increased levels of endogenous serum EXOs and EXO-reactive B cells.
Results
EXOs activate a subset of splenic B cells independent of T-cell help
Both the MIN6 and NIT-1 mouse insulinoma cell lines release EXOs that were immunostimulatory to splenocytes of NOD mice. MIN6 is used for EXO production mainly because it has less spontaneous apoptosis in culture than NIT-1, and also because only MIN6 expresses GAD65 protein that can be released into the culture supernatant [18]. Previously, we found that EXOs can activate antigen-presenting cells (APCs) including B cells [18]. By tracking the EXO-induced cell division with CFSE dye, we noticed that not only CD4+ T cells, but also many CD4- cells were proliferating when cultured with EXOs (Fig. 1A). These CD4- cells were found to be B220+ B cells, which divided as early as 48 h of culture (T-cell proliferation starts at 72 h). We thus purified B220+ cells (>95%) from NOD spleens using MACS beads, and stimulated them with EXOs. The purified splenic B220+ cells responded vigorously to EXOs in culture. At the peak time of B-cell proliferation (72 h), over one third of the cells are CFSE-low dividing cells (Fig. 1B). Thus, a subset of B cells can react to EXOs in the absence of T helper cells.
Fig. 1.
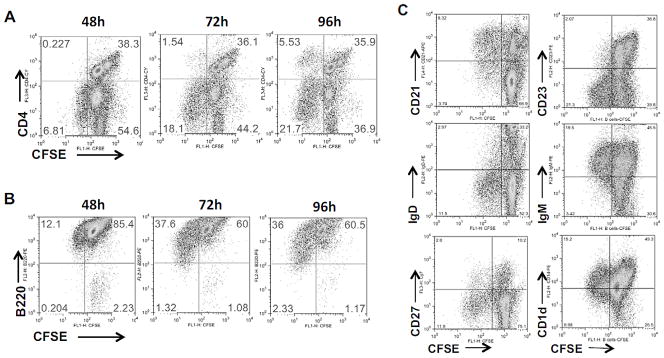
EXOs activate MZ B cells in the absence of T-cell help. (A) CFSE-labeled splenocytes (8×105/200 μl/well) of NOD females (8–10 weeks old) were stimulated with 20 μg/ml of EXOs in a flat-bottom 96-well plate. Cultured cells were harvested at different times and stained with CD4 or B220 antibodies. (B) B cells were purified from the CFSE-labeled NOD splenocytes with MACS beads coated with anti-B220, and then stimulated with EXOs. (C) CFSE-labeled splenocytes (left) or purified B cells (right) were stimulated with EXOs for 72 h, and stained with antibodies shown. Data shown are from one experiment representative of three performed.
EXO-reactive B cells express the surface markers of MZ B cells
To identify the B-cell subset(s) that contain the EXO-reactive B cells, we examined the expression of several B-cell markers on the CFSE-low, EXO-reactive B cells (Fig. 1C). These cells were IgM+/CD23-, excluding the subset of follicular B cells; they were IgD negative, which could be downregulated during the culture, and intermediate for CD1d. Thus, the majority of them could be defined as MZ B cells. Most of the dividing cells are CD21-high, but about one third of the dividing cells are CD21-low-to-intermediate (CD21-lo/int). In contrast, LPS stimulated only CD21-high MZ B cells to proliferate. About 20% of EXO-reactive cells were CD27+ (Fig. 1C), which is a marker for activated/memory B cells in humans [19]; whereas LPS activated far fewer (<3%) CD27+ cells. Therefore, the in vitro EXO-reactive B cells may include previously activated or memory B cells in addition to MZ B cells.
Increased EXO-reactive B cells and serum EXOs in NOD mice
To examine a possible correlation between the number of MZ B cells and the in vitro B-cell response to EXOs, we compared NOD and B6 mouse strains since adult NOD mice exhibit an abnormal increase in MZ B-cell numbers in comparison with B6 mice [20]. We confirmed that MZ B cells are increased in 8–12 week-old NOD mice (Fig. 2A); at the same age, B6 mice contain, on average, twofold fewer MZ B cells than NOD mice. When their splenocytes were stimulated in vitro by EXOs, the average number of EXO-reactive B cells in NOD was more than two-fold higher than that in B6 mice (Fig. 2B). In fact, serum EXO levels were found to be three-fold higher in adult (8–12 week-old) NOD mice, as measured by CD81 total protein in purified serum EXOs (Fig. 2C). These results indicate that the high levels of serum EXOs in NOD mice at the age of 8–12 week-old may cause expansion of memory-type autoreactive B cells, which can be recalled by in vitro stimulation with EXOs.
Fig. 2.
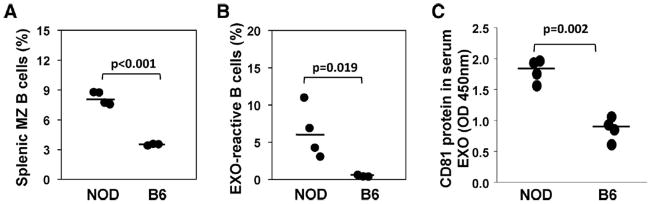
Comparison of NOD and B6 B-cell responses to EXOs. (A) Splenic MZ B cells in NOD or B6 mice (8–12 week-old) were identified as CD21-hi/CD23-lo. (B) CFSE-labeled splenocytes of NOD or B6 mice were stimulated with EXOs (20 μg/ml). After 72 h, proliferating B cells were identified as B220+/CFSE-low. Data shown are from one experiment representative of two performed. (C) Serum samples were collected from 8–12 week-old NOD or B6 mice. EXOs were precipitated using ExoQuick and EXO concentration was measured by ExoELISA and total CD81 protein in the EXO samples was presented as OD values. Each symbol represents a single mouse, bar represents the mean. Data shown are from one experiment representative of three performed. Statistical significance determined by student T-test.
Increased numbers of EXO-reactive MZ-like B cells in prediabetic NOD female mice
To further examine whether EXO-reactive B cells are associated with T1D development, we compared NOD female with NOD male mice, since only females are highly susceptible to T1D. The number of MZ B cells in 8–12 week-old NOD females is slightly higher, average 6.52% in females vs. 5.94% in males (Fig. 3A). However, the percentage of EXO-reactive B cells in the females was two-fold higher than that in age-matched males, 10.67% vs. 5.45% (Fig. 3B), suggesting an increased frequency of EXO-reactive, memory-type B cells in female mice. Among individual mice, the percentage of EXO-reactive B cells varies largely for both genders, from 2 to 16% of total splenic B cells, but the variation of MZ B-cell number in these mice was lower (from 4 to 9%). Thus, development of EXO-reactive B cells in NOD mice could not be predicted solely by the percentages of MZ B cells; additional marker(s) are required for their identification. Interestingly, 10 week-old females exhibited the strongest B-cell response to EXOs as compared with 7 or 14 week-old females (Fig. 3C), indicating a role of these EXO-reactive B cells in the early stage of the disease. The lack of response to EXOs in the older mice might be a result of the persistent high level of serum EXOs, which may lead to B-cell anergy or tolerance.
Fig. 3.
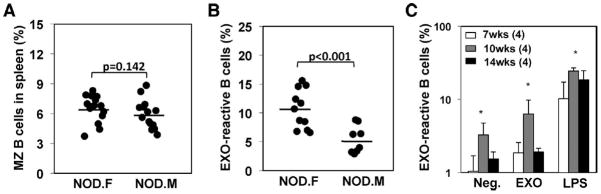
Increased EXO-reactive MZ B cells in prediabetic NOD female mice. (A) The percentages of MZ B cells in the spleens of 8–12 week-old NOD females or males were compared. (B) CFSE-labeled splencoytes of NOD females (NOD.F) or males (NOD.M) were stimulated with EXOs for 72 h, and the percentage of B220+/CFSE-low cells were shown. (A, B) Each symbol represents one mouse, bar represents mean. Data shown are pooled from three or four experiments performed. Statistical significance determined by T-test. (C) CFSE-labeled splenocytes from different ages of NOD females (4 mice per group) were stimulated with EXOs or 2 μg/ml of LPS for 72 h. The percentage of B220+/CFSE-low dividing cells are shown as mean + SD from one experiment representative of two performed. *p<0.05, T-test.
EXO immunization expands EXO-reactive B cells in young NOD mice
MZ B cells in young NOD mice (< 5 week-old) are not expanded, similar to the percentages in B6 mice. In agreement, serum level of EXOs was also undetectable in both mouse stains at this young age. As expected, the number of EXO-reactive B cells in both strains at this young age is low, less than 5%. To examine whether EXO immunization can induce specific B-cell expansion in these young mice, we injected NOD pups (one week-old) with the EXOs and examined the mice at 4 weeks of age. Fig. 4A showed that EXO-reactive B cells increased to 14.9% in the injected mice from 4.7% in control mice; however, the percentages of MZ B cells were comparable between the control and EXO-injected mice (Fig. 4B), suggesting that the early EXO immunization may induce EXO-reactive memory B cells, which may not completely exhibit MZ B-cell phenotype.
Fig. 4.
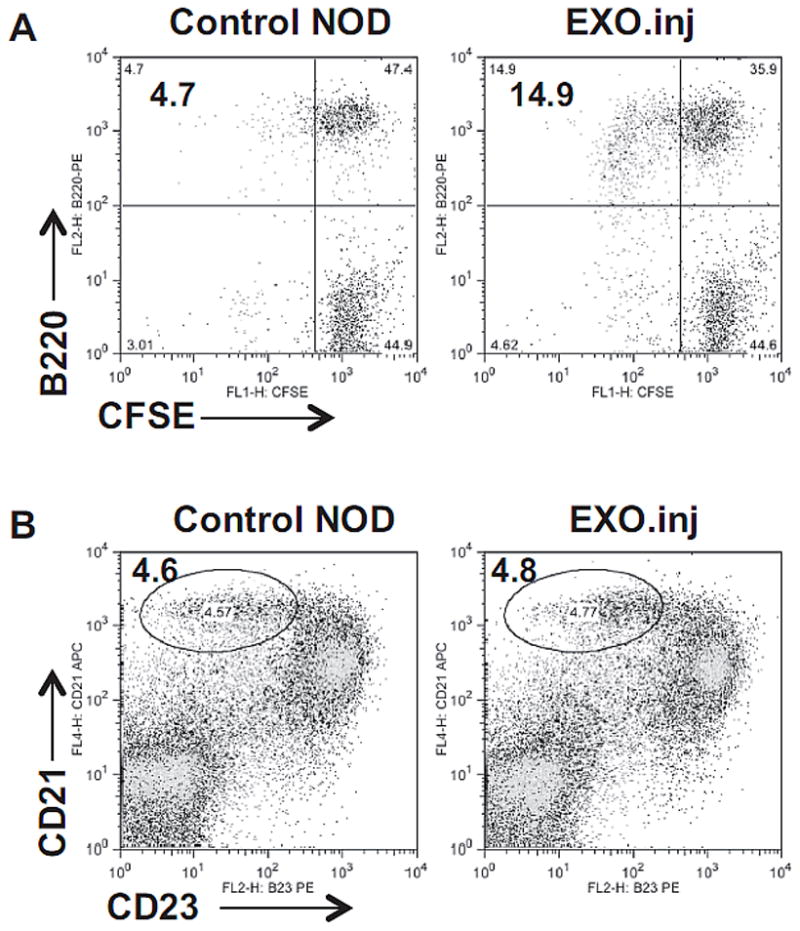
EXO immunization expands EXO-reactive B cells in young NOD mice. NOD pups (1 week-old) were injected intraperitoneally with 3 μg of EXOs in 50μl of PBS, and boosted once with 10 μg of EXOs one week after. The mice were examined at 4 weeks of age. (A) CFSE-labeled splenocytes from the injected or control mice were stimulated by EXOs. EXO-reactive B cells are shown as CFSE-low. (B) Percentages of MZ B cells in the freshly isolated splencoytes are shown. Data shown are from one experiment representative of three performed.
EXO-induced B-cell activation requires innate TLR signals
EXO-induced innate immune responses require the MyD88-mediated signaling pathway. In the absence of MyD88, NOD splenocytes failed to produce inflammation cytokines after EXO stimulation (Fig. 5A). Also, there was no B-cell proliferation in the NOD.MyD88−/− splenocytes. To address which TLR(s) are involved, we first examined Unc93b1 mutant mice. Unc93b1 is an adaptor molecule required for endosomal TLRs (TLR3, 7/8 and 9), and a single mutation 3d completely destroys Unc93a1 function and innate responses by these TLRs [21, 22]. We found that EXO-induced secretion of inflammatory cytokines and lymphocyte proliferation was intact in the 3d mutant mice (Fig. 5B&5C). This suggests that cell surface TLRs may be crucial for the EXO-induced innate response. We thus examined each of the TLR2, 3 and 4 deficient mice for their responses to EXO stimulation in 48 h cytokine secretion, 72 h CFSE-labeled B-cell division and 96 h 3H-thymidine incorporation assays. We found that both TLR2 and TLR4 partially contributed to the EXO-induced responses (Table I).
Fig. 5.

EXO-induced B-cell activation depends on MyD88 but not Unc93b1-mediated innate signaling pathways. (A) Splenocytes (8×105/200 μl/well) from 8–10 week-old female NOD or NOD.MyD88−/−mice were stimulated with EXOs. Cytokines in the 48 h culture supernatants were measured by CBA assay. (B, C) Splenocytes were collected from B6.lpr−/−/Unc93b1.WT (B6.lpr) or B6.lpr−/−/Unc93b1 3d mutant (Unc93b1-3d). (B) Cytokine release at 48 h and (C) B-cell proliferation at 72 h were examined by CBA or CFSE assays respectively. Data shown are from one experiment representative of three performed.
Table I.
TLR requirements by EXO-induced immune responses
| Immune responses | TLR2−/− | TLR3−/− | TLR4−/− | MyD88−/− | |
|---|---|---|---|---|---|
| EXO (20μl/well) | IL-6 | 21.6 | 185.5 | 52.2 | 9.2 |
| IFNγ | 12.7 | 122.3 | 31.8 | 7.0 | |
| TNFα | 12.0 | 34.5 | 22.1 | 5.3 | |
| Proliferation | (SI) | 4.8 | 7.9 | 4.0 | 1.6 |
| CFSE-low** | (%) | 4.31 | 8.86 | 7.64 | 0.34 |
Splenocytes from TLR or MyD88 deficient NOD mice were stimulated with EXOs. Cytokines in the supernatants of 48h culture were measured by CBA assay.
Proliferation of total splenocytes was monitored by incorporation of 3H-thymidine at the last 18h of total 96h culture.
CFSE-low, proliferating cells within total CFSE-labeled splenocytes were calculated after 72h of stimulation by EXOs.
Insulin-specific transgenic B cells can react to EXOs
To examine whether EXOs can stimulate antigen-specific autoreactive B cells, we tested a BCR-transgenic mouse strain (Tg125), which expresses both H- and L-chains of an insulin-specific Ig [23]. A control mouse strain (VH125) that expresses only the H-chain of the insulin-specific Ig was used. Tg125 has two-fold more MZ B cells than that in VH125 mice (Fig. 6A), whereas the number of CFSE-low/CD21+ dividing B cells was five-fold higher in Tg125 than that in VH125 (Fig. 6B). B cells from the Tg125 mice were not hyper-responsive in culture and produced a similar background as VH125 cells. MIN6-derived EXOs contain insulin protein as found in previous mass spectrometry analysis [18], which may contribute to the response of Tg125 B cells to EXOs.
Fig. 6.
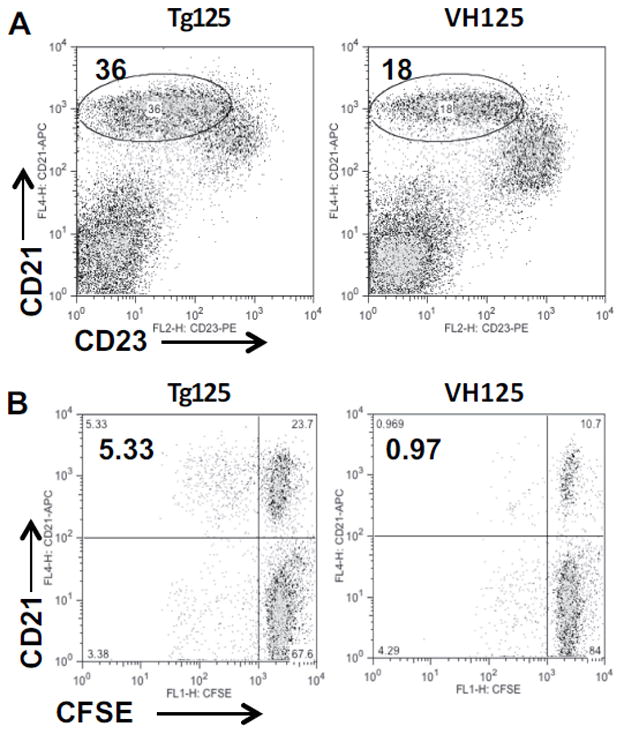
Activation of insulin-specific BCR transgenic B cells by EXOs. (A) Increased MZ B cells in transgenic mice expressing both H- and L- chains of Insulin-specific Ig (Tg125) as compared with single H-chain transgenic mice (VH125). (B) CFSE-labeled splenocytes from the Tg125 or VH125 mice were stimulated by EXOs for 72 h, followed by staining with anti-CD21 and analyzed by flow cytometry. Data shown are from one experiment representative of two performed.
FK506 inhibits the EXO-induced B-cell response
FK506 is a calcineurin inhibitor that blocks calcium flux, and can inhibit anti-IgM-induced B-cell response [24]. As shown in Fig. 7A, FK506 can inhibit the EXO-induced proliferation response. In contrast, it has no effect on LPS-induced B-cell proliferation at the same dose range (1–10 μM). At a lower dose of FK506 (200 nM), both anti-IgM and EXOs, but not LPS –induced B-cell proliferation remained sensitive to the inhibition (Fig. 7B), confirming this calcineurin-dependent activation of B cells by EXO stimulation. However, we found that phosphorylation of Syk was only induced by anti-IgM, but not by EXO stimulation (Fig. 7C), although a weak phosphorylation of ERK was detected after EXO stimulation. Alternative signaling pathway(s), independent of Syk phosphorylation, may be activated by EXOs.
Fig. 7.
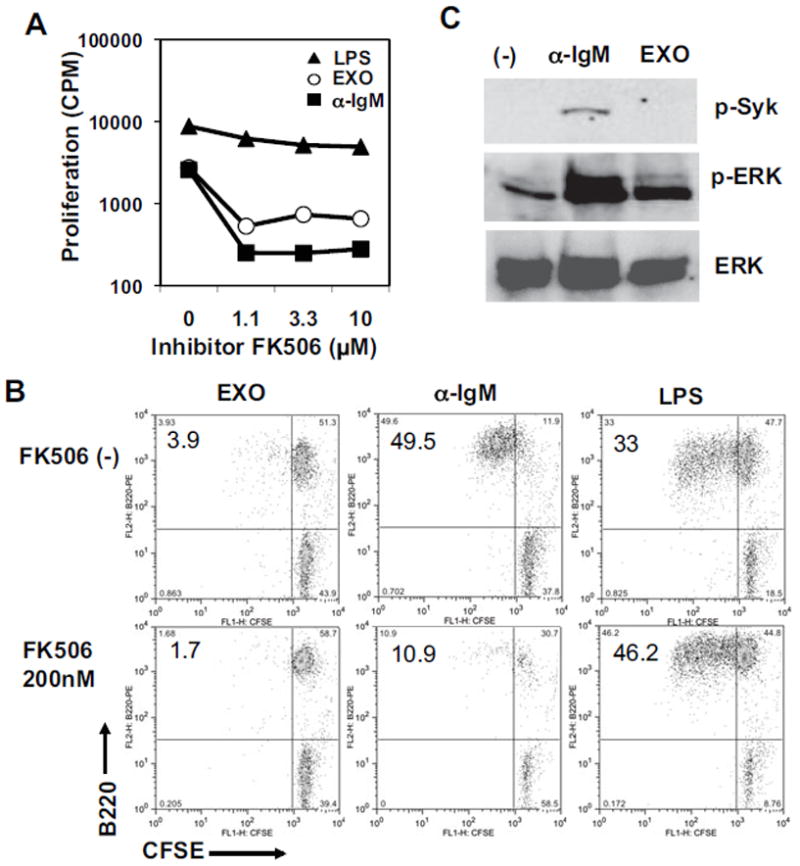
FK506 inhibits EXO-induced B-cell response. (A, B) Splenocytes from NOD females were stimulated with 20 μg/ml of EXOs, 3 μg/ml of anti-IgM, or 2 μg/ml of LPS, in the presence of FK506 in (A) a proliferation assay or (B) a CFSE-dilution assay. Similar results were observed in three experiments. (C) Splenocytes (107 cells) were stimulated by 4 μg/ml of anti-IgM for 2 minutes, or 50 μg/ml of EXOs for 30 minutes at 37ºC, followed by western blotting assay using antibodies for phosphorylated Sky (p-Syk), phosphorylated ERK (p-ERK), or total ERK kinases. Data shown are from one experiment representative of two performed.
Discussion
The reason behind the expansion of MZ B cells in the NOD mouse strain, particularly in adult females, is unknown. In this study, we found that EXOs can stimulate autoreactive MZ-like B cells. Since natural autoantibodies recognize a large array of different autoantigens [25], and are also abundant in NOD mouse strain [26], it is possible that EXOs may contain some of the autoantigens recognized by the natural autoantibodies. The interaction between exosomal antigens and the autoreactive B cells is one of low affinity since there was no phosphorylation of Syk tyrosine kinase induced; instead, it requires MyD88-mediated TLR signals. Whether this strong dependence on the innate signals is also true in vivo remains to be addressed. Nevertheless, the weak antigen-specific signals are sufficient to allow us to distinguish the NOD females from males, or diabetes-susceptible from resistant mice in their responses to EXOs, confirming an autoreactive nature of the EXO-reactive B cells that are expanded in prediabetic mice. Although it is not clear how this natural autoimmune response to a widely expressed antigen source, EXOs, leads to a highly tissue-specific autoimmune response in the pancreatic islets; with their unique adjuvant activity and expression of candidate autoantigens, EXOs appear to be an excellent tool for studying tissue-specific autoimmunity and endogenous pathways priming effector autoreactive T cells.
MZ B cells are responsible for primary immune response to T-independent antigens. They express germ-line-encoded, partially autoreactive antigen receptors and may play an important role in natural immunity and/or autoimmunity to stress-induced self antigens [27]. It has been shown that adult NOD mice have increased expansion of MZ B cells in their spleens as compared with B6 mice [28]. However, the expansion of MZ B cells in NOD mouse strain occurs only in the adult stage [20], and the percentage of MZ B cells is comparable with B6 mouse strain at a younger age (<5 week-old). This suggests that the expansion of MZ B cells is not due to dysfunction of B-cell development; instead, it is more likely that a continuous stimulation by some endogenous antigens may expand MZ B cells. It is unclear why increased MZ B cells associate with autoimmunity. One possibility is that MZ B cells may function as APCs to activate T helper cells [29]; but the antigens that stimulate MZ B cells are mostly T-independent ones, and more problematically, T cells rarely appear in the MZ area of the spleen. One related observation by Bachmann and colleagues [30] is that natural IgM antibodies that are mostly produced by MZ B cells preferentially bind to particulate, but not soluble antigens to facilitate antigen-capture by follicular dendritic cells. It might be helpful to examine the percentage of plasma B cells and the levels of serum Ig in prediabetic NOD mice if immune complex-mediated antigen-capture or APC activation is the key function attributed by the expanded MZ B cells.
Cross-linking BCR with TLRs induces functional properties of B cells that cannot be induced by engaging either receptors alone [31], suggesting a role for innate signals in breaking B-cell tolerance. TLR stimulation of B cells upregulates the expression of MHC and costimulatory molecules, and increased secretion of proinflammatory cytokines [32]. In fact, IL-6-producing B cells are the key mediators for the pathogenesis in experimental autoimmune encephalomyelitis [33], demonstrating that at least a subset of autoreactive B cells can be activated in the presence of innate stimuli. Early autoreactive B cells are believed to be low affinity and have broad antigen specificity [34, 35], which could be induced by EXOs that carry both innate and antigen specific stimuli.
With respect to the differences between female and male mice in terms of autoimmune responses, it has been shown that NOD females (>5 week-old) have increased anti-insulin titer [36], but this titer drops in older female mice [37]. Although NOD splenocytes responded to LPS three-fold stronger than B6 cells, the difference between female and male NOD mice in response to LPS stimulation was insignificant (data not shown). However, the gender difference became obvious (two-three-fold higher in the females) when EXOs were used to stimulate the splenocytes. The different response to EXOs could not be solely explained by the amount of MZ B cells since the females contain slightly higher percentages with an average of 6.52% and 5.93% for the males. More likely, the autoreactive B cell precursors or memory cells accumulated or expanded in the prediabetic females accounted for the gender difference in their response to EXOs. Interestingly, the EXO-reactive MZ B cells decreased in older female mice (14 week-old), possibly due to anergy or tolerance imposed on these B cells at the late prediabetic stage. This decrease in the B-cell response fits with the drop of anti-insulin titer in these mice [37], and also agrees with an observation by a non-invasive imaging method that islet inflammation peaks at 10 week-old, not at the onset of diabetes [38]. Likely, in 8–10 week-old female mice, the battle between effectors and regulatory cells might be accelerated, at which immune interventions would be the most effective.
We found that the serum level of EXOs is dramatically increased and persist in adult NOD but not in B6 mice, which may be responsible for expanding the MZ B cells in NOD mice. The source of serum EXOs in NOD mice is unknown, but likely do not originate from the islets; in addition to endo/epithelial cells, both T and B cells can also release EXOs to regulate immune responses [39, 40]. It remains to be examined whether the increase of serum EXOs is triggered by the initial islet autoimmune response, but it cannot be excluded that abnormal EXO production may occur systemically with or without the islet-specific autoimmune responses, possibly due to a dysregulation of EXO secretion in this autoimmune-prone mouse strain. Interestingly, Epstein-Barr virus (EBV) -transformed B lymphoma cells can release EXOs carrying the viral gp350 protein, which targets B cells specifically via binding to CD21 [41]. Thus, not only genetic abnormalities but also environmental triggers may also affect EXO secretion and the immune responses. The consequence of the increased serum EXOs to the islet-specific autoimmune response remains to be studied. It could cause expansion of autoreactive MZ B cells as we found in the spleen of prediabetic NOD mice, but a persistently high level of serum EXOs might also induce tolerance to both B and T antigens expressed by the EXOs. Additional antigenic and/or costimulatory signals may have to be provided locally within the islets in order to activate effector functions of the EXO-reactive B and T cells.
The antigens inducing the innate and the B-cell response are unknown. We found that MIN6 cells express insulin, GAD65 and several Heat shock proteins (HSP) [18]. Likely, the insulin-specific Tg125 transgenic B cells recognize insulin carried by the EXOs. HSP can stimulate TLRs to induce innate response [42]. The difficulty separating EXOs from other microvesicles such as apoptotic bodies hinders the studies specifically addressing innate or antigen-specific responses to EXOs. Considering the large number of EXO-reactive B cells in NOD female mice (over 10% in some mice), it is unlikely that all of these B cells recognize one islet antigen such as insulin; rather some common cross-reactive exosomal antigens may also attribute to the low affinity B cell recognition. It is noteworthy that phospholipid content in EXOs is unique -- EXOs contain more phophatidylserine (PS) but less phosphatidylcholine (PC) as compared with that in the cell membrane, and a higher ratio of saturated PC [43]. Also, EXO formation or budding was reduced after inhibiting sphingomyelinase [43].
Interestingly, anti-PC antibodies are known to contribute to natural immune defense, and are also present in autoimmune responses [44]; more interestingly some HIV neutralization antibodies can also cross-react to lipid antigens [45]. Future research will aim at understanding how this low affinity autoantibodies and/or autoreactive B cells participate in T-cell-mediated autoimmunity, which has much higher tissue and/or antigen specificity and is difficult to achieve tolerance.
Materials and methods
Mice
NOD/ShiLtJ and C57BL/6J (B6) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained as inbred strains at the animal facility of the Torrey Pines Institute for Molecular Studies (TPIMS). Splenocytes from NOD.MyD88−/−, NOD.TLR2−/−, NOD.TLR3−/− and NOD.TLR4−/−mouse strains were provided by Li Wen at Yale University (New Haven, CT). Splenocytes from B6.lpr−/−.Unc93b1 3d mutant and B6.lpr−/− mouse strains were provided by Dwight Kono at the Scripps Research Institute (La Jolla, CA). Splenocytes from NOD mice expressing transgenes of anti-insulin Ig H+L-double-chains (Tg125) or H-single-chain (VH125) were provided by James Thomas at Vanderbilt University (Nashville, TN). Experimental protocols were conducted with approvals from the Ethical Review Committee of TPIMS.
EXO Preparation
MIN6 insulinoma cell line [46] maintained in high glucose DMEM with 10% fetal calf serum (FCS) was used for EXO preparation following a previously described method [18]. Briefly, FCS was pre-centrifuged at 100,000 g × 90 min to remove serum EXOs and other microparticles. Culture supernatants were harvested every 2–3 days. When the cells reached confluence, trypsin–EDTA treatment was applied and one-third of the cells were inoculated for subculture. The supernatants were centrifuged at 3000 rpm × 15 min, followed by membrane filtration (0.22 μm). EXOs were collected by spinning the filtered supernatant in an ultracentrifuge (Sorvall Discovery 90SE; Hitachi) at 100,000 g × 90 min. After washing once with PBS, protein concentration was determined by Bradford protein assay (Bio-Rad, Hercules, CA). One liter of MIN6 culture supernatant yields 0.5–1.0 mg of EXOs. EXOs were examined by electromicroscope and mass spectrometry as previously described [18], and expression of exosomal signature proteins was further confirmed by flow cytometry analysis using anti-CD63/CD81-coated beads and detected by fluorescence-labeled anti-CD81 (see supplementary Fig.s1).
Flow cytometry
Flow cytometry was performed on FACSCalibur flow cytometer (BD Biosciences). Fluorescent antibodies against mouse B-cell surface markers, CD21, CD23, CD27 and IgD antibodies were purchased from BioLegend (San Diego, CA), IgM, B220/CD45R, CD4, and CD1d antibodies from BD Biosciences (San Jose, CA) or eBioscience (San Diego, CA).
CBA Cytokine Assay
A cytometric bead array (CBA)-based flow cytometry method (BD Biosciences) was used to analyze six different inflammatory cytokines or chemokines, IL-6, IL-10, MCP-1, IFN-γ, TNF-α, and IL-12p70, according to the manufacturer’s protocol with modification as previously described [18]. The concentration of each cytokines was extrapolated from the standard curves by testing with the respective recombinant proteins of the cytokines or chemokines.
Carboxyfluorescein Succinimidyl Ester (CFSE) Cell Proliferation Assay
Total splenocytes (107 cells/ml PBS) were labeled with 5 μM of CFSE (Sigma-Aldrich) at 37ºC for 10 minutes, and then cultured in a 96-well flat bottom plate at 8 × 105 cells per well in 200 μl of RPMI complete medium containing antigens. After 72 hours, the cells were harvested and stained with fluorescence-labeled B220 and CD4 antibodies to identify CFSE-low, proliferating B220+ B cells and CD4+ T cells by flow cytometry.
Measuring serum EXOs by Enzyme-linked Immuno-sorbent Assay (ELISA)
Serum EXO concentration was measured using an ExoQuick kit (System Biosciences, Mountain View, CA). Briefly, 250 μl of serum was incubated with 63 μl of ExoQuick polymer solution for 30 min to precipitate EXOs. EXO pellets were lysed by 200μl EXO-binding buffer, followed by centrifugation at 1500 g for 5 min. Then, 50 μl of the lysed supernatant were coated on Microtiter plates for overnight at 37°C. After three washes, plates were incubated with anti-CD81 for 1 h, and then HRP-conjugated secondary antibody. Substrate 3,3′,5,5′-tetramethylbenzidine was used for color reaction. The optical density (OD) was read at 450 nm. The levels of CD81 protein in serum EXOs are shown as OD values.
3H-Thymidine Incorporation Assay
Total splenocytes (6 × 105 cells/200 μl/ well) were cultured in RPMI complete medium with or without antigens in 96-well flat bottom plates for 78 h, followed by pulsing with 0.5 μCi/well 3H-thymidine (Moravek Biochemicals, Brea, CA) for 18 h. Cells were harvested with a Micro Cell Harvester (Skatron Instruments, Sterling, VA), and incorporation of [3H]-thymidine was measured on a Wallac MicroBeta Trilux counter (Perkin Elmer, Boston, MA). Stimulation indexes (SI) were calculated as counts per minute (cpm) of stimulated cells / cpm of the medium control.
SDS-PAGE and western blotting analysis
Cell lysates of stimulated splenocytes were similarly prepared and separated on SDS-PAGE as previously described [18]. After transferring the separated proteins onto a nitrocellulose membrane (Amersham, GE Healthcare Life Sciences, Piscataway, NJ), immuno-blotting was performed using 1 μg/ml of primary antibodies, followed by a respective secondary HRP-labeled anti-IgG (Amersham). The protein bands were visualized with an enhanced chemiluminescence (ECL) detection system (Amersham). Primary antibodies specific for phosphorylated Syk (Tyr323), ERK (Erk1/2) or total ERK were purchased from Cell Signaling Technology (Danvers, MA).
Supplementary Material
Acknowledgments
This work was supported by grants to Y.D.D. from the National Institutes of Health (R01DK091663) and the Diabetes National Research Group (DNRG). We thank Dr. Linda Wicker at the University of Cambridge UK and Dr. Linda Sherman at the Scripps Research Institute (San Diego, CA) for their guidance. We thank Dr. James Thomas at the Vanderbilt University, Dr. Li Wen at the Yale University (New Heaven, CT) and Dr. Dwight Kono at the Scripps Research Institute (San Diego, CA) for providing mouse strains and discussion.
Abbreviations
- T1D
type 1 diabetes
- NOD
non-obese diabetic
- MZ
marginal zone
- EXO
exosome
- TLR
Toll-like receptor
Footnotes
Conflict of interest: The authors declare no financial or commercial conflict of interest.
References
- 1.Sercarz EE. Driver clones and determinant spreading. J Autoimmun. 2000;14:275–277. doi: 10.1006/jaut.2000.0380. [DOI] [PubMed] [Google Scholar]
- 2.Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435:220–223. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pugliese A, Brown D, Garza D, Murchison D, Zeller M, Redondo M, Diez, et al. Self-antigen-presenting cells expressing diabetes-associated autoantigens exist in both thymus and peripheral lymphoid organs. J Clin Invest. 2001;107:555–564. doi: 10.1172/JCI10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gotter J, Brors B, Hergenhahn M, Kyewski B. Medullary Epithelial Cells of the Human Thymus Express a Highly Diverse Selection of Tissue-specific Genes Colocalized in Chromosomal Clusters. J Exp Med. 2004;199:155–166. doi: 10.1084/jem.20031677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quinn A, McInerney B, Reich EP, Kim O, Jensen KP, Sercarz EE. Regulatory and effector CD4 T cells in nonobese diabetic mice recognize overlapping determinants on glutamic acid decarboxylase and use distinct V beta genes. J Immunol. 2001;166:2982–2991. doi: 10.4049/jimmunol.166.5.2982. [DOI] [PubMed] [Google Scholar]
- 6.Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S A. 107:10978–10983. doi: 10.1073/pnas.1006545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med. 208:2375–2383. doi: 10.1084/jem.20111502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mannering SI, Harrison LC, Williamson NA, Morris JS, Thearle DJ, Jensen KP, Kay TW, et al. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med. 2005;202:1191–1197. doi: 10.1084/jem.20051251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wicker LS, Clark J, Fraser HI, Garner VE, Gonzalez-Munoz A, Healy B, Howlett S, et al. Type 1 diabetes genes and pathways shared by humans and NOD mice. J Autoimmun. 2005;25 (Suppl):29–33. doi: 10.1016/j.jaut.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Neonatal beta-cell apoptosis: a trigger for autoimmune diabetes? Diabetes. 2000;49:1–7. doi: 10.2337/diabetes.49.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, Lehuen A. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. 19:65–73. doi: 10.1038/nm.3042. [DOI] [PubMed] [Google Scholar]
- 12.Winer S, Tsui H, Lau A, Song A, Li X, Cheung RK, Sampson A, et al. Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat Med. 2003;9:198–205. doi: 10.1038/nm818. [DOI] [PubMed] [Google Scholar]
- 13.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 14.Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, Ricciardi-Castagnoli P, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4:594–600. doi: 10.1038/nm0598-594. [DOI] [PubMed] [Google Scholar]
- 15.Chen W, Wang J, Shao C, Liu S, Yu Y, Wang Q, Cao X. Efficient induction of antitumor T cell immunity by exosomes derived from heat-shocked lymphoma cells. Eur J Immunol. 2006;36:1598–1607. doi: 10.1002/eji.200535501. [DOI] [PubMed] [Google Scholar]
- 16.Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, Cheng Z, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. 2009;124:2621–2633. doi: 10.1002/ijc.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol. 2009;183:3720–3730. doi: 10.4049/jimmunol.0900970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheng H, Hassanali S, Nugent C, Wen L, Hamilton-Williams E, Dias P, Dai YD. Insulinoma-released exosomes or microparticles are immunostimulatory and can activate autoreactive T cells spontaneously developed in nonobese diabetic mice. J Immunol. 187:1591–1600. doi: 10.4049/jimmunol.1100231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klein U, Rajewsky K, Kuppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998;188:1679–1689. doi: 10.1084/jem.188.9.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marino E, Batten M, Groom J, Walters S, Liuwantara D, Mackay F, Grey ST. Marginal-zone B-cells of nonobese diabetic mice expand with diabetes onset, invade the pancreatic lymph nodes, and present autoantigen to diabetogenic T-cells. Diabetes. 2008;57:395–404. doi: 10.2337/db07-0589. [DOI] [PubMed] [Google Scholar]
- 21.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 22.Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, Arnold CN, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U S A. 2009;106:12061–12066. doi: 10.1073/pnas.0905441106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodward EJ, Thomas JW. Multiple germline kappa light chains generate anti-insulin B cells in nonobese diabetic mice. J Immunol. 2005;175:1073–1079. doi: 10.4049/jimmunol.175.2.1073. [DOI] [PubMed] [Google Scholar]
- 24.Wicker LS, Boltz RC, Jr, Matt V, Nichols EA, Peterson LB, Sigal NH. Suppression of B cell activation by cyclosporin A FK506 and rapamycin. Eur J Immunol. 1990;20:2277–2283. doi: 10.1002/eji.1830201017. [DOI] [PubMed] [Google Scholar]
- 25.Quintana FJ, Cohen IR. The natural autoantibody repertoire and autoimmune disease. Biomed Pharmacother. 2004;58:276–281. doi: 10.1016/j.biopha.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 26.Corte-Real J, Duarte N, Tavares L, Penha-Goncalves C. Autoimmunity triggers in the NOD mouse: a role for natural auto-antibody reactivities in type 1 diabetes. Ann N Y Acad Sci. 2009;1173:442–448. doi: 10.1111/j.1749-6632.2009.04661.x. [DOI] [PubMed] [Google Scholar]
- 27.Bendelac A, Bonneville M, Kearney JF. Autoreactivity by design: innate B and T lymphocytes. Nat Rev Immunol. 2001;1:177–186. doi: 10.1038/35105052. [DOI] [PubMed] [Google Scholar]
- 28.Rolf J, Motta V, Duarte N, Lundholm M, Berntman E, Bergman ML, Sorokin L, et al. The enlarged population of marginal zone/CD1d(high) B lymphocytes in nonobese diabetic mice maps to diabetes susceptibility region Idd11. J Immunol. 2005;174:4821–4827. doi: 10.4049/jimmunol.174.8.4821. [DOI] [PubMed] [Google Scholar]
- 29.Attanavanich K, Kearney JF. Marginal zone, but not follicular B cells, are potent activators of naive CD4 T cells. J Immunol. 2004;172:803–811. doi: 10.4049/jimmunol.172.2.803. [DOI] [PubMed] [Google Scholar]
- 30.Link A, Zabel F, Schnetzler Y, Titz A, Brombacher F, Bachmann MF. Innate immunity mediates follicular transport of particulate but not soluble protein antigen. J Immunol. 188:3724–3733. doi: 10.4049/jimmunol.1103312. [DOI] [PubMed] [Google Scholar]
- 31.Busconi L, Bauer JW, Tumang JR, Laws A, Perkins-Mesires K, Tabor AS, Lau C, et al. Functional outcome of B cell activation by chromatin immune complex engagement of the B cell receptor and TLR9. J Immunol. 2007;179:7397–7405. doi: 10.4049/jimmunol.179.11.7397. [DOI] [PubMed] [Google Scholar]
- 32.Barr TA, Brown S, Ryan G, Zhao J, Gray D. TLR-mediated stimulation of APC: Distinct cytokine responses of B cells and dendritic cells. Eur J Immunol. 2007;37:3040–3053. doi: 10.1002/eji.200636483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, Fan B, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med. 209:1001–1010. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Martin F, Forbush KA, Perlmutter RM, Kearney JF. Evidence for selection of a population of multi-reactive B cells into the splenic marginal zone. Int Immunol. 1997;9:27–41. doi: 10.1093/intimm/9.1.27. [DOI] [PubMed] [Google Scholar]
- 35.Kanayama N, Cascalho M, Ohmori H. Analysis of marginal zone B cell development in the mouse with limited B cell diversity: role of the antigen receptor signals in the recruitment of B cells to the marginal zone. J Immunol. 2005;174:1438–1445. doi: 10.4049/jimmunol.174.3.1438. [DOI] [PubMed] [Google Scholar]
- 36.Nakayama M, Castoe T, Sosinowski T, He X, Johnson K, Haskins K, Vignali DA, et al. Germline TRAV5D-4 T-cell receptor sequence targets a primary insulin peptide of NOD mice. Diabetes. 61:857–865. doi: 10.2337/db11-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abiru N, Yu L, Miao D, Maniatis AK, Liu E, Moriyama H, Eisenbarth GS. Transient insulin autoantibody expression independent of development of diabetes: comparison of NOD and NOR strains. J Autoimmun. 2001;17:1–6. doi: 10.1006/jaut.2001.0530. [DOI] [PubMed] [Google Scholar]
- 38.Fu W, Wojtkiewicz G, Weissleder R, Benoist C, Mathis D. Early window of diabetes determinism in NOD mice, dependent on the complement receptor CRIg, identified by noninvasive imaging. Nat Immunol. 13:361–368. doi: 10.1038/ni.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang GJ, Liu Y, Qin A, Shah SV, Deng ZB, Xiang X, Cheng Z, et al. Thymus exosomes-like particles induce regulatory T cells. J Immunol. 2008;181:5242–5248. doi: 10.4049/jimmunol.181.8.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qazi KR, Gehrmann U, Domange Jordo E, Karlsson MC, Gabrielsson S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood. 2009;113:2673–2683. doi: 10.1182/blood-2008-04-153536. [DOI] [PubMed] [Google Scholar]
- 41.Vallhov H, Gutzeit C, Johansson SM, Nagy N, Paul M, Li Q, Friend S, et al. Exosomes containing glycoprotein 350 released by EBV-transformed B cells selectively target B cells through CD21 and block EBV infection in vitro. J Immunol. 186:73–82. doi: 10.4049/jimmunol.1001145. [DOI] [PubMed] [Google Scholar]
- 42.Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. 120:457–471. doi: 10.1172/JCI40483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- 44.Shaw PX, Goodyear CS, Chang MK, Witztum JL, Silverman GJ. The autoreactivity of anti-phosphorylcholine antibodies for atherosclerosis-associated neo-antigens and apoptotic cells. J Immunol. 2003;170:6151–6157. doi: 10.4049/jimmunol.170.12.6151. [DOI] [PubMed] [Google Scholar]
- 45.Matyas GR, Wieczorek L, Beck Z, Ochsenbauer-Jambor C, Kappes JC, Michael NL, Polonis, et al. Neutralizing antibodies induced by liposomal HIV-1 glycoprotein 41 peptide simultaneously bind to both the 2F5 or 4E10 epitope and lipid epitopes. AIDS. 2009;23:2069–2077. doi: 10.1097/QAD.0b013e32832faea5. [DOI] [PubMed] [Google Scholar]
- 46.Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, et al. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.