

Abstract
Dopamine D2 receptors (D2R) are G protein–coupled receptors that modulate synaptic transmission and play an important role in various brain functions including affect learning and working memory. Abnormal D2R signaling has been implicated in psychiatric disorders such as schizophrenia. Here we report a new function of D2R in dendritic spine morphogenesis. Activation of D2R reduces spine number via GluN2B– and cAMP–dependent mechanisms in mice. Notably, this regulation takes place only during adolescence. During this period, D2R overactivation caused by mutations in the schizophrenia–risk–gene dysbindin leads to spine deficiency, dysconnectivity within the entorhinal–hippocampal circuit and impairment of spatial working memory. Notably, these defects can be ameliorated by D2R blockers administered during adolescence. These findings uncover a novel age–dependent function of D2R in spine development, provide evidence that D2R dysfunction during adolescence impairs neuronal circuits and working memory, and suggest that adolescent interventions of aberrant D2R activity protect against cognitive impairment.
D2R belongs to the D2–like (D2, D3 and D4 type) subfamily of dopamine receptors. D2R Dysfuntions has long been recognized and targeted for the therapy in schizophrenia, a debilitating mental disorder1. An increase in D2R density is consistently found in schizophrenic brains1, and all antipsychotics antagonize D2R2. Genetic studies also show that some genes associated with increased risks of schizophrenia encode proteins that regulate D2R, such as dysbindin which controls trafficking of D2R to the cell surface3, 4.
Although effective for psychosis, treatment with D2R antagonist has little effect on cognitive impairment, a core symptom of schizophrenia and a major determinant of disability2, 5. Proper synaptic connections are essential for cognition. In schizophrenic brains, however, interneuronal connections are impaired6. In both the prefrontal cortex and hippocampus of schizophrenic patients, for instance, there is a reduction in the number of dendritic spines7–10, small dendritic protrusions accomodating most excitatory synapses in the brain11. Also, neurons derived from iPS (induced pluripotent stem) cells from schizophrenic patients exhibit severe impairments of interneuronal connections12. The pathogenic mechanisms underlying synaptic dysconnectivity, however, is still largely unknown.
D2R activation is coupled to a number of signaling pathways. By coupling with Gi/o proteins, activated D2R negatively regulates the cAMP–PKA pathway13. Activated D2R also induces formation of the β–arrestin–2/Akt/protein phosphatase 2A signaling complex13. In hippocampal, cortical, and striatal neurons, brief activation of D2R inhibits currents mediated by N–methyl–D–aspartate receptors (NMDAR)14–17. Alteration of synaptic transmission is often accompanied by structural modification of synapses, such as formation, elimination and morphological changes of dendritic spines. Whether D2R regulates the structure of synapses, however, has not been experimentally tested.
Here we show that D2R modulates morphogenesis of dendritic spines in hippocampal neurons via GluN2B– and cAMP–dependent mechanisms. Intriguingly, D2R regulates spines only during postnatal week 3–6,, and in mice with deficient expression of the schizophrenia–risk–gene dysbindin, adolescent D2R hyperactivity causes a reduction in spine number. Importantly, even transient suppression of spine development during adolescence by hyperactive D2R adversely affects the entorhinal cortex–hippocampal connectivity and working memory in adulthood. These findings identify a novel function of D2R in spine development and a critical period during which dendritic spines are regulated by D2R.
RESULTS
D2R regulates the morphogenesis of dendritic spines
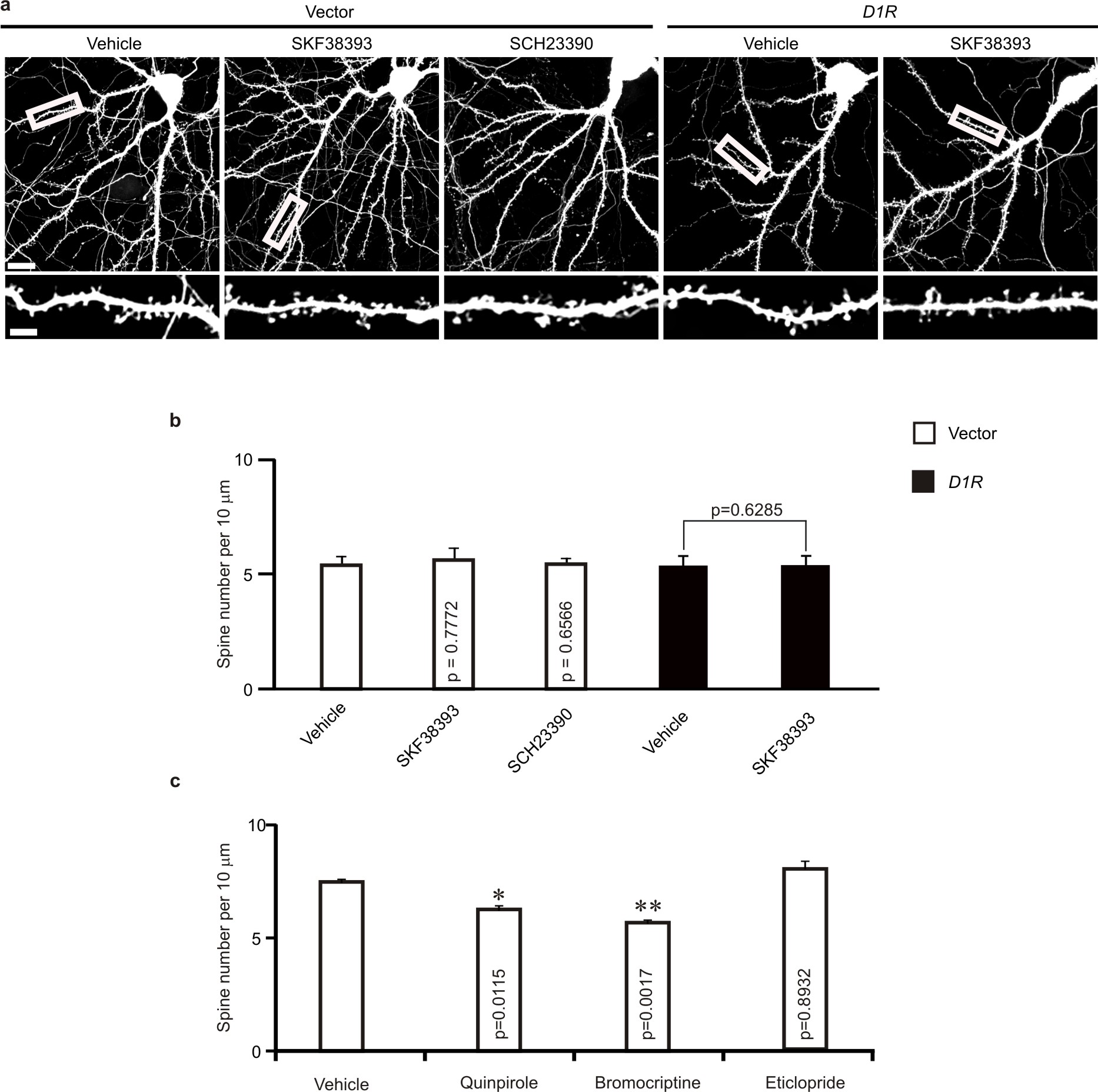
To determine whether D2R regulates the development of neuronal connections, we intraperitoneally injected male C57BL/6 mice (P21, during a period of active spine growth and synaptogenesis18) with vehicle, the D2R agonists quinpirole or bromocriptine, or the D2R antagonist eticlopride. Hippocampal slices were prepared at 24 h after injection for diolistic labeling of neurons. Notably, in mice injected with D2R agonists, spine density of hippocampal CA1 neurons was reduced (Fig. 1a, c). Spine density of CA1 neurons was unaffected by a single injection of eticlopride (data not shown), but was significantly increased by 5 daily injections of eticlopride (Fig. 1a, c). Thus, D2R activation inhibits spine development, while prolonged blockade of D2R activity promotes it.
Fig. 1.

D2R regulates spine development in hippocampal neurons in vivo. Hippocampal slices were prepared for diolistic labeling from 3–week–old C57BL/6 mice intraperitoneally injected with vehicle or with agonists or antagonists of D2R and D1R (a, c), or 4–week–old C57BL/6 mice injected with lentivirus expressing EGFP, D1R, D2R, D1R siRNA or D2R siRNA (b, d, e, f). (a, b) Representative images of DiI–labeled (a) or virus transduced (b) basal dendrites of hippocampal CA1 neurons. (c) Quantification of spine density for (a). (d) Quantification of spine density for (b). n = 3 mice for each condition, and 15 neurons from 3 slices of each animal were imaged for spine analysis in (c) and (d). (e) Sample trace of mEPSCs. (f) Analysis of mEPSC frequency; 5–8 slices from 3–5 animals were used for each condition; n = the total number of recorded neurons. Quinpirole: 0.5 mg/kg; bromocriptine: 10 mg/kg; eticlopride: 0.5 mg/kg; SKF38393: 1 mg/kg; SCH23390: 1 mg/kg. Data are presented as mean ± SEM. Scale bars, 5 μm. Two–tailed Mann–Whitney test was used to calculate p–values.
To test whether the D1–like subfamily of dopamine receptors (D1R–like) also regulate spine development, we injected vehicle, the D1R–like agonist SKF38393 or the D1R–like antagonist SCH23390 into P21 male C57BL/6 mice, and prepared hippocampal slices for diolistic labeling at 24 h after injection. Spine density of CA1 neurons was comparable in mice injected with these chemicals (Fig. 1a, c). Thus D1R–like receptors do not regulate spine development in hippocampal neurons.
To corroborate the results of our pharmacological experiments, we overexpressed or knocked down D2R in vivo. The CA1 region of male C57BL/6 mice (P21) were injected with lentivirus expressing enhanced green fluorescent protein (EGFP) along with siRNAs against D2R (D2R siRNA–1) or D1R (Supplementary Fig. 1a–e), or co–injected with the EGFP virus and the D1R or D2R virus. At 7 d after injection, brain sections were prepared from injected mice. In CA1 pyramidal neurons transduced with D2R virus, spine density was reduced, while in those transduced with D2R siRNA virus, it was increased (Fig. 1b, d). Transduction of virus expressing D1R or D1R siRNA, by contrast, left the spine number intact (Fig. 1b, d). These results confirm the findings from our pharmacological experiments.
The change in spine number may affect synaptic transmission. To test this possibility, we measured miniature excitatory postsynaptic currents (mEPSCs) in mice injected with the D2R or D2R siRNA lentivrus. While mEPSC amplitude was not changed by overexpressing or knocking down D2R, mEPSC frequency (which positively correlates with synapse number) was reduced in D2R virus transduced, but increased in D2R siRNA virus transduced neurons (Fig. 1e, f; Supplementary Fig. 1f). The change in mEPSC frequency is consistent with that in spine number in viral injected mice.
Taken together, these results indicate that D2R activation inhibits spine development.
D2R regulates maturation and growth dynamics of spines
Dendritic spines are generally categorized into three groups: mushroom spines with a large head and a constricted neck, thin spines with a small head and a long neck and stubby spines without constriction between the tip and the neck19. Mushroom and thin spines are the major type of spines in adult brains, while stubby spines are primarily found in immature neurons18, 20. To determine the effect of D2R activation on spine morphology, we conducted a detailed spine analysis in primary hippocampal neurons.
To visualize spines, we transfected neurons at 14 days in vitro (DIV14) with a construct expressing Venus (a mutant of yellow fluorescent protein21). At 3 d after transfection, neurons were treated with D2R agonists quinpirole or bromocriptine, the D2R antagonist eticlopride, the D1R–like agonist SKF38393 or the D1R–like antagonist SCH23390 for 24 h. Consistent with our in vivo results, the density of total spines (including mushroom, thin and stubby spines) was not changed by treatment with the D1R–like agonist or antagonist (Supplementary Fig. 2a, b), but was decreased in both quinpirole and bromocriptine treated cells (Supplementary Fig. 2c). Unlike in vivo, treatment with the D2R antagonist eticlopride did not affect spine density in vitro (Supplementary Fig. 2c), likely because of the lack of dopamine neurons and hence endogenous D2R activity in cultured hippocampal neurons.
We further analyzed the effect of D2R agonists on different types of spines and also on filopodia, thin and pointy dendritic protrusions that may be spine precursors22, 23. Due to confocal microscopy’s limited spatial resolution, we classfied all spines with a narrow neck and a head into a combined “mushroom/thin” spine group. Notably, quinpirole and bromocriptine treatment reduced the density of mushroom/thin spines and increased filopodium density, but did not affect stubby spine density (Fig. 2a, b). The neck of mushroom/thin spines in neurons treated with quinpirole or bromocriptine, moreover, was elongated (Fig. 2a, c).
Fig. 2.

D2R regulates the morphogenesis and growth dynamics of dendritic spines in cultured hippcampal neurons. Cultured hippocampal neurons were transfected with the the Venus or indicated constructs at DIV14 and imaged at DIV17. (a) Representative images of transfected neurons (top) and dendrites at a higher magnification (bottom). (b, c) Quantification for (a). (d) Representative images of cultured hippocampal neurons. (e, f) Quantification for (d). (g). Representative time–laspse images of neurons (top) and dendrites at a higher magnification (bottom) before (0′) and 60 min (60′) after quinpirole treatment. (h) Quantification for the number of added and retracted spines (normalized to the total number of spines) during the 60–min–treatment period. (i) Quantification for the conversion between different types of dendritic protrusions during the 60–min–imaging period. M: mushroom spines; T: thin spines; F: filopodia; S: stubby spines. The results were replicated in three independent experiments. Histograms show one of the three replicates (n = 15 neurons for each condition). Scale bar, 20 μm for images of neurons and 5 μm for images of dendrites. Data are presented as mean ± SEM. Two–tailed Mann–Whitney test was used to calculate p–values for comparison to vehicle treated cells in (b, c, h, i) and between vehicle and quinpirole treated cells transfected with the same construct in (e, f). Quinpirole: 1 μM; bromocriptine: 2.5 μM; eticlopride: 1 μM.
To confirm that the effects of quinpirole and bromocriptine on spines are mediated by D2R, we transfected neurons (DIV14) with a construct expressing D2R siRNA along with the Venus construct. At 3 d after transfection, we treated neurons with quinpirole for 24 h and analyzed dendritic protrusions in transfected neurons. Only protrusions that did not contact axons of transfected neurons were analyzed. While transfection with the D2R siRNA construct did not influence spine density, spine dimension, filopodium density, or quinpirole–induced elevation of filopodium density, it did abolish quinpirole’s effects on both the density and length of mushroom/thin spines (Fig. 2d–f).
To test whether the effects of siRNAs are caused by D2R knockdown, we co–transfected neurons with the D2R siRNA construct and a construct expressing D2R with silent mutations in the siRNA binding region making it resistant to D2R siRNA (D2R–M, Supplementary Fig. 1a, b). The effect of quinpirole on spines was restored in co–transfected cells (Fig. 2d–f), confirming that quinpirole reduces spine density by activating D2R. Because only D2R in postsynaptic neurons were knocked down, moreover, these results indicate that postsynaptic D2R mediates quinpirole’s effects on mushroom/thin spines (but not on filopodium).
D2R overexpression alone enhanced quinpirole’s effects on spine density (Fig. 2d–f), suggesting that the level of D2R positively correlates with the effect size of quinpirole. Spine density in neurons overexpressing D1R, however, was not affected by D1R agonist SKF38393 (Supplementary Fig. 2a, b). Thus the ineffectiveness of SKF38393 on spines is not due to inadequate D1R on hippocampal neurons.
Since long spines and filopodia are usually found in immature neurons18, their increase by D2R agonist suggests that D2R activation inhibits spine maturation. To test this possibility, we imaged the same neurons (DIV17) before and 1 hr after quinpirole treatment. Quinpirole treatment increased the rate of both spine addition and retraction and the conversion of mushroom/thin spines to filopodia, but reduced the conversion of filopodia to mushroom/thin spines (Fig. 2g–i). Thus D2R activation enhances the dynamics of spine growth, inhibits the conversion of dendritic protrusions from immature to mature appearance, and destabilizes mature spines. Taken together, our results from both in vivo and in vitro experiments demonstrate that D2R activation restricts spine number during spine development by inhibiting spine maturation.
D2R regulates spines via GluN2B and cAMP
To explore the mechanisms by which D2R regulates spines, we first tested whether D2R activation alters synaptic protein expression. Primary hippocampal neurons (DIV17) were treated with quinpirole for 24 h, then stained for the AMPA receptor subunits GluA1 and GluA2, and the presynaptic protein Bassoon and Synaptophysin. Expression of these proteins was not affected by quinpirole (Supplementary Fig. 3a, b).
We then tested whether NMDAR is involved in the regulation of spines by D2R, as NMDAR is required for spine maturation in hippocampal neurons11, 23. We treated primary hippocampal neurons with quinpirole along with the NMDAR antagonist (2R)–amino–5–phosphonovaleric acid (D, L–AP5). While AP5 did not affect either spine density or size, it did abolish quinpirole–induced changes (Fig. 3a–c). For D2R to inhibit spine morphogenesis, therefore, NMDAR activity is required.
Fig. 3.

GluN2B is required for D2R–mediated regulation of dendritic spines. (a–f) Cultured hippocampal neurons transfected with the Venus construct were treated with vehicle or chemicals as indicated. Representative images of transfected neurons (top) and dendrites at a higher magnification (bottom) are shown in (a) and (d). (b, c) Quantification for (a). (e, f) Quantification for (d); the results were replicated in three independent experiments; histograms show one of the three replicates (n = 15 neurons for each condition). (g, h) Hippocampal slices were prepared from C57BL/6 mice (P21) intraperitoneally injected with chemicals as indicated and diolistically labeled. Representative images are shown in (g), and quantification is in (h); n= 15 neurons from 3 slices of 3 animals for each condition. (i, j) Cultured cortical neurons (DIV17) were treated with quinpirole, then harvested for immunoblotting. Representative cropped immunoblots for GluN1, GluN2A and GluN2B are shown in (i); the full–length images are shown in Supplementary Fig. 7a, b. (j) Quantification for (i); n = 3 experiments for each condition. Scale bar, 20 μm for images of neurons and 5 μm for images of dendrites. Data are presented as mean ± SEM. Two–tailed Mann–Whitney test was used to calculate p–values for comparison between cells treated with indicated chemicals and those treated with the same chemical and quinpirole. Quinpirole: 1 μM; AP5: 50 μM; Ifenprodil: 3 μM; Ro 25–6981: 1μM; TCN201: 10 μM.
To determine how NMDAR assists D2R to regulate spines, we tested whether prolonged D2R activation (elicited in our experiments) modulates NMDAR–mediated synaptic transmission, as brief D2R activation does15–17, 24. We analyzed evoked NMDAR–mediated excitatory postsynaptic currents (EPSCNMDA) in hippocampal slices taken from 3–week–old mice intraperitoneally injected with quinpirole, bromocriptine or vehicle. EPSCs were recorded from CA1 neurons by stimulating the Schaffer collateral pathway. The input–output relationship of EPSCNMDA was indistinguishable between vehicle– and D2R–agonist–injected mice (Supplementary Fig. 3c). Thus prolonged D2R activation does not alter NMDAR–mediated currents.
In hippocampal neurons, NMDAR is composed of GluN1, GluN2A and GluN2B subunits25. To determine whether a specific NMDAR subunit mediates D2R’s effects on spines, we treated primary hippocampal neurons (DIV17) with quinpirole along with the GluN2A antagonist TCN201 or the GluN2B antagonist ifenprodil or Ro 25–6891. While ifenprodil and Ro 25–6891 obliterated quinpirole–induced changes in dendritic protrusions, TCN201 did not (Fig. 3d–f). Likewise, quinpirole–induced spine reduction was abolished in mice (male, C57BL/6, P21) intraperitoneally injected with ifenprodil or Ro 25–6891, but not TCN201 (Fig. 3g, h). Thus GluN2B, but not GluN2A, cooperates with D2R to control spine number.
To determine how GluN2B assist D2R to regulate spines, we first tested whether chronic D2R activation, like acute D2R activation16, induces GluN2B dephosphorylation at Ser1303. The level of phosphorylated GluN2B was comparable in quinpirole treated and control cells (Supplementary Fig. 3d–e, 7c). Likewise, after prolonged D2R activation, phosphorylation of GluA1 at Ser845, which is also reduced by acute D2R activation26, remained unchanged (Supplementary Fig. 3d, e).
We next examined whether D2R activation influences GluN2B expression. Total GluN2B expression and that of GluN1 and GluN2A was indistinguishable between quinpirole treated and control cells (Fig. 3i, j). A surface biotinylation assay, however, showed that quinpirole treatment greatly reduced surface GluN2B, but not GluN1 or GluN2A (Fig. 3i–j, Supplementary 7a, b).
To confirm the result of the biotinylation assay, we transfected cultured hippocampal neurons (DIV17) with GFP–tagged GluN2B and GluN2A, treated them with quinpirole for 24 h, and stained surface–expressed, GFP–tagged NMDAR with an anti–GFP antibody. On the surface of quinpirole–treated cells, we detected normal GluN2A but less GluN2B (Supplementary Fig. 3f–i). This change was likely due to excessive GluN2B endocytosis, because it was obliterated by treatment with dynasore, a dynamin inhibitor that blocks GluN2B endocytosis17 (Supplementary Fig. 3h, i). GluN2B antagonist ifenprodil and Ro 25–6891 also blocked quinpirole–induced decrease in surface GluN2B (Supplementary Fig. 3h, i). To test whether the change in surface GluN2B is required for the effects of quinpirole on spines, we treated hippocampal neurons (DIV17, transfected with the Venus construct for 3 d) with quinpirole along with dynasore. The effect of quinpirole on spines was abolished by dynasore (Supplementary Fig. 3j, k). Hence, for D2R to regulate spine development, GluN2B internalization is required.
Since D2R activation reduces cAMP formation and Akt activity13, we also tested whether these molecular changes contribute to the effects of D2R activation on spines. We treated primary hippocampal neurons (DIV17, 3 d after transfection with the Venus construct) with quinpirole along with the adenyl cyclase activator forskolin or the PI3K activator 740 Y–P. While forskolin blocked quinpirole–induced changes in dendritic protrusions, 740 Y–P did not (Fig. 4). Treatment with either forskolin or 740 Y–P alone did not affect dendritic protrusions (Fig. 4). Hence, D2R regulates spine development through the cAMP but not the Akt cascade. Taken together, these results show that to affect spine development, both GluN2B internalization and cAMP down–regulation are required.
Fig. 4.

cAMP mediates D2R’s effects on spine density. Cultured hippocampal neurons transfected with the Venus construct were treated with vehicle or chemicals as indicated. (a) Representative images of transfected neurons (top) and dendrites at a higher magnification (bottom). (b, c) Quantification for (a). The results were replicated in three independent experiments. Histograms show one of the three replicates (n = 15 neurons for each condition). Scale bar, 20 μm for images of neurons and 5 μm for images of dendrites. Quinpirole: 1μM; forskolin: 50 μM; 740Y–P: 50 μg/ml. Two–tailed Mann–Whitney test was used to calculate p–values for comparison between cells treated with indicated chemicals and those treated with the same chemical and quinpirole. Data are presented as mean ± SEM.
D2R–dependent spine deficiency in dysbindin mutant mice
Disturbances of the density and functions of D2R are consistently found in the brains of schizophrenics1. To test whether D2R pathology contributes to spine abnormalities, we examined dendritic spines in sandy (Sdy) mice, which harbor a spontaneous deletion in the schizophrenia–risk–gene dysbindin, express no dysbindin protein, and have increased D2R on the cell surface3, 4. We analyzed spines in both cultured hippocampal neurons and slices from sandy mice. While the density of mushroom/thin spines was decreased in primary hippocampal neurons from sandy mice (DIV17, 3 d after transfection with the Venus plasmid), that of filopodia was increased, and mushroom/thin spines were enlongated (Fig. 5a–c). Likewise, in the CA1 region of sandy mice injected with lentivirus expressing EGFP (P28, 1 week after injection), spine density was less than in their wild–type (WT) littermates (Fig. 5d, e).
Fig. 5.

Spine deficiency in sandy mice is caused by D2R hyperactivity. (a–c) Primary hippocampal neurons were prepared from wild–type and sandy mice, transfected with the Venus construct at DIV14, and imaged at DIV17. Representative images of transfected neurons and dendrites at a higher magnification are shown in (a), and quantification is shown in (b, c); the results were replicated by three independent experiments, and one of the three replicates (n = 15 for each condition) is shown in the histograms. (d–i) At 3 weeks of age, sandy mice and their wild–type littermates were injected with lentivirus expressing EGFP or D2R siRNA, or intraperitoneally injected with vehicle or forskolin (4 mg/kg), then used for preparation of hippocampal slices for spine analysis (d, e), mEPSC analysis (f, g) or diolistic labeling (h, i). (d, h) Representative images of basal dendrites from CA1 neurons. (e) Quantification of spine density for (d); 15 neurons from 3 slices of each animal and 3 mice for each condition were imaged and analyzed. (i) Quantification of spine density for (h); 15 neurons from 3 slices of each animal and 3 mice for each condition were imaged and analyzed. (f) Sample trace of mEPSCs. (g) Quantificaiton for (f); 5–8 slices of 3–5 animals for each condition; n= the total number of recorded neurons. Scale bar, 20 μm for images of neurons and 5 μm for images of dendrites. Data are presented as mean ± SEM. Mann–Whitney test was used for statistical analysis.
To test whether elevated D2R activity in sandy mice contributes to the alteration in spines, we injected lentivirus expressing D2R siRNA into the CA1 region of sandy mice. Spine density was increased in transduced cells (Fig. 5d, e). Thus the reduction in spine density in sandy mice stems from overactive D2R.
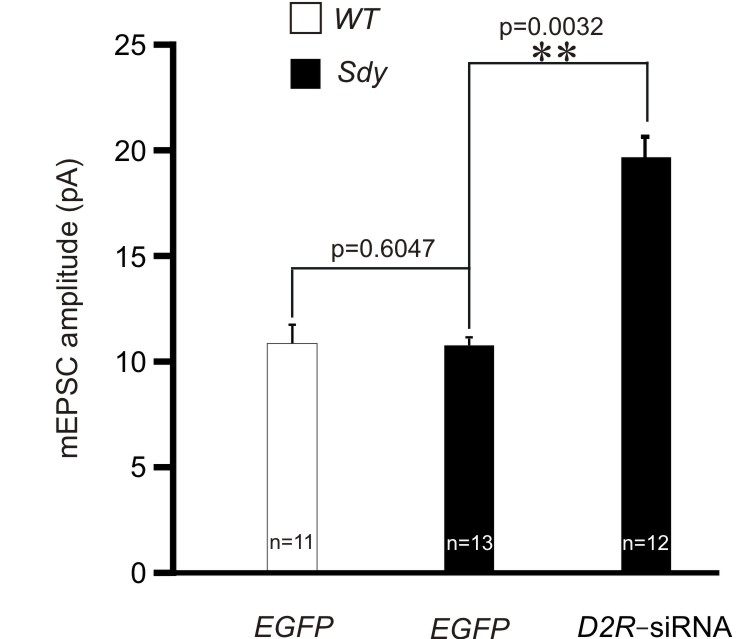
Since mEPSC is affected by D2R activation in wild–type mice (Fig. 1e, f), we tested whether it is also changed in sandy mice. mEPSCs were recorded in the CA1 region of hippocampal slices. In sandy mice, mEPSC frequency was reduced, while mEPSC amplitude was comparable to that in wild–type mice (Fig. 5f, g; Supplementary Fig. 4). These results are consistent with previous findings from sandy mice27. The change in mEPSC frequency in sandy mice was abolished by injecting lentivirus expressing D2R siRNA (Fig. 5f, g), indicating that it is caused by D2R overactivation. Moreover, D2R knockdown in sandy mice caused an increase in mEPSC amplitude (Supplementary Fig. 4). This effect is likely due to the intereaction of D2R with other molecules or signaling pathways altered in sandy mice, because in wild–type mice, D2R knockdown did not change mEPSC amplitude (Supplementary Fig. 1f).
Consistent with elevated surface expression of D2R in sandy mice4, the level of cAMP in the hippocampus of sandy mice was reduced by 30.0% ± 10.6 (p = 0.045). Moreover, enhancing cAMP production by injecting forskolin rescued the spine deficiency in sandy mice (Fig. 5i, j). Forskolin injection did not change spine density in wild–type mice (Fig. 5i, j). These findings suggest that cAMP reduction in sandy mice contributes to spine defects.
Taken together, these results suggest that overactivation of D2R caused by mutations of the dysbindin gene impairs spine development.
D2R regulates spine number in an age–dependent manner
To test whether D2R also regulates spine number in mature hippocampal neurons, we injected vehicle or quinpirole into 2– to 12–week–old male C57BL/6 mice, and prepared hippocampal slices for diolistic labeling at 24 h after injection. In vehicle–injected mice, spine density of CA1 neurons showed an upward trajectory from 2 to 8 weeks of age, then dropped slightly at 12 weeks (Fig. 6a, b). Notably, quinpirole injection caused a reduction of spine density only in mice aged 3–6 weeks, but not in those aged 2, 8, or 12 weeks (Fig. 6a, b). To confirm the effect of quinpirole in adult mice, we injected 8–week–old C57BL/6 mice with lentivirus expressing EGFP alone or along with D2R, and analyzed spines in hippocampal slices prepared from injected mice 1 week after injection. Spine density was unaffected by overexpressing D2R in adult mice (Supplementary Fig. 5a, b). These results indicate that D2R regulates spine number in an age–dependent manner.
Fig. 6.

The critical period for D2R to regulate dendritic spine number and reversal of the spine deficiency by antipsychotics treatment in sandy mice. Hippocampal slices for diolistic labeling were prepared from C57BL/6 mice of various ages intraperitoneally injected with vehicle or with the D2R agonist 0.5 mg/kg of quinpirole (a, b), sandy mice and their wild–type littermates of various ages (c, d), or sandy mice and their wild–type littermates at 3 weeks of age intraperitoneally injected with vehicle, 5 mg/kg of loxapine or 8 mg/kg of clozapine (e, f). Representative images of DiI–labeled basal dendrites of hippocampal CA1 neurons are shown in (a, c, e). Quantification of spine density is shown in (b, d, f). 15 neurons from 3 slices of each animal were imaged and analyzed. n = 3 mice for each condition. Scale bar, 5 μm. Data are presented as mean ± SEM. Mann–Whitney test was used for comparison between wild–type and sandy mice in (b, d), and between vehicle and drug treated mice of the same genotype in (f).
Next, we examined whether spine deficiency in sandy mice is also age–dependent. While spine density of CA1 neurons was lower in 3–6–week–old sandy mice than in their wild–type littermates, at ages 2, 8, and 12 weeks, spine density in the two groups was comparable (Fig. 6c, d). To test whether spines in adult sandy mice are also regulated by D2R as in adolescent sandy mice (Fig. 5d, e), we injected lentivirus expressing D2R siRNA along with EGFP into the CA1 region of 8–week–old sandy mice, and analyzed spines in hippocampal slices at 1 week after injection. Spine density was comparable in virus–injected and control mice (Supplementary Fig. 5a, b). Thus, the effect of genetic activation of D2R on spines is also age–dependent.
As all antipsychotics are D2R blockers28, we tested whether antipsychotic treatment can ameliorate the spine deficiency in sandy mice. We injected the typical antipsychotic loxapine (a potent D2R blocker)29, 30 and the atypical antipsychotic clozapine (a potent serotonin receptor antagonist and a weak D2R blocker)30, 31 into sandy mice and their wild–type littermates at P21. Hippocampal slices were prepared at 24 h after injection for diolistic labeling of neurons. Remarkably, loxapine injection, which had no effect on spine density in wild–type mice, increased spine density of CA1 neurons in sandy mice to a level comparable to that in their wild–type littermates (Fig. 6e, f). Clozapine injection, by contrast, had no effects on spine density in either sandy or wild–type mice (Fig. 6e, f). These results indicate that typical antipsychotics acting on D2R, but not atypical antipsychotics with less affinity for D2R, can rescue spine defects in sandy mice if applied during the period when spines are sensitive to D2R activity.
Taken together, these findings indicate that the effect of D2R activation on dendritic spines is age–dependent, and that adolescence is a critical period when D2R hyperactivity reduces spine number.
GluN2B determines the age–dependency of D2R’s function
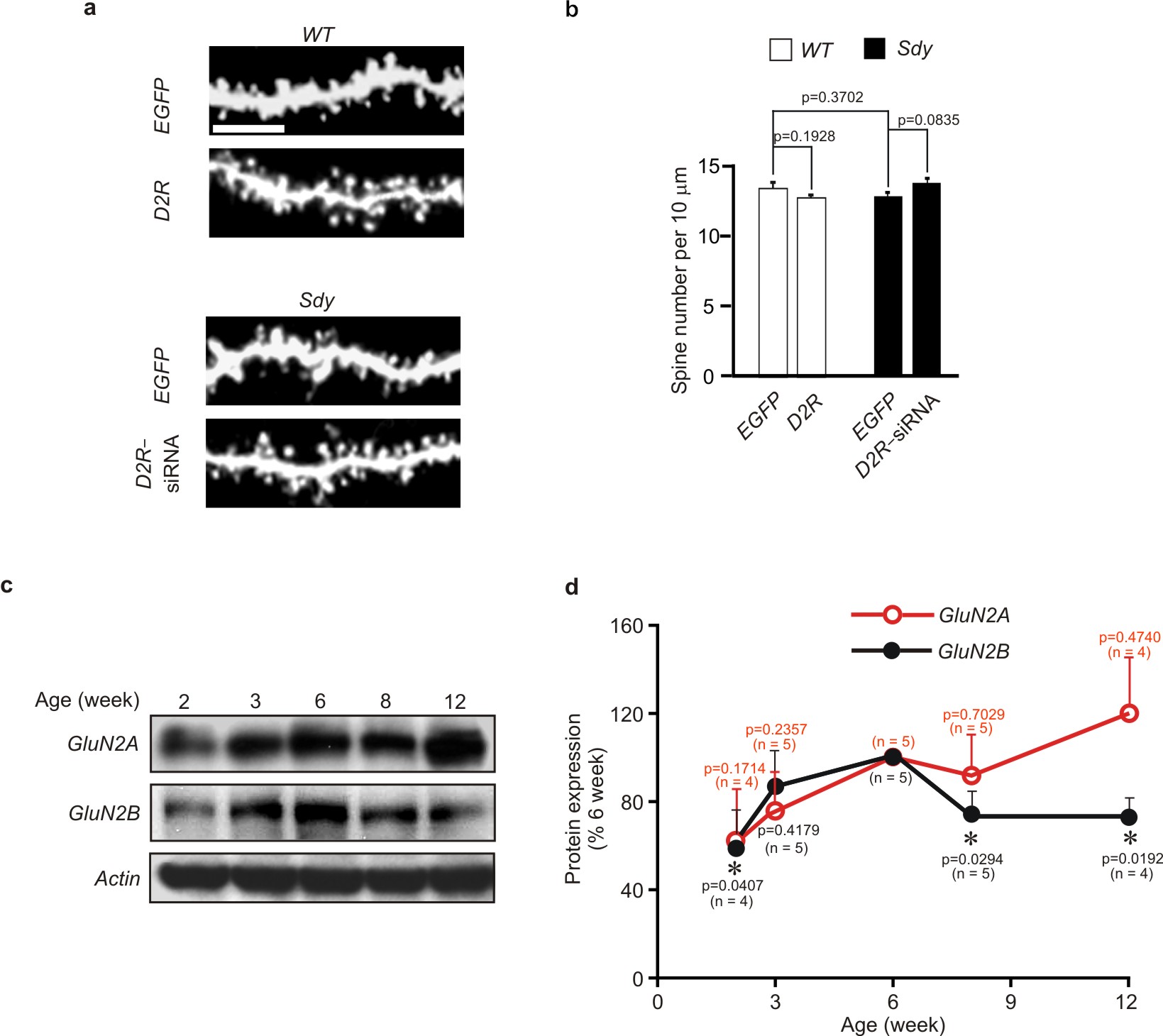
During brain development, NMDAR in the forebrain is switched from predominant GluN2B–containing to GluN2A–rich receptors25. Since we found that GluN2B is required for D2R–mediated spine regulation, we hypothesized that the developmental change in NMDAR composition might account for the fact that D2R no longer affects spine number in mature brains. To test this possibility, we first analyzed the temporal pattern of GluN2B expression in the hippocampus. Immunoblotting of hippocampal lysates showed that GluN2B was decreased and GluN2A was increased in adulthood (Supplementary Fig. 5c–d, 7d).
To test whether the changes in GluN2A and GluN2B expression influence the effects of D2R activation on spines, we tranfected young hippocampal neurons (DIV18, expressing more GluN2B and less GluN2A) with GluN2A, and mature hippocampal neurons (DIV 56, expressing less GluN2B and more GluN2A) with GluN2B. While mature neurons no longer changed spine density following quinpirole treatment, overexpression of GluN2B restored their ability to respond to quinpirole (Fig. 7a–c). By contrast, deviating GluN2A expression from its normal developmental trajectory, either by overexpressing GluN2A in young neurons, or by blocking GluN2A acivity with TCN201 in mature neurons, left quinpirole’s effects on spines unchanged (Fig. 7). These results indicate that it is the decrease in GluN2B rather than the increase in GluN2A expression that makes mature neurons unable to change spines following D2R activation.
Fig. 7.

The age–dependency of D2R–mediated regulation of dendritic spines requires the developmental change in GluN2B expression. Cultured young (DIV18) or mature (DIV56) hippocampal neurons were transfected with the Venus construct alone or along with GFP–GluN2A or GFP–GluN2B and treated with quinpirole (1 μM) alone or along with GluN2A inhibitor TCN201 (10 μM). (a) Represenative images of mature neurons. (b, c) Quantification for (a). (d) Representative images of young neurons. (e, f) Quantification for (d). n = 15 neurons for each condition. Scale bar, 20 μm for images of neurons and 5 μm for images of dendrites. Data are presented as mean ± SEM. Mann–Whitney test was used for comparison between vehicle and quinpirole treated cells.
Adolescent D2R hyperactivity impairs neural connectivity
Although spine density returns to normal in adult sandy mice which experienced spine loss during adolescence due to D2R hyperactivity, given that adolescence is a period for the refinement and maturation of neuronal circuits32, 33, perturbation of dendritic spines during this period may permanently alter these circuits. To test this possibility, we examined the entorhinal cortex (EC)–CA1 connection, as neurons in the layer III of the EC send mono–synaptic projections to CA1 neurons34, 35.
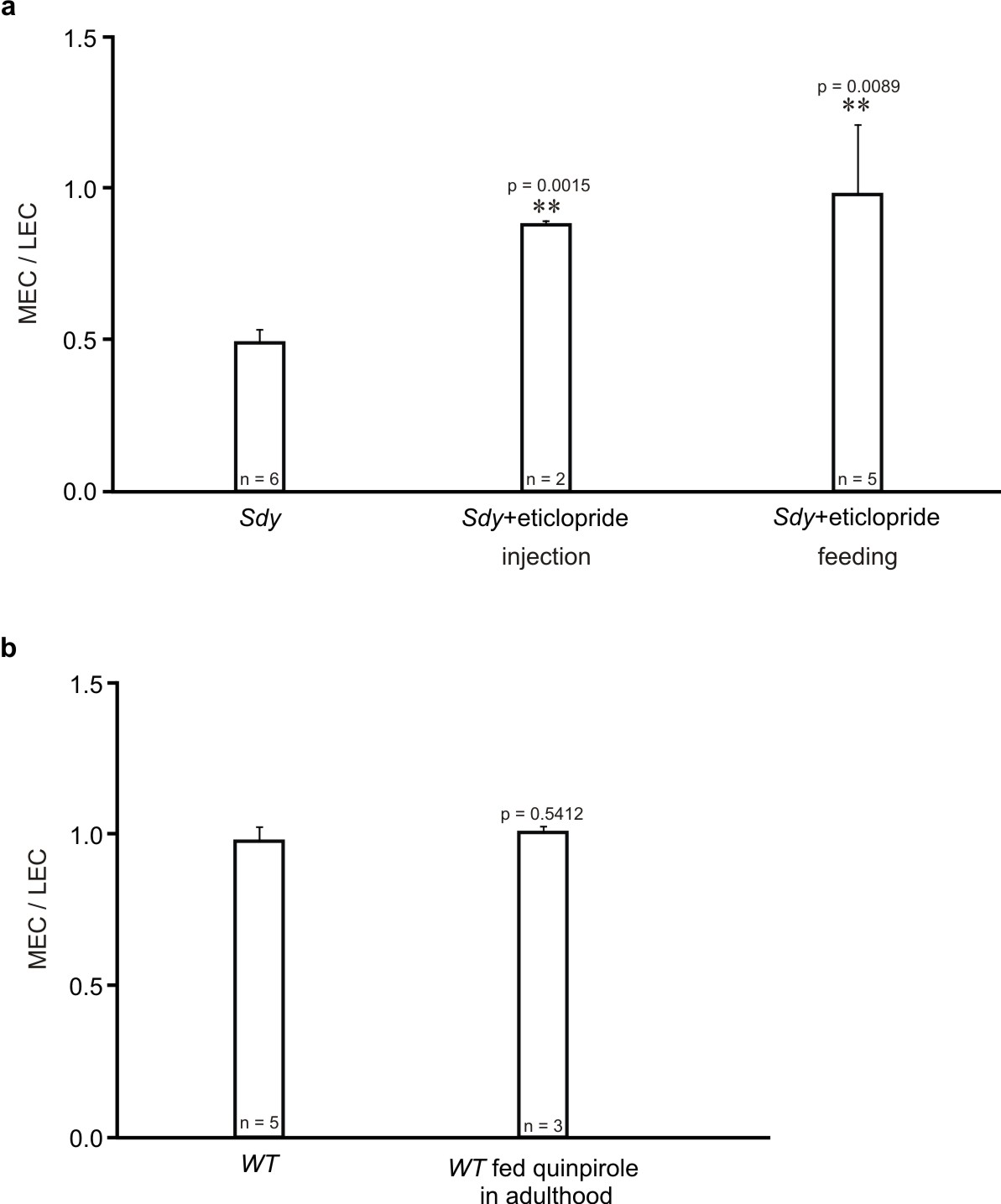
The retrograde tracer cholera toxin subunit B (CTB) was injected into the CA1 region of the right hippocampus. At 24 h after injection, horizontal brain sections containing the EC were prepared. In wild–type mice, neurons retrogradely labeled by CTB were found in both the medial EC (MEC) and lateral EC (LEC) (Fig. 8a). By contrast, in sandy mice the number of labeled neurons was drastically reduced in the MEC, but increased in the LEC, and thereby the ratio of labeled MEC to LEC neurons was decreased (Fig. 8a–c). To test whether these changes are caused by D2R hyperactivity during adolescence, sandy mice were intraperitoneally injected with eticlopride from P21 to P35 (once daily), and injected with CTB at 8 weeks of age. Notably, eticlopride treatment restored the ratio of labeled MEC to LEC neurons (Fig. 8a, b). We also tested the effect of feeding sandy mice water supplemented with eticlopride (ad libitum from P21 to P35). Since feeding and injecting eticlopride had comparable effects on the ratio of labeled MEC to LEC neurons (Supplementary Fig. 6a), we merged the data from the injected and fed groups (Fig. 8b, c).
Fig. 8.

D2R hyperactivity during adolescence impairs the entorhinal–hippocampal circuit and spatial working memory. Sandy mice were treated with eticlopride (5 μg/ml) during either adolescence (P21–35) or adulthood (P56–70). Wild–type mice were treated with quinpirole (2.5 μg/ml) from P21 to P28. At 3–4 weeks after treatment, mice were used for the retrograde tracing experiment and behavioral tests. (a) Representative images of retrogradely labeled neurons in the EC. Scale bar, 100 μm. (b) The ratio of retrogradely labeled MEC to LEC neurons. (c) The number of retrogradely labeled neurons in each section (150 μm in length along the MEC–LEC axis) of the EC normalized to the total number of labeled neurons in the EC. 0 in the x–axis indicates the border between the MEC and LEC. n = 5 mice for each condition in (b) and (c). (d) Alternation scores from the Y–maze test. (e) Total distance traveled during 30 min in the open field test. Pooled data from two independent experiments are shown in (d) and (e). Data are presented as mean ± SEM. Mann–Whitney test was used for statistical analysis. p–values less than 0.05 for comparison between untreated and eticlopride–fed sandy mice (blue), between sandy and wild–type mice (yellow), and between untreated and quinpirole–fed wild–type mice (orange) are shown in (c); p–values less than 0.05 for comparison between sandy and wild–type mice are shown in (e); the complete list of p–values for (c, e) is provided in Supplementary Table 1.
To test whether the effect of eticlopride on neuronal connectivity in sandy mice is age–dependent, we fed adult sandy mice eticlopride from P56 to P70, and injected them with CTB at 13 weeks of age. The ratio of labeled MEC to LEC neurons was comparable in treated and untreated sandy mice (Fig. 8a–c). Hence, eticlopride treatment in adolescence but not in adulthood can correct connectivity within the EC–hippocampal circuit in sandy mice. These findings indicate that adolescent D2R hyperactivity causes perturbations of adult neural circuits.
To corroborate the results obtained with sandy mice, we examined the effect of adolescent D2R hyperactivity on the EC–hippocampal connection in wild–type mice. Wild–type mice were fed qunipirole from P21 to P28, and injected with CTB at 8 weeks of age. Quinpirole treatment caused a decrease in the number of retrogradely labeled cells in the MEC, and in the ratio of labeled MEC to LEC neurons (Fig. 8a–c, Supplementary Table 1). By contrast, quinpirole treatment in adult mice (from P56 to P63 and injected with CTB at 12 weeks of age) did not change the ratio of labeled MEC to LEC neurons (Supplementary Fig. 6b). These results confirm that adolescent D2R hyperactivity adversely affects the EC–hippocampal connectivity.
Taken together, these findings indicate that adolescent D2R hyperactivity disturbs the establishment of adult patterns of neural connectivity, and that treatments need to be administrated during adolescence to alleviate this effect.
Adolescent D2R hyperactivity impairs working memory
The EC–hippocampal circuit is essential for spatial working memory34, 36–38. To test whether D2R hyperactivity affects spatial working memory, we conducted behavioral tests with sandy mice fed eticlopride and wild–type mice fed quinpirole during adolescence. At 8 weeks of age, we conducted the spontaneous Y–maze test to analyze spontaneous spatial working memory, and the open field test to measure locomotor activity. Wild–type and sandy mice given untreated water were used as controls. In the Y–maze test, sandy mice had a lower alternation score, corresponding to poorer spatial working memory, than wild–type control mice (Fig. 8d). The performance of wild–type mice fed quinpirole in the Y–maze was also reduced (Fig. 8d), suggesting that overactivation of D2R impairs working memory. Intriguingly, the alternation score of sandy mice was improved by eticlopride treatment during adolescence (Fig. 8d).
In the open field test, sandy mice traveled longer distances than wild–type mice (Fig. 8e), consistent with their reported hyperactivity39, 40. Eticlopride treatment during adolescence, however, did not change sandy mice’s locomotor activity (Fig. 8e). For wild–type mice, locomotion tested at 4 weeks after feeding with quinpirole during adolescence was comparable to that in untreated mice (Fig. 8e, Supplementary table 2), suggesting that hyperactivity might be caused by acute but not long–lasting effects of D2R activation on brains.
To test whether eticlopride treatment in adult sandy mice can also improve working memory, we fed 8–week–old sandy mice eticlopride–supplemented water for 2 weeks, and conducted behavioral tests at 13 weeks of age. In contrast to the improvement seen when eticlopride was applied in adolescence, working memory performace was not changed (Fig. 8d). Hyperactivity in sandy mice, by contrast, was ameliorated by eticlopride treatment in adulthood (Fig. 8e), excluding the possibility that eticlopride treatment in adulthood was ineffective on behavior.
Taken together, these findings indicate that overactivation of D2R during adolescence causes impairments of spatial working memory in adulthood. Blocking D2R during this critical period in sandy mice, moreover, improves their spatial working memory thereafter.
DISCUSSION
In this study, we uncovered an age–dependent function of D2R in controlling spine development. We provided evidence that D2R hyperactivity during the critical period when spines are subject to regulation by D2R impairs the establishement of the entorhinal–hippocampal circuit and working memory in adult mice.
Our results from a series of pharmacological, molecular genetic, and gene knockout experiments consistently confirmed that overactivation of D2R during adolescence impairs spine development, neural circuits and working memory. We excluded D2R and D1R knockout mice from our study for fear that their retarded growth, reduced body weight, smaller brains, and shorter dendrites in cortical neurons41–44 indicate global developmental defects.
The effect of intraperitoneal injection of D2R agonist on spine density of hippocampal neurons could be caused by activation of D2R on postsynaptic neurons, on presynaptic neurons or in extra–hippocampal areas. Our results from transfection of D2R siRNA into postsynaptic hippocampal neurons in vitro and D2R overexpression or knockdown in vivo indicate that the spine phenotype is caused by D2R on postsynaptic neurons. Filopodia, however, might be regulated by presynaptic D2R, because postsynaptic D2R knockdown does not change D2R agonist–induced filopodium overgrowth.
Our results indicate that for D2R to induce spine changes, NMDAR, particularly GluN2B, is required. Unlike brief D2R activation, however, chronic D2R activation does not affect NMDAR–mediated currents or GluN2B phosphorylation. It is D2R activation–induced GluN2B endocytosis that is required for the effect of D2R on spines. In addition to GluN2B, we show that forskolin blocks the effect of D2R activation on spines, consistent with the known role of cAMP in spine morphogenesis45.
In both wild–type and sandy mice, we found that D2R regulate spines only during postnatal week 3–6, the period of active spine growth, synaptogenesis, and adolescent development in mice46. Moreover, we found that it is the developmental decrease in GluN2B expression that makes spines in mature brains no longer change in respons to D2R activaiton. Regulation of spines by D2R during adolescence has robust effects on the shaping of neuronal circuits. These effects are retained and manifested in adulthood. Adolescence precedes early adulthood, the usual age of onset for schizophrenia in humans47. By extrapolation, D2R–dependent spine morphogenesis may play an important role in the pathophysiology of schizophrenia.
Our finding that D2 blockers administered during adolescence in mice can prevent D2R hyperactivity–induced impairments in spine development and working memory suggests that young human subjects with D2R hyperactivity, perhaps from mutations in the dysbindin gene, might benefit from prodromal medication that targets D2R to protect spine development and cognition. Further studies in humans are needed to test this possibility.
METHODS
Animals, plasmids and reagents
All animal procedures followed the NIH Guidelines “Using Animals in Intramural Research” and were approved by the National Institutes of Mental Health Animal Care and Use Committee. Four mice were housed in one cage in a room with a 12h light/dark cycle. Sandy mice which had been backcrossed with the C57BL/6 mice for more than 10 generations were purchased from the Jackson Laboratory. Animals were intraperitoneally injected with various drugs at a volume of 10 ml/kg. Annealed oligos containing siRNA sequences targeted to rat D2R (D2R siRNA–1: 1138′–CTCGGTGTGTTCATCATCT–1156′— a region conserved between rats and mice; D2R siRNA–2: 1269′–CCCCATCATCTACACCACC–1; D2R siRNA–3: 530′–CAGACCAGAATGAGTGTAT–548′) and mouse D1R (1075′–GAGACTGTAAGCATCAACA–1093′), and the cDNAs of myc–tagged D1R and HA–tagged D2R were inserted into the pSuper and the pRRLsin lentiviral vectors. cDNAs for D2R, Beclin1, ATG5 and siRNA–resistant D2R (C1140G, G1146T, C1149T, C1155T, generated by mutagenesis using the KOD kit, Novagen) were cloned into the pGW1 vector behind the HA sequence. The N–terminal GFP – tagged GluN2A and GluN2B constructs are kind gifts from Dr. Katherine Roche’s lab. The D1R cDNA was obtained by PCR from a construct purchased from Origene and cloned into the pGW1 vector behind the myc tag. The following reagents were obtained commercially: quinpirole, eticlopride, loxapine, ifenprodil, Ro 25–6981, dynasore and anti–actin antibody (1:10000, Cat# A 4700) from Sigma; bromocriptine, SKF38393, SCH23390, forskolin, 740 Y–P, clozapine and TCN201 from TOCRIS; anti–HA antibody (1:100, Cat# MMS–101P), anti–GluN2A(1:500, Cat# 07–63248), anti–synaptophysin (1:1000, Cat# AB927249), anti–pGluA1–ser845 (1:1000, Cat# 04–107350), anti–pGluN2B–ser1303 (1:1000, Cat# 07–398) and anti–GluA1 (1:500, Cat# PC24651) antibody from Millipore; anti–GluN1 antibody from BD Pharmingen (1:500, Cat# 55630852); anti–GluN2B antibody from Upstate (1:1000, Cat# 06–60048); anti–Bassoon antibody from Assay designs (1:1000, Cat# VAM–PS00353); anti–GluA2 antibody from Chemicon (1:1000, Cat# MAB39753).
Acute hippocampal slices
Mice were decapitated after anesthetization with an overdose of isoflurane. The brain was placed in ice–cold artificial cerebrospinal fluid (ACSF, pH 7.4, bubbled with 95% O2/5% CO2), which consisted in mM of 124 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 CaCl2 1.3 MgSO4, and 10 D–glucose. Transverse hippocampal slices were sectioned in ice–chilled, oxygenated ACSF with a Leica VT1000S vibratome.
Diolistic labeling
Diolistic labeling was performed as reported54. Briefly, immediately after sectioning, 200 μm thick acute hippocampal slices were fixed in PBS containing 4% formaldehyde and 4% sucrose for 30 min. After rinse, the fixed slices were shot with DiI–coated tungsten bullets by using a Bio–Rad gene gun at 50–100 psi, kept in PBS overnight and mounted for imaging. DiI was purchased from Sigma (Cat# D282)
Electrophysiology
Hippocampal slices prepared from 3–4–week–old mice were placed in a chamber perfused with 95% O2/5% CO2 bubbled ACSF (30°C) at the rate of 2 ml/min. The stimulating electrode was placed on the stratum radiatum in the CA2 area, and the recording electrode was placed on the cell body of CA1 neurons. Stimuli were delivered to the electrode at an interval of 20 s. Bicuculline (10 μM) was added to the ACSF to block GABAA receptors. The patch pipette (3–5 MΩ) solution was composed of (in mM) 130 cesium methanesulfonate, 8 NaCl, 4 Mg–ATP, 0.3 Na–GTP, 0.5 EGTA, 10 HEPES and 5 QX–314 at pH 7.3. EPSCNMDA was recorded at a holding potential of +40 mV. EPSCNMDA was measured as the peak amplitude of EPSCs in the presence of the AMPA receptor blocker NBQX (20 μM), or measured at 50–70 ms after the peak of EPSCAMPA without NBQX55, 56. Since EPSCNMDA measured with the NBQX and the kinetics method was comparable, we pooled data from both methods for statistical analysis. For mEPSC analysis, action potentials were blocked by adding 1 μM tetrodotoxin (TTX) to ACSF. The series resistance and input resistance were monitored on–line and analyzed with the Clampex program off–line. Only cells with a series resistance of < 25 MΩ and a < 10% drift in both series resistance and input resistance during the recording period were included.
Primary hippocampal neuron culture, transfection and immunocytochemistry
Hippocampal and cortical neuron cultures were prepared from embryonic day 18–19 rat embryos as previously described56. Hippocampal neurons were seeded on coverslips coated with 30 μg/ml of poly–D–lysine and 5 μg/ml of laminin at a density of 330 cells/mm2. Cortical neurons were seeded on plates coated with 30 μg/ml poly–D–lysine at a density of 650 cells/mm2. Neurons were grown in Neurobasal medium (Invitrogen) supplemented with 2% B27 and 1% glutamax. Neurons were transfected with Lipofectamine 2000 (Invitrogen). For immunocytochemistry of synaptic proteins, neurons were fixed with 4% formaldehyde in PBS containing 4% sucrose, and incubated with primary antibodies in blocking buffer containing 0.3% Triton X–100 overnight at 4°C, followed by staining with Alex 488– or Alex 555–conjugated secondary antibodies. For surface staining of GFP–GluN2A/GluN2B, neurons were incubated with an anti–GFP antibody in blocking buffer without Triton X–100 overnight at 4 °C, then incubated with an Alex 488–conjugated secondary antibody to label surface GFP–GluN2A/GluN2B. After rinse, cells were permeabilzed and incubated with the anti–GFP antibody again at room temperature for 2 h, then with an Alex 555–conjugated secondary antibody to label intracellular GFP–GluN2.
cAMP assay
Hippocampi were removed from the mouse brain and homogenized with a motor–driven homogenizer for 20 strokes in the homogenization buffer provided by the cAMP assay kit (R&D Systems Cat# KGE002B). The homogenates were sonicated with a Virsonic cell disrupter (Model 16–850) and measured for protein concentration. 100 μg proteins were used for each cAMP assay according to the manufacture’s manual.
Surface biotinylation assay
Primary cortical neurons were cultured on 10–cm plates and treated with quinpirole at DIV17. At 24 h after treatment, neurons were chilled on ice, incubated with 0.25 mg/ml EZ–link sulfo–NHS–LC–LC–Biotin for 45 min on ice. After rinse with ice–cold PBS (containing 0.1 mM Ca2+, 4 mM Zn2+), extra EZ–link sulfo–NHS–LC–LC–Biotin was quenched with 10 mM glycine. Neurons were washed with cold PBS, lysed in RIPA buffer and analyzed for protein concentration. Cell lysates containing the same amout of proteins were incubated with streptavidin for 2 h at 4°C and centrifuged. Precipitates were washed three times with RIPA buffer, resuspended in 2× SDS–PAGE gel–loading buffer, and boiled at 95 °C for 5 min for immunoblotting. Image J was used for quantification of immunoblots.
Image acquisition and dendritic spine analysis
Confocal images were acquired by using a Zeiss LSM510 confocal microscope with a 63 X (NA1.4) objective for cultured neurons and an Olympus FV1000 confocal microscope with a 60 X (NA 1.35) objective for hippocampal slices and time–lapse imaging. Images were collapsed into 2D projections with LSM 5 browser and Olympus fluoview version 2.1c viewer and analyzed with Metamorph software (Molecular Devices) for spine density, spine dimension and integrated fluorescence intensity of stained protein. Images were acquired at 1024×1024 p.p.i. resolution. 2–3 secondary or tertiary dendrites (50–200 μm long, 20–100 μm away from the soma) from each neuron were analyzed for spine analysis. Dendrites, the width of spine head (the diameter of spine heads’ largest section that is perpendicular to spine necks) and the length of spine neck (from the bottom of spine heads to the junction between spines and dendritic shafts) were traced manually and measured by Metamorph. The number of dendritic protrusions were counted manually. Images were taken and analyzed “blind” to the treatment.
Reverse transcription polymerase chain reaction (RT–PCR)
Cultured cortical neurons were lysed in Trizol. The mRNA was extracted and transcribed into cDNAs with the oligo(dT) 20 primer. The following primers (D1R, 5′ primer: CATTCTGAACCTCTGCGTGA, 3′ primer: GTTGTCATCCTCGGTGTCCT; D2R, 5′ primer: CATTGTCTGGGTCCTGTCCT and 3′ primer: GACCAGCAGAGTGACGATGA; Actin: 5′ GCTCCTCCTGAGCGCAAGTACTC, 3′ CTCATCGTACTCCTGCTTGCTG) were used for RT–PCR.
Brain injection and sectioning
Mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg), and mounted in a stereotaxic apparatus. 1 μl lentivirus or 200 nl CTB–Alexa555 (1 mg/ml, Invitrogen) was injected into the CA1 area of the right hippocampus using a Hamilton syringe (1.8 mm caudal to bregma, 1.5 mm lateral to the midline, 2.1mm below the surface of the cerebral cortex). At 7 d (for lentivirus injection) or 24 h (for CTB injection) after injection, mice were perfused with 4% paraformaldehyde in PBS and cut coronally (for spine analysis) or horizontally (for CTB tracing), using a cryostat, into 50 μm sections. Brain sections were mounted onto slides in mounting media with DAPI (for CTB injection) or without DAPI (for lentivirus injection), and imaged with a Zeiss confocal microscope (63× for lentivirus injection and 10× for CTB inection).
Behavioral tests
Mice for behavior tests were housed under a reversed 12 hr light/dark cycle and tested during the dark phase. Animals were transferred to the test room at 1 hr before the behavioral test for acclimatization. The behavioral apparatus was cleaned with 70% ethanol and dried with paper towels after each trial. For the Y–maze test, mice were placed at the end of one of the three arms of a Y–maze with three identical arms which were 120° apart, and allowed to explore the arena freely for 8 minutes. The number and sequence of arm entries were recorded manually. A consecutive entry into 3 different arms was counted as an alternation. The alternation score was computed with the formula: [(total number of alternations)/(total number of arm entries)–2]×100. Mice staying in one arm for longer than 2 consecutive minutes or with less than 12 arm entries were excluded from the statistical analysis. The open field test was conducted at one day after the Y–maze test. Mice were placed in the center of the test chamber (W49 × D49 × H40 cm) illuminated at 20 lux, and allowed to explore the chamber for 30 min. Mice were videotaped during the test. The total travel distance was analyzed with the TopScan software (Clever System). Animals were randomly assigned to the various experimental groups. Behavioral tests were video–recorded and analyzed blind to the treatment.
Statistical analysis
No Statistical methods were used to predetermine, but our sample sizes are similar to those reported in previous publications53, 56, 57. The data were analyzed with the Kolmogorov–Smirnov and Chi–Squared test for distribution. As all our data are not normally distributed, two–tailed Mann–Whitney test was used to calculate p–values. The results of statistical tests were not corrected for multiple comparisons.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Heinz Arnheiter (NINDS/NIH) for critical discussion of the manuscript, Dr. Elizabeth J. Sherman for editing the manuscript. This work is supported by the NIMH Intramural Research Program.
Footnotes
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
J–M Jia conducted the experiments and data analysis. J Zhao analyzed mEPSCs. D Lindberg contributed to the CTB experiment. Z Li and J–M Jia designed the experiments and wrote the manuscript. Z Hu generated the constructs and lentivirus expressing D1R, D2R and siRNAs against them.
References
- 1.Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III––the final common pathway. Schizophr Bull. 2009;35:549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyamoto S, Duncan GE, Marx CE, Lieberman JA. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry. 2005;10:79–104. doi: 10.1038/sj.mp.4001556. [DOI] [PubMed] [Google Scholar]
- 3.Iizuka Y, Sei Y, Weinberger DR, Straub RE. Evidence that the BLOC–1 protein dysbindin modulates dopamine D2 receptor internalization and signaling but not D1 internalization. J Neurosci. 2007;27:12390–12395. doi: 10.1523/JNEUROSCI.1689-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ji Y, et al. Role of dysbindin in dopamine receptor trafficking and cortical GABA function. Proc Natl Acad Sci U S A. 2009;106:19593–19598. doi: 10.1073/pnas.0904289106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green MF. Stimulating the development of drug treatments to improve cognition in schizophrenia. Annu Rev Clin Psychol. 2007;3:159–180. doi: 10.1146/annurev.clinpsy.3.022806.091529. [DOI] [PubMed] [Google Scholar]
- 6.Stephan KE, Baldeweg T, Friston KJ. Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry. 2006;59:929–939. doi: 10.1016/j.biopsych.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Garey LJ, et al. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 1998;65:446–453. doi: 10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 9.Kolomeets NS, Orlovskaya DD, Rachmanova VI, Uranova NA. Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: a postmortem morphometric study. Synapse. 2005;57:47–55. doi: 10.1002/syn.20153. [DOI] [PubMed] [Google Scholar]
- 10.Kolluri N, Sun Z, Sampson AR, Lewis DA. Lamina–specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry. 2005;162:1200–1202. doi: 10.1176/appi.ajp.162.6.1200. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Sheng M. Some assembly required: the development of neuronal synapses. Nat Rev Mol Cell Biol. 2003;4:833–841. doi: 10.1038/nrm1242. [DOI] [PubMed] [Google Scholar]
- 12.Brennand KJ, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonci A, Hopf FW. The dopamine D2 receptor: new surprises from an old friend. Neuron. 2005;47:335–338. doi: 10.1016/j.neuron.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 14.Beazely MA, et al. D2–class dopamine receptor inhibition of NMDA currents in prefrontal cortical neurons is platelet–derived growth factor receptor–dependent. J Neurochem. 2006;98:1657–1663. doi: 10.1111/j.1471-4159.2006.04064.x. [DOI] [PubMed] [Google Scholar]
- 15.Kotecha SA, et al. A D2 class dopamine receptor transactivates a receptor tyrosine kinase to inhibit NMDA receptor transmission. Neuron. 2002;35:1111–1122. doi: 10.1016/s0896-6273(02)00859-0. [DOI] [PubMed] [Google Scholar]
- 16.Liu XY, et al. Modulation of D2R–NR2B interactions in response to cocaine. Neuron. 2006;52:897–909. doi: 10.1016/j.neuron.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 17.Li YC, Xi D, Roman J, Huang YQ, Gao WJ. Activation of glycogen synthase kinase–3 beta is required for hyperdopamine and D2 receptor–mediated inhibition of synaptic NMDA receptor function in the rat prefrontal cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:15551–15563. doi: 10.1523/JNEUROSCI.3336-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyer C, Schikorski T, Stevens CF. Comparison of hippocampal dendritic spines in culture and in brain. J Neurosci. 1998;18:5294–5300. doi: 10.1523/JNEUROSCI.18-14-05294.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peters A, Kaiserman–Abramof IR. The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. The American journal of anatomy. 1970;127:321–355. doi: 10.1002/aja.1001270402. [DOI] [PubMed] [Google Scholar]
- 20.Harris KM. Structure, development, and plasticity of dendritic spines. Curr Opin Neurobiol. 1999;9:343–348. doi: 10.1016/s0959-4388(99)80050-6. [DOI] [PubMed] [Google Scholar]
- 21.Nagai T, et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell–biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 22.Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- 23.Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tseng KY, O’Donnell P. Dopamine–glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci. 2004;24:5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Traynelis SF, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological reviews. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hakansson K, et al. Regulation of phosphorylation of the GluR1 AMPA receptor by dopamine D2 receptors. Journal of neurochemistry. 2006;96:482–488. doi: 10.1111/j.1471-4159.2005.03558.x. [DOI] [PubMed] [Google Scholar]
- 27.Chen XW, et al. DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. The Journal of cell biology. 2008;181:791–801. doi: 10.1083/jcb.200711021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arranz MJ, de Leon J. Pharmacogenetics and pharmacogenomics of schizophrenia: a review of last decade of research. Mol Psychiatry. 2007;12:707–747. doi: 10.1038/sj.mp.4002009. [DOI] [PubMed] [Google Scholar]
- 29.Glazer WM. Does loxapine have “atypical” properties? Clinical evidence. J Clin Psychiatry. 1999;60 (Suppl 10):42–46. [PubMed] [Google Scholar]
- 30.Giovanni GD, Matteo VD, Esposito E, editors. Seritonin–dopamine interaction: experimental evidenceand therapeutic relevance. Elsevier Science; 2008. [Google Scholar]
- 31.Meltzer HY, Massey BW. The role of serotonin receptors in the action of atypical antipsychotic drugs. Curr Opin Pharmacol. 2011;11:59–67. doi: 10.1016/j.coph.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Tau GZ, Peterson BS. Normal development of brain circuits. Neuropsychopharmacology. 2010;35:147–168. doi: 10.1038/npp.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaaro–Peled H, et al. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin–1–ErbB4 and DISC1. Trends Neurosci. 2009;32:485–495. doi: 10.1016/j.tins.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suh J, Rivest AJ, Nakashiba T, Tominaga T, Tonegawa S. Entorhinal cortex layer III input to the hippocampus is crucial for temporal association memory. Science. 2011;334:1415–1420. doi: 10.1126/science.1210125. [DOI] [PubMed] [Google Scholar]
- 35.Andersen P, Morris R, Amaral D, Bliss T, O’Keefe J, editors. The Hippocampus Book. Oxford University Press; 2007. [Google Scholar]
- 36.Schultz H, Sommer T, Peters J. Direct evidence for domain–sensitive functional subregions in human entorhinal cortex. J Neurosci. 2012;32:4716–4723. doi: 10.1523/JNEUROSCI.5126-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hasselmo ME, Stern CE. Mechanisms underlying working memory for novel information. Trends Cogn Sci. 2006;10:487–493. doi: 10.1016/j.tics.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho YH, Jaffard R. Spatial location learning in mice with ibotenate lesions of entorhinal cortex or subiculum. Neurobiol Learn Mem. 1995;64:285–290. doi: 10.1006/nlme.1995.0011. [DOI] [PubMed] [Google Scholar]
- 39.Papaleo F, et al. Dysbindin–1 modulates prefrontal cortical activity and schizophrenia–like behaviors via dopamine/D2 pathways. Mol Psychiatry. 2012;17:85–98. doi: 10.1038/mp.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talbot K. The sandy (sdy) mouse: a dysbindin–1 mutant relevant to schizophrenia research. Prog Brain Res. 2009;179:87–94. doi: 10.1016/S0079-6123(09)17910-4. [DOI] [PubMed] [Google Scholar]
- 41.Baik JH, et al. Parkinsonian–like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995;377:424–428. doi: 10.1038/377424a0. [DOI] [PubMed] [Google Scholar]
- 42.Drago J, et al. Altered striatal function in a mutant mouse lacking D1A dopamine receptors. Proc Natl Acad Sci U S A. 1994;91:12564–12568. doi: 10.1073/pnas.91.26.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang HD, Stanwood GD, Grandy DK, Deutch AY. Dystrophic dendrites in prefrontal cortical pyramidal cells of dopamine D1 and D2 but not D4 receptor knockout mice. Brain research. 2009;1300:58–64. doi: 10.1016/j.brainres.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu M, et al. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine–mediated behavioral responses. Cell. 1994;79:729–742. doi: 10.1016/0092-8674(94)90557-6. [DOI] [PubMed] [Google Scholar]
- 45.Ji Y, Pang PT, Feng L, Lu B. Cyclic AMP controls BDNF–induced TrkB phosphorylation and dendritic spine formation in mature hippocampal neurons. Nat Neurosci. 2005;8:164–172. doi: 10.1038/nn1381. [DOI] [PubMed] [Google Scholar]
- 46.Fox JG, et al., editors. The Mouse in Biomedical Research. Academic Press; 2007. [Google Scholar]
- 47.Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- 48.Tabuchi K, et al. A neuroligin–3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fiorentino H, et al. GABA(B) receptor activation triggers BDNF release and promotes the maturation of GABAergic synapses. J Neurosci. 2009;29:11650–11661. doi: 10.1523/JNEUROSCI.3587-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai YQ, et al. Central amygdala GluA1 facilitates associative learning of opioid reward. J Neurosci. 2013;33:1577–1588. doi: 10.1523/JNEUROSCI.1749-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fukata Y, et al. Local palmitoylation cycles define activity–regulated postsynaptic subdomains. The Journal of cell biology. 2013;202:145–161. doi: 10.1083/jcb.201302071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilkie MP, et al. The relationship between NMDA receptor function and the high ammonia tolerance of anoxia–tolerant goldfish. The Journal of experimental biology. 2011;214:4107–4120. doi: 10.1242/jeb.057513. [DOI] [PubMed] [Google Scholar]
- 53.Li Z, et al. Caspase–3 activation via mitochondria is required for long–term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Brien JA, Lummis SC. Diolistic labeling of neuronal cultures and intact tissue using a hand–held gene gun. Nat Protoc. 2006;1:1517–1521. doi: 10.1038/nprot.2006.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan L, Terashima T, Urabe H, Lin F, Kojima H. Pathogenesis of diabetic neuropathy: bad to the bone. Annals of the New York Academy of Sciences. 2011;1240:70–76. doi: 10.1111/j.1749-6632.2011.06309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiao S, Li Z. Nonapoptotic function of BAD and BAX in long–term depression of synaptic transmission. Neuron. 2011;70:758–772. doi: 10.1016/j.neuron.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Belforte JE, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia–like phenotypes. Nature neuroscience. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.