

Abstract
Background
Most molecular studies of plant stress tolerance have been performed with Arabidopsis thaliana, although it is not particularly stress tolerant and may lack protective mechanisms required to survive extreme environmental conditions. Thellungiella salsuginea has attracted interest as an alternative plant model species with high tolerance of various abiotic stresses. While the T. salsuginea genome has recently been sequenced, its annotation is still incomplete and transcriptomic information is scarce. In addition, functional genomics investigations in this species are severely hampered by a lack of affordable tools for genome-wide gene expression studies.
Results
Here, we report the results of Thellungiella de novo transcriptome assembly and annotation based on 454 pyrosequencing and development and validation of a T. salsuginea microarray. ESTs were generated from a non-normalized and a normalized library synthesized from RNA pooled from samples covering different tissues and abiotic stress conditions. Both libraries yielded partially unique sequences, indicating their necessity to obtain comprehensive transcriptome coverage. More than 1 million sequence reads were assembled into 42,810 unigenes, approximately 50% of which could be functionally annotated. These unigenes were compared to all available Thellungiella genome sequence information. In addition, the groups of Late Embryogenesis Abundant (LEA) proteins, Mitogen Activated Protein (MAP) kinases and protein phosphatases were annotated in detail. We also predicted the target genes for 384 putative miRNAs. From the sequence information, we constructed a 44 k Agilent oligonucleotide microarray. Comparison of same-species and cross-species hybridization results showed superior performance of the newly designed array for T. salsuginea samples. The developed microarrays were used to investigate transcriptional responses of T. salsuginea and Arabidopsis during cold acclimation using the MapMan software.
Conclusions
This study provides the first comprehensive transcriptome information for the extremophile Arabidopsis relative T. salsuginea. The data constitute a more than three-fold increase in the number of publicly available unigene sequences and will greatly facilitate genome annotation. In addition, we have designed and validated the first genome-wide microarray for T. salsuginea, which will be commercially available. Together with the publicly available MapMan software this will become an important tool for functional genomics of plant stress tolerance.
Keywords: Arabidopsis thaliana, Cold acclimation, Gene annotation, LEA proteins, MAP kinases, Microarray design, microRNAs, Protein phosphatases, Thellungiella salsuginea, Transcriptome sequencing
Background
The majority of molecular studies of plant stress tolerance have been performed with Arabidopsis thaliana, although it is not an extremophile and can therefore be expected to lack many protective mechanisms required to survive extreme environmental conditions. Thellungiella salsuginea has therefore attracted increasing interest as an alternative plant model species that possesses the characteristics of an extremophile, i.e. high tolerance of salinity, freezing, nitrogen-deficiency and drought stress [1-7]. The genus Thellungiella is part of the Brassicaceae family and therefore related to Arabidopsis[8,9]. The species name has sometimes been used synonymously with Thellungiella halophila, but it is clear now that three distinct Thellungiella species (T. halophila, T. salsuginea, and T. botschantzevii) have to be distinguished [10]. T. salsuginea is also referred to as Eutrema salsugineum, however, we prefer to stay with the older and better established species name as that is not in conflict with the taxonomy [10]. On the other hand, Thellungiella parvula is not as closely related to these other species as previously thought and should be called Schrenkiella parvula[10]. However, to avoid unnecessary confusion in the assignment of sequence information, we have used the species names used in the respective publications in all comparisons with our data.
T. salsuginea resembles Arabidopsis in many important features such as short life cycle, self-fertility and the possibility to transform plants by the floral-dip method [4]. While the genomes of T. salsuginea[11,12] and T. parvula[13] have recently been sequenced, important functional genomics tools such as dedicated microarrays are still lacking for these species.
Arabidopsis-based microarrays [2,4,14-17] and a cDNA array comprising 3,628 unique T. salsuginea ESTs [18] have been used to profile the effects of abiotic stress conditions on the T. salsuginea transcriptome. However, T. salsuginea is not in the Arabidopsis genus and therefore has several distinguishing genetic and developmental properties [1,4]. Identity between the Arabidopsis genome sequence and T. salsuginea ESTs (Expressed Sequence Tags) is between 95% and 60%, but there are also specific ESTs in T. salsuginea[4,7,19,20]. Importantly, analysis of T. salsuginea mRNA using the Affymetrix Arabidopsis GeneChip ATH1 produced signals significantly above background for only 20% of the probe sets [17], indicating the necessity for a dedicated T. salsuginea microarray for cost-effective transcriptome studies.
Transcriptome sequencing is playing an increasingly important role in functional genomic studies of organisms without full genome sequences (e.g. [21-25]). The 454 technology is an efficient and cost effective way to generate high-throughput EST data for such organisms [26-30], providing the possibility for de novo assembly of transcriptomes from uncharacterized genomes [31,32].
Large scale isolation of ESTs is a powerful approach for the discovery of novel genes, the investigation of genes of unknown function, comparative genomic studies and identification of exon/intron boundaries [33]. There are currently 44,559 T. salsuginea EST sequences in the GenBank dbEST (as of June 19, 2012; [34]) and 19,429 full-length cDNAs are available from RIKEN [35]. These numbers are far smaller than for Arabidopsis (1,529,700 ESTs as of June 19, 2012), indicating the need for further gene discovery and characterization in T. salsuginea.
Here, we have sequenced, assembled and annotated the transcriptome of the T. salsuginea Yukon accession using 454 pyrosequencing. On the basis of these transcriptome sequences, we have developed a dedicated T. salsuginea microarray using the Agilent long oligonucleotide array platform. This array has been benchmarked against the corresponding Arabidopsis array and tested in comparative cold acclimation experiments [6].
Results
454 sequencing and transcriptome assembly
We constructed non-normalized and normalized cDNA libraries of T. salsuginea from various tissues derived from soil- and hydroponically-grown plants, including rosette and cauline leaves, roots, flowers, and siliques of plants treated with cold, drought and salt to achieve comprehensive transcriptome information. Using the GS FLX 454 sequencer, we generated 400,631 and 811,683 reads from a normalized and non-normalized library, respectively, comprising 562.67 Mbp with an average read length of 464 bp (Table 1). The average read length was greater for ESTs derived from the non-normalized library (566 bp) than from the normalized library (257 bp) and the proportion of reads shorter than 257 bp was larger for the normalized library compared to the non-normalized library (Figure 1), as a result of cDNA fragmentation in the procedure for constructing normalized libraries.
Table 1.
Summary of T. salsuginea ESTs generated from 454 sequencing and assembled contigs
| Library | Non-normalized | Normalized | Combined 1 |
|---|---|---|---|
| Number of reads |
811,683 |
400,631 |
1,212,314 |
| Number of contaminated reads2 |
4,496 |
2,506 |
7,002 |
| Average read length (bp) |
566 |
257 |
464 |
| Total reads (Mbp) |
459.54 |
103.13 |
562.67 |
| Number of assembled reads |
712,262 |
376,509 |
1,060,666 |
| Number of contigs3 |
33,870 |
28,928 |
46,220 |
| Number of contigs with only 454 reads4 |
21,662 |
16,949 |
33,147 |
| Average coverage |
11.99 |
6.74 |
12.63 |
| N50 (bp) |
665 |
632 |
646 |
| Average contig length (bp) |
621 |
502 |
567 |
| Number of contigs with ORF5 |
33,625 |
28,416 |
45,583 |
| Number of Arabidopsis peptides found6 | 23,787 | 23,220 | 24,457 |
1Combined 454 reads derived from normalized and non-normalized library.
2Best hits with E-value < 1e-10, non streptophyta sequences (NCBI, nr, nt, as of February 12, 2010).
3Includes publicly available T. salsuginea ESTs (as of April 19, 2010).
4Number of contigs minus number of contigs assembled with at least one publicly available EST.
5Open reading frames (ORFs) predicted including partial ORFs (Min et al. [83]).
6Any hits with E-value < 0.001 from BLASTX search against TAIR9 database (27,379 peptides).
Figure 1.
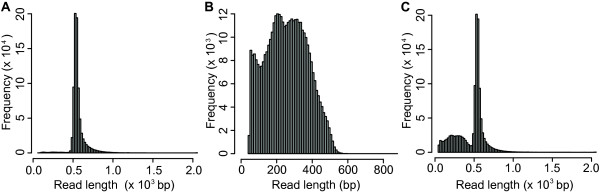
Expressed sequence tags of T. salsuginea generated using 454 pyrosequencing. Frequency distribution of 454 read lengths derived from (A) non-normalized and (B) normalized library, and (C) combined data from both libraries.
We combined the 454 reads with 44,551 publicly available T. salsuginea ESTs and performed a transcriptome assembly with MIRA [36]. This de-novo assembly of the combined libraries generated 46,220 contigs with an average length of 567 bp (Table 1, Figure 2C). The contigs showed a similar trend of frequency distribution of average contig coverage (reads per contig length) for three different assemblies (Figure 2D, E, F). Pearson correlation analysis revealed a significant positive correlation (p < 10-6) between contig length and contig average coverage (Figure 2G, H and I). This result suggests that the consensus nucleotide sequences of long contigs are confidently supported with more reads than the shorter contigs. The assembly of the combined reads increased the number of contigs and average coverage (12.63 reads per nucleotide in the template), but the average contig length was smaller when compared to the non-normalized library (Table 1, Figure 2A and C). Of 46,220 contigs, 33,147 (71.7%) were unique and had not been reported before.
Figure 2.

Size distribution and characteristics of assembled contigs. Frequency distribution of contigs assembled from the pool of publicly available T. salsuginea ESTs and 454 reads derived from (A) non-normalized library, (B) normalized library or (C) combined libraries. Frequency distribution of contig average coverage for the assembly of publicly available T. salsuginea ESTs pooled with 454 reads derived from (D) non-normalized library, (E) normalized library or (F) combined libraries. Pearson’s correlations between contig length and contig average coverage for the assembly of publicly available T. salsuginea ESTs pooled with 454 reads derived from (G) non-normalized library, (H) normalized library or (I) combined libraries.
We further examined whether sequencing both a normalized and a non-normalized library increased the number of unique transcripts that were detected. The normalization procedure produced about 24% (6898) unique contigs that were not found in the non-normalized library, while the non-normalized library yielded about 20% (6755) unique contigs using BLASTN with E value threshold of 10-50. This indicates that to approach full transcriptome information, the sequencing of both types of libraries is necessary. No consensus number was found for the matched contigs between the two libraries using BLASTN in reciprocal searches (Figure 3A).
Figure 3.

Similarity of T. salsuginea (Yukon) unigenes to Arabidopsis and T. parvula orthologs and T. salsuginea/Eutrema salsugineum (Shandong) gene models. Venn diagrams depicting the number of unique unigenes derived only from (A) normalized or non-normalized libraries, (B)T. salsuginea or Arabidopsis,(C)T. salsuginea or T. parvula (Schrenkiella parvula) orthologs, and T. salsuginea (Eutrema salsugineum) Yukon uniges or Shandong gene models (D). Vertical dividing lines indicate non-matched overlap values for both libraries and species due to the nature of the BLAST search algorithm.
To assess the quality of the assembly, we predicted open reading frames (ORFs) of the contigs and used BLASTX to align all contigs to the TAIR9 Arabidopsis annotated protein database using an E-value threshold of 10-3. Of 27,379 Arabidopsis peptides, 24,457 unique peptides were matched to contigs generated from the combined libraries (Table 1), indicating a high quality of the transcriptome assembly.
Gene discovery
The transcriptome assembly yielded 4,020 contigs sharing more than 99% sequence identity and coverage with at least one other contig in the assembly, likely constituting alleles of the same genes (or very close paralogs) with single nucleotide polymorphysms (SNPs). Because the seed material was not derived from single seed descent, the plants used in the experiments were not genetically identical, making the presence of SNPs in this plant population likely. We therefore condensed these 4,020 contigs into 610 clusters with ambiguous nucleotides at the SNP sites, reducing the final number of unigenes to 42,810. Since T. salsuginea and Arabidopsis are both members of the Brassicaceae family, we estimated the number of T. salsuginea unigenes that were orthologous to Arabidopsis genes using BLASTX. This identified 32,029 unigenes as orthologs of Arabidopsis genes, while approximately 25% were unique to T. salsuginea. No consensus overlap was found for the number of matched sequences between both species when a reciprocal search was performed, because every given reference sequence may hit more than one paralog in the other species, while also more than one homologous sequence in one species may hit a single sequence in the other species (Figure 3B). The genome sequence of T. parvula was published recently [13] and although T. salsuginea and T. parvula are related, about 26% of T. salsuginea unigenes have no counterparts in T. parvula, while 21% of the T. parvula genes have no orthologs in T. salsuginea (Figure 3C), probably reflecting the fact that the relationship between T. salsuginea and T. parvula (Schrenkiella parvula[10]) is not as close as originally assumed. On the other hand, approximately 88% (37,622 of 42,810) of the T. salsuginea unigenes could be aligned to 3,703 T. salsuginea genomic contig sequences comprising 135.6 Mbp of genome sequence [11] using BLASTN with a stringent threshold (E-value < 10-50).
More recently the T. salsuginea (Shandong accession) genome sequence obtained by Sanger sequencing technology has been published [12]. We estimated that 72% (18,970 out of 26,351) of the gene models predicted from the genome were expressed under our experimental conditions (BLASTN, E-value < 10-10). On the other hand, 37,447 T. salsuginea Yukon unigenes were best matched (BLASTN, E-value < 10-10) with the Shandong gene models (Figure 3D). Therefore, 5,363 unigenes from the Yukon accession have no counterpart among the predicted gene models from the Shandong accession and may be Yukon-specific transcripts (Figure 3D). Approximately 56.7% of matched unigenes (21,228 out of 37,447) were estimated to be full length in relation to the published gene models from the genome sequence (E-value <10-10 and total query coverage > 90%).
We further investigated how many Yukon unigenes were likely derived from gene duplication events in relation to Shandong gene models. We assessed the sequence relationships between Yukon accession unigenes and Shandong accession gene models by BLASTN analysis (E-value < 10-10 and query coverage >90%). Based on these criteria (21,228 Yukon unigenes matching Shandong gene models), we assessed whether the relationships were one-to-one (single copy number conserved in both accessions), one-to-many (one Yukon transcript matches more than one Shandong gene model), many-to-one (more than one Yukon transcript uniquely matches a single Shandong gene model), or many-to-many (larger homologous groups with several members from both accessions). 376 putatively duplicated Yukon unigenes were identified in relation to Shandong gene models. These unigenes were enriched with genes involved in protein synthesis, nucleotide metabolism, DNA synthesis, light reaction of photosynthesis, regulation of transcription, redox and storage proteins (Table 2).
Table 2.
Enrichment of unigenes in MapMan terms for 12,272 duplicated Yukon unigenes
| Mapman bin code | Mapman term | Number of duplicated Yukon unigenes | Fisher’s exact test P -value (Bonferroni-corrected)* |
|---|---|---|---|
| 29.2.1 |
protein.synthesis.ribosomal protein |
558 |
1.48E-18 |
| 11.1 |
lipid metabolism – Fatty Acid synthesis and Fatty Acid elongation |
89 |
1.56E-06 |
| 29.5 |
protein.degradation |
652 |
3.28E-05 |
| 34 |
transport |
433 |
1.25E-04 |
| 30 |
signalling |
488 |
2.93E-04 |
| 1.1.1.01 |
PS.lightreaction.photosystem II.LHC-II |
52 |
3.98E-04 |
| 20.1.7 |
stress.biotic.PR-proteins |
29 |
1.00E-03 |
| 20.1 |
stress.biotic |
93 |
1.86E-03 |
| 29.3.4 |
protein.targeting.secretory pathway |
95 |
3.52E-03 |
| 1.1.2.01 |
PS.lightreaction.photosystem I.LHC-I |
41 |
4.32E-03 |
| 23.4.010 |
nucleotide metabolism.phosphotransfer and pyrophosphatases.nucleoside diphosphate kinase |
21 |
8.96E-03 |
| 20.2.3 |
stress.abiotic.drought/salt |
63 |
1.17E-02 |
| 30.3 |
signalling.calcium |
129 |
1.20E-02 |
| 21 |
redox.regulation |
167 |
2.28E-02 |
| 11.1.012 |
lipid metabolism.FA synthesis and FA elongation.ACP proteins |
18 |
2.49E-02 |
| 17.6.3 |
hormone metabolism.gibberelin.induced-regulated-responsive-activated |
18 |
2.49E-02 |
| 27.3.25 |
RNA.regulation of transcription.MYB domain transcription factor family |
14 |
2.54E-02 |
| 27.3.52 |
RNA.regulation of transcription.Global transcription factor group |
16 |
2.70E-02 |
| 29.1 |
protein.aa activation |
59 |
2.72E-02 |
| 4.02 | glycolysis.PGM | 10 | 3.22E-02 |
*The Bonferroni-corrected Fisher’s exact test P-value that was extracted from MapMan term enrichment analysis using MEFISTO software [101].
The single nucleotide polymorphisms (SNPs) between Shandong gene models and Yukon unigenes were called using VarScan v2.3.5 [37] as allelic variation may contribute to the variation of gene expression and the phenotypes of the accessions [38-40]. Using Shandong gene models as reference sequences, we identified 16,209 putative non-ambiguous SNPs between Yukon and Shandong in 4,020 Shandong gene models (Table 3, Additional file 1). Whether the identified SNPs contribute to differences in gene expression and/or phenotypic variation, e.g. in stress responses, remains to be investigated.
Table 3.
Non-ambiguous SNPs identified between Shandong gene models and Yukon unigenes
| SNP frequency* | Number of Shandong gene models |
|---|---|
| 1 |
1,119 |
| 2 |
854 |
| 3 |
590 |
| 4 |
374 |
| 5 |
302 |
| 6 |
179 |
| 7 |
122 |
| 8 |
101 |
| 9 |
69 |
| ≥ 10 |
310 |
| Total | 4,020 |
*Detected nonambiguous SNPs between Shandong gene models and Yukon unigenes. The Yukon unigenes were aligned to the reference sequence of the Shandong gene models and called for variations using VarScan v2.3.5 [37].
Functional annotation
Sequence-based functional annotation of the 42,810 unigenes was performed using Mercator [41] and MapMan ontology [42] to predict and classify the protein coding unigenes. The Mercator tool predicts the function of input DNA sequences by searching a variety of reference databases (currently six are available: three BLAST-based, two RPSBLAST-based and InterProScan). Subsequently, each input gene is assigned to one or more functional MapMan bins based on the manually curated binning of the reference database entries [42,43]. We compared the distribution of gene functions and found that a large proportion of Arabidopsis (35.5%) and T. salsuginea unigenes (48.9%) could not be classified to any functional bin (Figure 4A). A large number of genes with known functions were classified in bins of protein, DNA, RNA, signaling, transport, stress, cell, development, photosynthesis and miscellaneous for Arabidopsis (53.8%) and T. salsuginea (40.0%). The distribution of gene functions was similar in both species with similar proportions of genes for each bin except gene functions related to DNA metabolism (9.5% in Arabidopsis and 1.5% in T. salsuginea) and differences of about 2% between Arabidopsis and T. salsuginea in bins for RNA, protein and photosynthesis (Figure 4A).
Figure 4.

Distribution of predicted T. salsuginea gene functions in comparison to Arabidopsis thaliana. (A) Functional classes of encoded proteins and (B) micro RNA prediction and distribution of their target genes.
microRNA prediction
Higher plant transcriptomes contain a large number of non-coding RNAs including microRNA (miRNA) that function in diverse regulatory processes that influence e.g. growth and development [44]. Since primary miRNA transcripts are polyadenylated [45-47] it seemed possible that the T. salsuginea unigenes contain primary miRNA transcripts that code for one or more miRNA stem loops that could be further processed to mature miRNAs. We scanned all unigenes using BLASTN with a threshold E-value of 10-10 against the complete set of miRNA precursor transcripts from the plant miRNA database [48] and obtained 384 unigenes that putatively encode miRNA precursor transcripts. The predicted target transcripts of the corresponding miRNAs were classified according to their functions using MapMan ontology and the predicted targets were compared with those of Arabidopsis miRNAs (Figure 4B). The proportion of predicted targets in the different functional classes was generally lower for T. salsuginea than for Arabidopsis except for target functions related to protein metabolism (54.5% vs. 20.1%) and mitochondrial electron transport (1.3% vs. 0.2%), where T. salsuginea showed higher proportions compared to Arabidopsis.
Genes encoding protein kinases and protein phosphatases
Mitogen activated protein kinases (MAPK) and MAPK phosphatases are important components that mediate abiotic stress signaling in plant cells [49]. Members of the families of protein tyrosine phosphatases (PTPs), including dual specificity phosphatases (DSPs) and protein phosphatases 2C (PP2Cs) have been characterized as plant MAPK phosphatases that regulate stress responses [50,51]. In the PP2C gene family the A-type PP2C phosphatases are generally upregulated in response to abscisic acid (ABA), whereas B-type PP2Cs (AP2Cs) have been characterized as MAPK phosphatases [51]. Since drought or salt stress induces ABA biosynthesis in plants and MAPK cascades have been related to salt stress tolerance [49], it was of high interest to identify putative orthologous proteins of PP2Cs and PTPs/DSPs, as well as MAPKs and MAPK kinases (MAPKKs) in the extremophile T. salsuginea.
Individual T. salsuginea putative MAPKK, MAPK, PP2C, DSP and PTP proteins were designated according to the corresponding orthologous protein annotations in Arabidopsis (Additional files 2, 3 and 4) and their phylogenetic relationships were visualized in unrooted phylogenetic trees (Additional files 5, 6 and 7). Using 80 PP2Cs from Arabidopsis[51-53] as well as 74 and 75 PP2Cs encoded in the T. salsuginea and T. parvula genomes [11-13], respectively, led to the identification of 118 PP2C unigenes in the sequenced T. salsuginea ESTs that could be translated into 59 PP2C proteins (Table 4, Additional file 8).
Table 4.
Number of putative protein tyrosine phosphatase (PTP), dual specificity phosphatase (DSP), protein phosphatase 2C (PP2C), mitogen activated protein kinase (MAPK) and MAPK kinase (MAPKK) unigenes and proteins in the T. salsuginea ( T.s. ), T. parvula ( T.p. ) and A. thaliana ( A.t. ) genomes
| Unigenes | Proteins from unigenes | Proteins from genome | |
|---|---|---|---|
|
PTP
T.s.
|
1 |
1 |
1 |
|
PTP
T.p.
|
na |
na |
2 |
|
PTP
A.t.
|
na |
na |
1 |
|
DSP
T.s.
|
28 |
18 |
23 |
|
DSP
T.p.
|
na |
na |
20 |
|
DSP
A.t.
|
na |
na |
22 |
|
PP2C
T.s.
|
118 |
59 |
75 |
|
PP2C
T.p.
|
na |
na |
74 |
|
PP2C
A.t.
|
na |
na |
80 |
|
MAPK
T.s.
|
25 |
13 |
18 |
|
MAPK
T.p.
|
na |
na |
19 |
|
MAPK
A.t.
|
na |
na |
20 |
|
MAPKK
T.s.
|
8 |
6 |
10 |
|
MAPKK
T.p.
|
na |
na |
11 |
| MAPKK A.t. | na | na | 10 |
na = not applicable.
All plant species studied so far revealed a single gene encoding a PTP [54]. Correspondingly, a single unigene of a PTP was identified in the T. salsuginea transcriptome. However, two PTP genes were identified in the genome of T. parvula (Table 4). Reciprocal BLAST of 22 and 20 DSP sequences from Arabidopsis[54] and T. parvula[13], respectively, revealed 28 DSP encoding unigenes (Table 4) that code for 18 T. salsuginea DSP proteins (Additional file 8).
The genome sequences of T. salsuginea and T. parvula[11-13] revealed 18 and 19 MAPK encoding genes, respectively, in comparison to 20 MAPKs [55] found in Arabidopsis. Here, analysis of MAPKs in the transcriptome of T. salsuginea identified 25 unigenes, which were translated into 13 putative MAPK proteins (Table 4, Additional file 8). MAPKK proteins are represented by 10 and 11 members in the genomes of Arabidopis[55] and T. parvula[13], respectively. In the T. salsuginea trancriptome we identified eight ESTs representing MAPKK, corresponding to six MAPKK proteins (Table 4, Additional file 8).
Genes encoding Late Embryogenesis Abundant (LEA) proteins
The accumulation of LEA proteins (or their transcripts) has been associated with environmental stress conditions such as cold, drought, or desiccation in plants, as well as in some bacteria and invertebrates [56,57]. In Arabidopsis 51 genes encoding LEA proteins have been identified and all genes were shown to be expressed in some tissue and/or under particular stress conditions [58]. Most of the Arabidopsis LEA proteins were predicted to be highly hydrophilic, intrinsically disordered proteins. Since LEA proteins are generally assumed to play important roles in cellular stress tolerance, it was of interest to identify potential orthologous proteins in the extremophile T. salsuginea. Therefore, the 51 Arabidopsis LEA proteins were reciprocally compared by BLASTX (threshold E-value 10-10) with the translated unigenes. This resulted in 148 putative T. salsuginea LEA protein sequences (Additional file 9). They were aligned with the Arabidopsis LEA proteins using ClustalW and an unrooted dendogram was drawn (Additional file 10) assigning the putative LEA proteins to the corresponding Pfam domain/motif groups (Additional file 11). Due to high similarity at the protein sequence level, these proteins were condensed to 52 protein sequences that showed distinct models (Additional file 11). For example, one of the best characterized Arabidopsis LEA proteins is COR15A (LEA24) which clustered with three orthologous T. salsuginea proteins (73, 139, 168, Additional file 10) encoded by distinct unigenes. However, multiple protein sequence alignment showed that amino acid sequences translated from thellun_all_rep_c5293 (73) and thellun_all_rep_c26181 (139) were highly similar to thellun_all_rep_c36160 (168; Figure 5) which is the longest of the three translated polypeptides. We therefore chose translated frame +3 of thellun_all_rep_c36160 as the representative COR15A orthologous protein in T. salsuginea (Additional file 12).
Figure 5.

Multiple sequence alignment of COR15A orthologous proteins between T. salsuginea and Arabidopsis. Boxes indicate substitutions of amino acids among T. salsuginea COR15A orthologous proteins. Identical amino acids are marked with a star. Amino acids showing conserved substitutions are marked with two dots, and semi-conserved substitutions are marked with a single dot.
Following this strategy for all 148 putative T. salsuginea LEA proteins identified 15 Arabidopsis LEA proteins without an orthologous protein in the T. salsuginea transcriptome. However, a BLASTN search (E < 1e-50) revealed the presence of genes encoding 12 of these proteins in the T. salsuginea genome. For example, there were no apparent orthologs of LEA_5 proteins among the unigenes, but the BLASTN search of the gene sequences (At2g40170 and At3g51810) against the T. salsuginea genome retrieved two genomic sequences (AHIU01008450.1 and AHIU01008580.1), indicating that two LEA_5 proteins are encoded in the T. salsuginea genome, but probably not expressed under our experimental conditions. LEA_4 and dehydrin are the two largest groups of LEA proteins in Arabidopsis and this was also true for T. salsuginea with 18 LEA_4 proteins in both species. Interestingly, T. salsuginea contains more dehydrins (17 vs 10) than Arabidopsis. This is due to the fact that some Arabidopsis LEA proteins were represented by several T. salsuginea orthologs. For instance, LEA23 (At2g42530; COR15B) and LEA51 (At5g66400; RAB18) were each represented by four distinct protein sequences and several others by three (Additional file 12).
Overall, the majority of putative T. salsuginea LEA proteins showed negative GRAVY (grand average of hydropathy) values indicating the expected high hydrophilicity (Additional file 12). Since a precise in-vivo function has not been established for any Arabidopsis LEA protein, it is currently impossible to evaluate the functional significance of the multiple orthologous T. salsuginea proteins.
Design and performance of a dedicated T. salsuginea microarray
Between 2 and 12 60-mer oligonucleotide probes were designed for each T. salsuginea unigene, depending on the length of the predicted gene. With this oligonucleotide sequence information as input, 400 K arrays were produced by Agilent. A Pre-Selection Strategy (PSS) [59] was used to select the best-performing probes by hybridizing either cRNA derived from pooled total RNA (identical to the RNA used in cDNA library construction for sequencing) or genomic DNA to these arrays. On the basis of the hybridization signal strength, the 43,800 best-performing probes were selected. 40,952 probes were selected on the basis of their hybridization signals with the cRNA sample and 2,848 additional probes were derived from the hybridization with genomic DNA. Using the Agilent eArray Platform [60], 44 K arrays were produced from these oligonucleotide sequences. For 2,237 unigenes, the number of features on the array allowed the design of two different probes for the same contig, while for the remaining 39,326 unigenes only one probe was designed. The resulting 43,800 probes therefore represented 41,563 unigenes.
To test the newly designed arrays, they were hybridized with total RNA derived from control (NA) or 14-day cold acclimated (ACC) T. salsuginea plants from three independent biological experiments. Spot intensities between replicates were highly correlated with r-values of Pearson correlations between 0.985 and 0.993, p < 2.2 × 10-16 (Additional file 13). In addition, the log2-transformed fold change in hybridization signals between samples from non-acclimated and cold acclimated plants for the 2,237 pairs of probes targeting the same unigene were also highly correlated (Additional file 14, r = 0.951, p < 2.2 × 10-16). These results validate the probe design and selection with the PSS method.
To further examine the quality of the T. salsuginea array and to assess whether hybridization results from a dedicated array are superior to cross-species hybridization using the Arabidopsis array from the same manufacturer, we performed reciprocal array hybridizations. In this experiment, RNA from the Yukon accession of T. salsuginea and RNA from the Col-0 accession of Arabidopsis were both hybridized to the Agilent T. salsuginea and Arabidopsis arrays. For both species, RNA was extracted from acclimated and non-acclimated plants from three independent biological replicates. The comparison between the performance of the two arrays was limited to a set of probes that represent genes that are highly similar between the two species. These genes were identified using BLASTX with a threshold E-value of 10-10 that resulted in 15,557 orthologous genes. The signal intensities (median values) of the corresponding probes were significantly correlated (Figure 6A, B, D, E, r-values between 0.437 and 0.457, and p < 2.2 × 10-16) between the T. salsuginea and Arabidopsis arrays when hybridized with identical RNA samples. However, closer examination of these correlations revealed that a substantial proportion of probes yielded very low signal intensities in cross-species hybridizations. For example, when RNA from non-acclimated T. salsuginea plants was hybridized to Arabidopsis arrays (Figure 6A), this yielded 4,426 genes with log10 signal intensities below 1, while the log10 signal intensities were above 2 when identical RNA samples were hybridized to T. salsuginea arrays.
Figure 6.

Comparison of median raw signal intensities of 15,557 genes matched between T. salsuginea and Arabidopsis arrays.T. salsuginea genes were matched with A. thaliana genes using BLASTX, with a significance cut-off of E < 10-10 for a sequence similarity search against the Arabidopsis TAIR10 database. This resulted in 15,557 genes that matched between the two microarrays. Pearson’ s correlations (p < 2.2 × 10-16) are shown between log10-transformed signal intensities obtained from T. salsuginea and Arabidopsis arrays hybridized with identical leaf RNA samples from either non-acclimated Yukon (A) or Col-0 (B), or cold acclimated Yukon (D) or Col-0 (E). The median density signal distributions of all 15,557 probes after hybridization with non-acclimated (C) or cold acclimated RNA samples (F) are also shown. The green lines indicate the density distribution of median signal intensity of Yukon samples hybridized on the array designed for Arabidopsis, while the blue lines show the density distribution of Yukon samples hybridized on the array designed for T. salsuginea. The red lines show the density distribution of Col-0 samples hybridized on the Agilent array designed for T. salsuginea and the black lines show the density distribution of Col-0 samples hybridized on the array designed for Arabidopsis.
Significantly, the distribution of raw signal intensities from all probes on the arrays revealed a bimodal distribution [61,62] with a dominant density peak at high signal intensities (likely derived from perfect-match hybridizations) and a minor peak at low signal intensities (likely derived from noise and mismatch hybridization) for the same-species array hybridizations (black and blue lines, Figure 6C and F). In contrast, cross-species array hybridizations yielded predominantly low signal intensities (green and red lines, Figure 6C and F), suggesting that these hybridizations were dominated by non-specific signals.
In fact, approximately half of the probes were detected as background signals (Additional file 15) when T. salsuginea RNA was hybridized to Arabidopsis arrays and 58% (2,553 of 4,425) of the genes differentially expressed during cold acclimation that were detected using T. salsuginea arrays were not identified as significantly changed using Arabidopsis arrays (Figure 7). A similar situation was evident when Col-0 RNA was hybridized to both arrays, where 82% (422 out of 512) of the genes that were found significantly cold-regulated using the same-species arrays showed no significant regulation in cross-species array hybridizations (Figure 7).
Figure 7.

Comparison of significant gene expression changes determined using the Agilent Arabidopsis or Thellungiella salsuginea arrays. Venn diagrams show the overlap between genes with significant (linear model, FDR, p < 0.05) changes in expression detected using data from the Arabidopsis or T. salsuginea arrays hybridized with either Col-0 or Yukon RNA. To ensure valid comparisons, all T. salsuginea genes were matched with Arabidopsis genes using BLASTX.
To facilitate data visualization and biological interpretation of array hybridization results, a T. salsuginea ontology/mapping file (available freely on-line at [63]) was developed by transferring the Arabidopsis MapMan ontology [42] to the T. salsuginea transcripts for use with the MapMan software [42,43].
Mapping of cold-responsive genes from same-species microarrays to the MapMan ontology showed several changes in the expression of genes related to primary and secondary metabolisms, and photosynthesis (Figure 8). The proportion of cold-responsive genes in different functional groups was tested for significant deviation from the expected value for all genes in these groups represented on the arrays. MapMan functional groups that were significantly (FDR, p < 0.05) under- or over-represented were identified using Fisher’s exact test with false discovery rate corrected p-values [64]. Many functional groups were affected for either up- or down-regulated genes in T. salsuginea plants cold acclimated at 4°C for two weeks (Additional file 16). The functional groups that were most significantly affected when considering cold-repressed genes were photosynthesis (p = 1.49E-129), cell wall (p = 1.38E-26) and hormones (p = 4.45E-15). The most affected functional groups for cold-induced genes were glycolysis (p = 4.45E-12), stress (p = 6.16E-12) and biodegradation of xenobiotics (p = 1.58E-7). The genes in the stress bin include 26 genes classified into the sub-bin stress.abiotic.cold (p = 4.45E-12). These results suggest that the newly designed T. salsuginea array reliably detected genes or pathways that were known to be differentially affected on the transcript level in Arabidopsis during plant cold acclimation [18,65-69].
Figure 8.
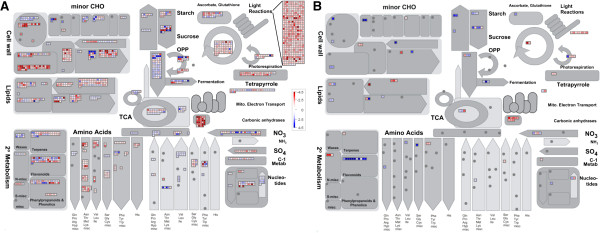
An overview of cold responsive genes annotated in MapMan functional groups for primary and secondary metabolism. Comparison of (A)T. salsuginea and (B)Arabidopsis genes encoding proteins involved in primary and secondary metabolisms. Samples derived from T. salsuginea Yukon were hybridized against T. salsuginea arrays, samples derived from Arabidopsis Col-0 to Arabidopsis arrays. Differential gene expression was determined in response to 14 days of cold acclimation at 4°C. Significance of expression changes was determined with a threshold of p < 0.05 after false discovery correction by fitting the data to a linear model. Blue indicates significantly up-regulated and red significantly down-regulated genes. Note the large difference in the number of regulated genes in the light reactions bin between the two species.
Discussion
T. salsuginea has many features similar to Arabidopsis and is in addition considered an “extremophile” with a higher tolerance to various abiotic stresses and has therefore been proposed as an alternative model species to Arabidopsis[9]. While the genomes of T. salsuginea and of the related species T. parvula have recently been sequenced [11-13], genome annotation is still far from complete and, due to the low number of ESTs available, evidence for transcriptional activity is also scarce for most genes. In addition, the effective use of T. salsuginea for functional genomics investigations of mechanisms of plant abiotic stress tolerance is severely hampered by a lack of affordable tools for genome-wide gene expression studies. We therefore used next generation sequencing to obtain comprehensive information about the T. salsuginea transcriptome and used this information for gene annotation and the development of a dedicated microarray for global expression studies. To cover the transcriptome as fully as possible, we performed 454 pyrosequencing on ESTs generated from both a non-normalized and a normalized library synthesized from RNA samples pooled from a wide range of plant samples from different developmental stages and from plants grown under different abiotic stress conditions. Although the number of reads obtained from the normalized library was only about half of that obtained from the non-normalized library, both libraries yielded a large number of unique sequences that were not detected in the other library, indicating that sequencing of normalized and non-normalized libraries is necessary to obtain optimal coverage of a transcriptome.
A combination of 44,551 publicly available ESTs and more than 1 million new sequence reads were assembled into 42,810 contigs for the prediction of gene functions. Of these contigs, 33,147 were novel because they were not covered by publicly available T. salsuginea ESTs. In total, the number of publicly available T. salsuginea unigenes increased more than 3-fold (from 13,073 to 42,810) with the present study. The number of genes estimated from the two draft genomes of T. salsuginea is 29,509 [11] and 26,351 [12], while the draft genome of T. parvula contains 30,419 genes [13]. This suggests that the unigenes identified in this study represent a comprehensive T. salsuginea transcriptome. The average contig/unigene length in our assembly is 567 bp, which is shorter than the length of the predicted Shandong gene models (1,224 bp; [12]). The likely reason for this discrepancy is the fragmentation procedure necessary for the construction of normalized cDNA libraries. Nevertheless, more than 50% of our unigenes are full length (21,228 out of 42,810) based on the BLASTN alignment (E-value < 10-10 and query coverage > 90% in relation to Shandong gene models). In addition, 87.5% of our unigenes (37,447 out of 42,810) were mapped to Shandong gene models and 94.6% (40,480 out of 42,810) were mapped to the Shandong genomic sequence [12]. These results indicate that most of our sequence assembly is correct based on its high similarity to a closely related accession.
Many SNPs were found between our Yukon unigenes and the Shandong gene models [12] when the Yukon unigenes were aligned with the reference Shandong gene models (Table 3, Additional file 1). The significance of the putative SNPs in the context of abiotic stress tolerance or other phenotypic variation between the Yukon and Shandong accessions remains to be determined.
Based on the MapMan ontology [42,43], functional annotation of both T. salsuginea unigenes and Arabidopsis genes revealed similar proportions of genes in each functional group, with the exception of genes annotated as encoding proteins with a function in photosynthesis, which were more abundant in T. salsuginea, and genes encoding proteins annotated as functioning in DNA metabolism, which were less abundant. A similar observation was also made when we identified 12,272 Yukon unigenes that were likely derived from gene duplication events relative to Shandong gene models (Additional file 17). These duplicated unigenes map for most of their length (coverage > 90%) over Shandong genes and are significantly enriched for functional categories of protein synthesis, lipid metabolism, photosynthesis and abiotic/biotic stress (Table 2). The higher abundance of photosynthesis-related genes, together with the large number of such genes that were found to be regulated during cold acclimation in the microarray study, may be related to the importance of an effective regulation of photosynthesis for the abiotic stress tolerance of plants to sustain growth and avoid oxidative damage (see [70] for a review).
The percentage of genes in different functional groups in T. salsuginea that were predicted to be targeted by miRNAs was generally lower than the corresponding percentage in Arabidopsis, except for target functions related to protein metabolism and mitochondrial electron transport, where T. salsuginea showed much higher proportions. Whether any of the relevant miRNAs play a role in abiotic stress tolerance, however, remains to be investigated. In addition, some genes encoding LEA proteins were present in higher copy numbers of orthologous genes in the T. salsuginea than in the Arabidopsis genome. This is in agreement with the observed higher frequency of gene duplications in the T. salsuginea[11] and T. parvula genomes [13]. This may suggest that both Thellungiella species retained a higher fraction of duplicated genes since the last whole genome duplication event [71,72] or after tandem duplications that diverged in function (neofunctionalization; [73]) as an adaptive strategy to survive in habitats with harsher environmental stress conditions than Arabidopsis[74].
On the other hand, the analysis of PP2C, PTP(DSP) and MAPK(K) in T. salsuginea, T. parvula and A. thaliana indicated very similar numbers of putative genes and proteins of these families in all three species. This may indicate that the retention of duplicated genes was highly selective, a hypothesis that was also put forward in the most recent T. salsuginea genome sequencing study [12].
The assembled unigenes were used for the construction of a 44 K Agilent microarray that showed superior specificity for T. salsuginea compared to Arabidopsis samples, high reproducibility and the ability to detect the complex cold-acclimation induced changes in transcript abundance. In addition, it showed interesting differences in the cold regulated transcript patterns between Arabidopsis and T. salsuginea. Interestingly, a previous paper provided evidence for possible differences in the metabolic adaptation strategies of the two species [6], based on targeted biochemical measurements of compatible solutes. In the future, the availability of microarrays will allow us to investigate such differences in adaptation strategies at the functional genomics level.
The T. salsuginea microarray consists of 43,800 oligonucleotide probes representing 41,563 unigenes. Based on a BLASTN search of all unigenes against the T. salsuginea genomic contig sequences [11], 37,622 T. salsuginea unigenes were aligned to the published genome sequence. The remaining 5,188 unigenes showed no significant BLAST hits and filtering out unigenes that were not represented on the array still left 4,092 unigenes that had produced above-background signal intensities in the hybridization experiments performed during the pre-selection procedure. Likely reasons for this discrepancy are that approximately 10% of the genome have not been sequenced yet [11], that the corresponding transcripts are encoded by genes that consist of short exons and long introns that may result in low BLASTN hit score values, or that these unigenes have relatively high sequencing or sequence assembly errors. The latter point may have resulted in the relatively high fraction of 71% (2,942 out of 4,092) among these unigenes that were annotated as having an unknown function, while for the whole unigene set this fraction was about 50%.
To assess whether the use of a dedicated T. salsuginea microarray really yields data of a higher quality than cross-species hybridization on an Arabidopsis array from the same manufacturer, we hybridized identical samples from either non- or cold-acclimated plants on same-species T. salsuginea and cross-species Arabidopsis arrays. In the reciprocal experiment, Arabidopsis samples were also hybridized to the same set of arrays. The results demonstrated the expected bimodal signal distribution [61,62]. However, only signals obtained from same-species array hybridizations showed a distribution consistent with a high proportion of specific hybridization reactions and a low proportion of non-specific hybridizations and noise. In fact, using the 25-mer oligonucleotide-based Affymetrix GeneChip ATH1, only 20% of the probes produced signals significantly above background with T. salsuginea samples [17]. In contrast, using longer oligonucleotide probes (70-mer) showed that approximately 80% of the probes on the Arabidopsis microarray [75] yielded significant signals after hybridization with T. salsuginea samples. However, the detection of differentially expressed T. salsuginea genes was limited to Arabidopsis homologs with an overall sequence identity of 92-95% with this type of array [2]. Obviously, the expression of T. salsuginea-specific unigenes could not be detected using Arabidopsis-based arrays and approximately 58% (2,553 out of 4,425) of the cold-responsive genes detected using the same-species array were not identified as differentially expressed from the Arabidopsis array, further stressing the need for a dedicated T. salsuginea microarray for the reliable identification of stress-regulated genes at the genomic level.
One of the challenges faced when using microarrays is to visualize the expression data in an efficient way [76]. To make the T. salsuginea microarray as useful as possible for the research community, we adapted the MapMan visualization [43,77] and the Robin [78] and mefisto statistical analysis tools to T. salsuginea by designing a mapping file for the annotated unigenes by transfer into the MapMan ontology [42]. To validate its performance, the T. salsuginea microarray was used to identify cold-responsive genes in comparison to Arabidopsis. The results were visualized using the MapMan tool. Further functional analysis was provided by statistical tests for the significant under- or over-represention of cold regulated genes in different functional groups. These results demonstrate that the newly designed T. salsuginea microarray reliably detects genes and pathways that are known to be differentially affected during cold acclimation in Arabidopsis and other plants. We are currently using the T. salsuginea array to investigate the cold and salt responsiveness of the T. salsuginea transcriptomes in the diverse set of accessions from all three Thellungiella species recently described [6].
Conclusions
While model species such as Arabidopsis are valuable tools to study many important traits in plants, additional models are needed to investigate the adaptation mechanisms to more extreme environmental conditions. T. salsuginea is such an emerging model species and the present study provides the first comprehensive transcriptome information for this species. The large number of additional publicly available unigene sequences will greatly facilitate genome annotation. In addition, the newly designed and validated genome-wide microarray for T. salsuginea, which will be commercially available, will be an important tool for future functional genomics investigations of plant stress tolerance. Also, we hope that the approaches developed in this study will help in the establishment of similar genomic tools for other non-model species with interesting physiological and ecological traits.
Methods
Plant material and abiotic stress treatments
Seeds of the Thellungiella salsuginea ((Pallas) O.E. Schulz) (Eutrema salsugineum) accession Yukon were kindly provided by Prof. Ray A. Bressan (Purdue University, West Lafayette, IN). For transcriptome sequencing, plants were either grown in soil or hydroponically under non-sterile conditions in a greenhouse at 16-h day length with light supplementation to reach at least 200 μE m-2 s-1 and a temperature of 20°C during the day, 18°C during the night. The nutrient solution for hydroponically-grown plants contained 1 mM MgSO4, 1 mM KH2PO4, 1 mM NH4NO3, 0.5 mM CaCl2, 0.1 mM FeNA-EDTA, 25 mM NaCl, 0.1 mM H3BO3, 1.5 μM CuSO4, 50 μm KCl, 10 μm MnSO4, 0.075 μM Na2MoO4, 0.1 mM Na2O3Si, and 2 μM ZnSO4. Six-week-old soil-grown and five-week-old hydroponically-grown plants were treated for 24 h or 2 weeks at 4°C in a growth cabinet at 16 h day length with 90 μE m-2 s-1, or stressed with 200 mM NaCl solution by irrigation or supplementation of the nutrient solution. For drought treatment, water was withheld from soil-grown plants for 5 days (mild drought with slight curling of leaves) or 10 days (severe drought with strong curling of leaves). Shoots and roots were collected from soil- and hydroponically-grown plants, except for soil-grown roots after stress treatments. In addition, rosette leaves, cauline leaves, opened flowers and siliques were collected from control plants grown in soil or hydroponically. For all samples, material from 5 to 10 individual plants was pooled. All samples were immediately frozen in liquid nitrogen and stored at −80°C.
For the microarray experiments, T. salsuginea seeds were sown in soil and exposed to 4°C under the conditions described above for one week to promote germination. Seedlings were transferred to a greenhouse as above for 8 weeks (nonacclimated plants). For cold acclimation, plants were transferred to a 4°C growth cabinet under the conditions described above for an additional 14 days [6]. Arabidopsis plants were grown and acclimated under identical conditions [79,80], but were grown under nonacclimating conditions for 6 weeks to reach the same developmental state [6].
RNA isolation, library construction and sequencing
Total RNA was isolated and on-column DNA digestion was performed with the RNeasy Plant mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Equal amounts of RNA from 23 individual samples obtained as described above were pooled and cDNA was synthesized with the Mint-Universal kit (Evrogen, Moscow, Russia). Half of the cDNA was normalized to equalize the abundance of transcripts by using the duplex-specific nuclease normalization approach [81] with the TRIMMER-DIRECT cDNA normalization kit (Evrogen) according to the manufacturer’s protocol. The cDNA in both libraries was size-fractionated using Chroma Spin-1000 columns (Clontech, Mountainview, CA). 454 libraries were constructed using custom-made adapters with MIDs (Multiplex Identifiers) following the Roche Technical Bulletin TCB 09004, introducing SfiI-sites for creating directed sequencing libraries. The 454 libraries were immobilized on beads and clonally amplified using the GS FLX Titanium LV emPCR Kit (Lib-L) (Roche, Basel, Switzerland). The libraries were then sequenced using the GS FLX Titanium Sequencing Kit XLR70 (Roche) and GS FLX Titanium PicoTiterPlate Kit (Roche) according to the manufacturer’s protocols.
Transcriptome assembly and annotation
Adapter and polyA sequences were removed from the data with the SFF Tools of the Genome Sequencer FLX System software package 2.3 (Roche) and the cross_match (http://www.phrap.org) algorithm. The clipped reads were pre-checked for contamination by non-plant sequences using BLAST [82] searches in the NCBI database of non-redundant sequences (nr; updated at 12-02-2010) with an E-value threshold of 10-10. All reads matching as best hit a sequence originating from an organism outside the Streptophyta phylum were considered “contaminant”. These reads were a minor part of the population and were thus included during the assembly process, but contigs including them were marked as “contaminant contigs”. The assembly was conducted using the MIRA assembler program version 3.1.15 [36] with default settings and the 454 reads from the normalized, the non-normalized and the combined libraries. To improve the accuracy of sequence assembly, we downloaded the 44,551 publicly available T. halophila and T. salsuginea ESTs from GenBank, NCBI (as of April 19, 2010) and included them in all assemblies. Reads not alignining to any other read (singletons) were not included in the final contig population. Average contig coverage was calculated as the mean number of reads per base per contig. The N50 was calculated as the contig length above which 50% of the sequence information is contained, i.e. 50% of the nucleotides in the output are contained in contigs of length N50 or longer.
All contigs were checked for presence of open reading frames (ORFs) as previously described [83]. To assess the completeness of the transcriptome assemblies and the degree of overlap between T. salsuginea and Arabidopsis, we used BLASTX to align the contig sequences to the 27,739 Arabidopsis Information Resource (TAIR9; [84]) peptide sequences [85]. We calculated the percentage of Arabidopsis proteins matching T. salsuginea contigs with a loose threshold to account for interspecies variation (E-value < 10-3). All following steps were conducted on the assembly based on the combined library only. Since the population of plants used for RNA extraction was not homozygous, SNPs generated several nearly identical contigs. We aligned each contig to all contigs in the population via BLAST, to identify clusters of contigs matching each other with a sequence coverage and identity higher than 99%. A multiple alignment was produced for each cluster using MUSCLE [86]. Consensus sequences for each cluster were extracted from the multiple alignments using the consambig tool from the EMBOSS suite [87]. Where disagreeing base pairs were found, the resulting cluster sequence was dubbed using the International Union of Pure and Applied Chemistry (IUPAC) code for nucleotide ambiguity.
Functional classification of the putative transcripts was performed using the Mercator pipeline [41]. Mercator aligns all sequences against five different databases: TAIR9 proteins [85], SwissProt/Uniprot plant proteins (PPAP) [88], Conserved Domain Database (CDD) [89], Clusters of Orthologous Groups (COG) [90], and InterProScan [91] and subsequently computes preliminary MapMan bin [43] codes/ontology based on manually curated reference classifications using a majority vote scheme. The programs used to perform the searches were RPSBLAST [92] for CDD and COG and BLASTX [82] for TAIR9 and PPAP. Hits with bit scores lower than 50 were ignored as not significantly similar. The sequencing procedure does not guarantee that the orientation of the original mRNAs is kept. Hence any of the 42,810 transcripts can be either 5″-3′ or 3′-5′ oriented. To unify the orientation, we used the protein models present in the NCBI nr database and, where available, we used the best BLAST hit (E-value < 10-3) to define the orientation of the transcript.
To estimate the number of T. salsuginea unigenes that were orthologs of Arabidopsis or T. parvula genes, we used BLASTX to search the unigenes with a stringent threshold (E-value < 10-10) against Arabdopsis TAIR10 database or coding sequence of T. parvula genome assembly version 2 [13]. To estimate the number of T. salsuginea unigenes/transcripts that were derived from the T. salsuginea genome, we used BLASTN to search the unigenes with a stringent threshold (E-value < 10-50) against the T. salsuginea genome assembly [11]. In addition, we took advantage of the recently published gene models of the Shandong accession of T. salsuginea (Eutrema salsugineum) [12] by comparing them to the unigenes from the Yukon accession used in our study, using BLASTN with an E-value < 10-10. We estimated the full length genes of Yukon by aligning the unigene sequences with Shandong gene models using BLASTN (E-value < 10-10) and > 90% coverage of the aligned query region with the nucleotide sequence length of the published gene models (query coverage). An estimate of single nucleotide variation between Shandong gene models and Yukon unigenes was obtained using VarScan version 2.3.5 with default parameters [37] with Shandong gene models [12] as reference sequences. Ambiguous single nucleotide variations (more than one nucleotide variation at a given nucleotide position) were filtered out.
Orthologs were identified to determine duplicated genes among Yukon unigenes relative to Shandong gene models based on BLASTN analysis (E-value < 10-10) and aligned query coverage > 90% compared to Shandong gene models.
All T. salsuginea contigs were searched by BLASTN against the complete set of miRNA precursor transcripts from the plant miRNA database [93] with an E-value < 10-10. The functions of the predicted miRNAs were inferred from the target transcripts that were extracted based on the tables provided as search results [48].These inferred functions were classified based on MapMan ontology. Bincodes were truncated at the primary level of the hierarchy. We aslo performed a similar miRNA prediction of Arabidopsis TAIR9 transcripts and classified their functions based on MapMan ontology with identical search parameter settings.
PP2C/DSP/PTP/MAPK or MAPKK protein sequences derived from the A. thaliana, T. parvula and T. salsuginea genomes (http://www.arabidopsis.org/, [11,13]) were compared to T. salsuginea unigenes using BLASTN (e < 10-20). BLASTX (e < 10-60) scans were performed using putative T. salsuginea protein models to verify BLASTN hits [94]. Unigenes were translated according to BLASTX results. Individual T. salsuginea putative MAPKK, MAPK, PP2C, DSP and PTP proteins were designated according to the corresponding orthologous protein annotations in Arabidopsis. Multiple alignments of Arabidopsis and T. salsuginea putative PP2Cs, PTPs, DSPs, MAPKKs and MAPKs were performed using MUSCLE [86,95] and were edited manually afterwards. Unrooted phylogenetic trees were computed with UGENE [96] using a JTT model with standard parameters (gamma distribution and a coefficient of variation of substitution rates among sites of 0.5).
Genes encoding putative LEA proteins of T. salsuginea were identified based on reciprocal BLASTX (E < 10-10) searches of the 51 Arabidopsis LEA genes [58] against all T. salsuginea unigenes. All resulting LEA proteins were aligned using ClustalW and an unrooted dendogram was drawn [97]. GRAVY (grand average of hydropathy) values were determined using protparam [98]. Subcellular localization was predicted from protein sequence analysis using TargetP [99].
Microarray design and hybridization
We designed a T. salsuginea microarray based on the custom gene expression 4 × 44 K platform from Agilent, consisting of four arrays per slide with 45,220 features, 43,803 of which are user-defined 60-mer oligonucleotide probes. Probes and microarrays were designed by imaGenes GmbH (Berlin, Germany). Briefly, 415,000 oligonucleotide probes were designed from the 42,810 contig sequences and printed as 2 × 400 K arrays. The same pooled RNA that was used for the sequencing libraries was used for the synthesis of Cy3-labeled cRNA that was hybridized to one of these arrays. Genomic DNA was extracted from rosette leaves of 6-week-old T. salsuginea plants using the DNeasy mini kit (Qiagen). Fragmented and labeled genomic DNA was hybridized to the other 400 K array. The 43,800 best-performing probes were selected and submitted to Agilent eArray platform to fabricate 4 × 44 K arrays. The arrays are commercially available from Agilent [60] with the design number 031554.
Microarray data analysis
The feature extraction files were analyzed using the Limma bioconductor package implemented in the Robin/RobiNA tool [78,100]. Background signals were subtracted and values were quantile-normalized before a linear model was fitted for statistical testing of differential expression. Transcripts showing a log2 fold change ≥ 1 or ≤ −1 with a p-value below 0.05 after false discovery rate correction [64] in the contrast between cold acclimated and nonacclimated samples were accepted as significantly changed. Non-acclimated replicate 1 of Col-0 hybridized to the Arabidopsis array was an outlier and was excluded from the analysis of differential gene expression. Otherwise, all RNA samples were included in all analyses. Statistical comparisons of functional groups of cold-responsive genes were made against all genes in a particular group represented on the array using Fisher’s exact test implemented in the mefisto software [101]. Significantly regulated genes were considered as significantly over- or under-represented in a MapMan functional group at p < 0.05 after false discovery rate correction [64]. Microsoft Excel 2010 and the R software [102] were used to perform general analyses.
Availability of supporting data
The 454 reads for Thellungiella salsuginea reported here have been submitted to NCBI sequence read archive (SRA, http://www.ncbi.nlm.nih.gov/sra) under the accession numbers SRX197603 and SRX197604. Microarray hybridization results are available at GEO (http://www.ncbi.nlm.nih.gov/geo) under the accession number GSE42156.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YPL and SK performed the experiments, YPL, RR and DKH designed the project, YPL, FMG, ML, KK, BU and IM performed data analysis and provided bioinformatic tools, YPL and DKH drafted the manuscript, all authors participated in manuscript writing. All authors read and approved the final manuscript.
Supplementary Material
A list of non-ambiguous single nucleotide variations identified between Shandong gene models [[12]] and Yukon unigenes using VarScan [[37]].
Arabidopsis MAPK and MAPKK genes and the corresponding T. salsuginea contigs. The indicated groups refer to the Arabidopsis MAPK and MAPKK protein nomenclature [103].
Arabidopsis PP2C genes and the corresponding T. salsuginea contigs. The indicated clusters refer to the Arabidopsis PP2C protein nomenclature [52] and the phylogenetic tree shown in Additional file 5.
Arabidopsis PTP and DSP genes and the corresponding T. salsuginea contigs.
Unrooted phylogenetic tree of all Arabidopsis and T. salsuginea PP2C proteins shown in Additional file3.
Unrooted phylogenetic tree of all Arabidopsis and T. salsuginea PTP and DSP proteins shown in Additional file4.
Unrooted phylogenetic tree of all Arabidopsis and T. salsuginea MAPK and MAPKK proteins shown in Additional file2. The indicated groups refer to the Arabidopsis MAPK and MAPKK protein nomenclature [103].
List of all genes encoding MAPK, MAPKK, PP2C, PTP and DSP proteins in Arabidospis and of all putative genes encoding orthologous proteins in Thellungiella salsuginea. The table contains the gene identifiers, the number of encoded amino acids for each gene and the derived protein sequences.
List of all genes encoding LEA proteins in Arabidospis and of all putative genes encoding orthologous LEA proteins in T. salsuginea. The table contains the gene identifiers, the number of encoded amino acids for each gene and the derived protein sequences.
Unrooted dendogram of all Arabidopsis and T. salsuginea LEA proteins. The sequence numbers and their corresponding protein sequences are listed in Additional file 9.
Assignment of putative T. salsuginea LEA proteins to different protein family domains based on multiple sequence alignment and phylogeny analyses of putative T. salsuginea and known Arabidopsis LEA proteins.
Characteristics of T. salsuginea LEA proteins.
Reproducibility of signal intensities of all hybridized probes among biological replicates of non-acclimated (NA) and cold acclimated leaf samples (ACC).
Correlation of fold change of different probes designed from the same contigs. Log2-transformed fold change of mean signal intensities for 2,237 probe pairs from the same contigs for the three biological replicates.
Number of probes identified from the reciprocal hybridization of RNA samples derived from non-acclimated or cold acclimated leaves to Arabidopsis and T. salsuginea arrays.
Statistical analysis showing over- and under-representation of MapMan functional groups among significantly cold regulated genes.
List of putative duplicated Yukon unigenes with respect to Shandong gene models. The annotations provided are derived from Mercator and Mapman software (Yukon unigenes) and from the Phytozome annotation (Shandong gene models).
Contributor Information
Yang Ping Lee, Email: yangp.lee@feldaglobal.com.
Federico M Giorgi, Email: fmg2117@c2b2.columbia.edu.
Marc Lohse, Email: lohse@mpimp-golm.mpg.de.
Kotryna Kvederaviciute, Email: kotryna.kvederaviciute@gmail.com.
Sven Klages, Email: klages@molgen.mpg.de.
Björn Usadel, Email: b.usadel@bio1.rwth-aachen.de.
Irute Meskiene, Email: irute.meskiene@univie.ac.at.
Richard Reinhardt, Email: rr@molgen.mpg.de.
Dirk K Hincha, Email: hincha@mpimp-golm.mpg.de.
Acknowledgements
We would like to thank Prof. Ray Bressan (Purdue University, West Lafayette, IN) for making his Thellungiella salsuginea seed collection available to us. YPL thanks the Swiss National Science Foundation and the Max Planck Society for post-doctoral fellowships.
References
- Bressan RA, Zhang C, Zhang H, Hasegawa PM, Bohnert HJ, Zhu JK. Learning from the Arabidopsis experience. The next gene search paradigm. Plant Physiol. 2001;127:1354–1360. doi: 10.1104/pp.010752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q, Li P, Ma S, Indu RS, Bohnert HJ. Salinity stress adaptation competence in the extremophile Thellungiella halophila in comparison with its relative Arabidopsis thaliana. Plant J. 2005;44:826–839. doi: 10.1111/j.1365-313X.2005.02587.x. [DOI] [PubMed] [Google Scholar]
- Griffith M, Timonin M, Wong ACE, Gray GR, Akhter SR, Saldanha M, Rogers MA, Weretilnyk EA, Moffatt BA. Thellungiella: an Arabidopsis-related model plant adapted to cold temperatures. Plant Cell Environ. 2007;30:529–538. doi: 10.1111/j.1365-3040.2007.01653.x. [DOI] [PubMed] [Google Scholar]
- Inan G, Zhang Q, Li P, Wang Z, Cao Z, Zhang H, Zhang C, Quist TM, Goodwin SM, Zhu J. et al. Salt cress. A halophyte and cryophyte Arabidopsis relative model system and its applicability to molecular genetic analyses of growth and development of extremophiles. Plant Physiol. 2004;135:1718–1737. doi: 10.1104/pp.104.041723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant S, Bi YM, Weretilnyk E, Barak S, Rothstein SJ. The Arabidopsis halophytic relative Thellungiella halophila tolerates nitrogen-limiting conditions by maintaining growth, nitrogen uptake, and assimilation. Plant Physiol. 2008;147:1168–1180. doi: 10.1104/pp.108.118125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YP, Babakov A, de Boer B, Zuther E, Hincha DK. Comparison of freezing tolerance, compatible solutes and polyamines in geographically diverse collections of Thellungiella spec. and Arabidopsis thaliana accessions. BMC Plant Biol. 2012;12:131. doi: 10.1186/1471-2229-12-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CE, Li Y, Whitty BR, Díaz-Camino C, Akhter SR, Brandle JE, Golding GB, Weretilnyk EA, Moffatt BA, Griffith M. Expressed sequence tags from the Yukon ecotype of Thellungiella reveal that gene expression in response to cold, drought and salinity shows little overlap. Plant Mol Biol. 2005;58:561–574. doi: 10.1007/s11103-005-6163-6. [DOI] [PubMed] [Google Scholar]
- Al-Shehbaz IA, O’Kane SL, Price RA. Generic placement of species excluded from Arabidopsis (Brassicaceae) Novon. 1999;9:296–307. doi: 10.2307/3391724. [DOI] [Google Scholar]
- Amtmann A. Learning from evolution: Thellungiella generates new knowledge on essential and critical components of abiotic stress tolerance in plants. Mol Plant. 2009;2:3–12. doi: 10.1093/mp/ssn094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, German DA. Taxonomy and systematics are key to biological information: Arabidopsis, Eutrema (Thellungiella), Noccaea and Schrenkiella (Brassicaceae) as examples. Front Plant Sci. 2013;4:267. doi: 10.3389/fpls.2013.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H-J, Zhang Z, Wang J-Y, Oh D-H, Dassanayake M, Liu B, Huang Q, Sun H-X, Xia R, Wu Y. et al. Insights into salt tolerance from the genome of Thellungiella salsuginea. Proc Natl Acad Sci USA. 2012;109:12219–12224. doi: 10.1073/pnas.1209954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Jarvis DJ, Chen H, Beilstein M, Grimwood J, Jenkins J, Shu SQ, Prochnik S, Xin M, Ma C. et al. The reference genome of the halophytic plant Eutrema salsugineum. Front Plant Sci. 2013;4:46. doi: 10.3389/fpls.2013.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassanayake M, Oh D-H, Haas JS, Hernandez A, Hong H, Ali S, Yun D-J, Bressan RA, Zhu J-K, Bohnert HJ. et al. The genome of the extremophile crucifer Thellungiella parvula. Nat Genet. 2011;43:913–918. doi: 10.1038/ng.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Mane SP, Sioson AA, Robinet CV, Heath LS, Bohnert HJ, Grene R. Effects of chronic ozone exposure on gene expression in Arabidopsis thaliana ecotypes and in Thellungiella halophila. Plant Cell Environ. 2006;29:854–868. doi: 10.1111/j.1365-3040.2005.01465.x. [DOI] [PubMed] [Google Scholar]
- Oh D, Gong Q, Ulanov A, Zhang Q, Li Y, Ma W, Yun D, Bressan RA, Bohnert HJ. Sodium stress in the halophyte Thellungiella halophila and transcriptional changes in a thsos1-RNA interference line. J Integr Plant Biol. 2007;49:1484–1496. doi: 10.1111/j.1672-9072.2007.00548.x. [DOI] [Google Scholar]
- Taji T, Seki M, Satou M, Sakurai T, Kobayashi M, Ishiyama K, Narusaka Y, Narusaka M, Zhu JK, Shinozaki K. Comparative genomics in salt tolerance between Arabidopsis and Arabidopsis-related halophyte salt cress using Arabidopsis microarray. Plant Physiol. 2004;135:1697–1709. doi: 10.1104/pp.104.039909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V, Wang B, Dominy PJ, Fricke W, Amtmann A. Thellungiella halophila, a salt-tolerant relative of Arabidopsis thaliana, possesses effective mechanisms to discriminate between potassium and sodium. Plant Cell Environ. 2003;27:1–14. [Google Scholar]
- Wong CE, Li Y, Labbe A, Guevara D, Nuin P, Whitty B, Diaz C, Golding GB, Gray GR, Weretilnyk EA. et al. Transcriptional profiling implicates novel interactions between abiotic stress and hormonal responses in Thellungiella, a close relative of Arabidopsis. Plant Physiol. 2006;140:1437–1450. doi: 10.1104/pp.105.070508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taji T, Sakurai T, Mochida K, Ishiwata A, Kurotani A, Totoki Y, Toyoda A, Sakaki Y, Seki M, Ono H. et al. Large-scale collection and annotation of full-length enriched cDNAs from a model halophyte, Thellungiella halophila. BMC Genomics. 2008;11:262. doi: 10.1186/1471-2229-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li P, Fredricksen M, Gong Z, Kim CS, Zhang C, Bohnert HJ, Zhu J, Bressan RA, Hasegawa PM. et al. Expressed sequence tags from Thellungiella halophila, a new model to study plant salt-tolerance. Plant Sci. 2004;166:609–616. doi: 10.1016/j.plantsci.2003.10.030. [DOI] [Google Scholar]
- Hahn D, Ragland G, Shoemaker D, Denlinger D. Gene discovery using massively parallel pyrosequencing to develop ESTs for the flesh fly Sarcophaga crassipalpis. BMC Genomics. 2009;10:234. doi: 10.1186/1471-2164-10-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchman T, Geist K, Grahnen J, Benkman C, Buerkle CA. Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics. 2010;11:180. doi: 10.1186/1471-2164-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Li Y, Wu Q, Luo H, Sun Y, Song J, Lui E, Chen S. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genomics. 2010;11:262. doi: 10.1186/1471-2164-11-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulza E, Shin M, Blanc G, Audic S, Laabir M, Collos Y, Claverie J, Grzebyk D. Gene expression in proliferating cells of the dinoflagellate Alexandrium catenella (Dinophyceae) Appl Environ Microbiol. 2010;76:4521–4529. doi: 10.1128/AEM.02345-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I, Marden JH. Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol Ecol. 2008;17:1636–1647. doi: 10.1111/j.1365-294X.2008.03666.x. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Sequencing goes 454 and takes large-scale genomics into the wild. Mol Ecol. 2008;17:1629–1635. doi: 10.1111/j.1365-294X.2008.03699.x. [DOI] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Novaes E, Drost D, Farmerie W, Pappas G, Grattapaglia D, Sederoff R, Kirst M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics. 2008;9:312. doi: 10.1186/1471-2164-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaghi M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2001;11:3–11. doi: 10.1101/gr.11.1.3. [DOI] [PubMed] [Google Scholar]
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Emrich SJ, Barbazuk WB, Li L, Schnable PS. Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res. 2007;17:69–73. doi: 10.1101/gr.5145806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvie T, Harkins T. Transcriptome sequencing with the Genome Sequencer FLX system. Nat Meth. 2008;5:v–viii. [Google Scholar]
- Parkinson J, Blaxter M. Expressed sequence tags: an overview. Meth Mol Biol. 2009;533:1–12. doi: 10.1007/978-1-60327-136-3_1. [DOI] [PubMed] [Google Scholar]
- GenBank dbEST. http://www.ncbi.nlm.nih.gov/
- RIKEN full length Thellungiella cDNAs. http://www.brc.riken.jp/lab/epd/Eng/catalog/halophila.shtml.
- Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, Suhai S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–1159. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Rupe MA, Zinselmeier C, Habben J, Bowen BA, Smith OS. Allelic variation of gene expression in maize hybrids. Plant Cell. 2004;16:1707–1716. doi: 10.1105/tpc.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovan A, Keszei A, Foley W, Kulheim C. Differences in gene expression within a striking phenotypic mosaic Eucalyptus tree that varies in susceptibility to herbivory. BMC Plant Biol. 2013;13:29. doi: 10.1186/1471-2229-13-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastry BS. In: Single Nucleotide Polymorphisms. 578. Komar AA, editor. Totowa, NJ: Humana Press; 2009. SNPs: impact on gene function and phenotype; pp. 3–22. [Google Scholar]
- Mercator. http://mapman.gabipd.org/web/guest/app/mercator.
- Usadel B, Poree F, Nagel A, Lohse M, Czedik-Eysenberg A, Stitt M. A guide to using MapMan to visualize and compare omics data in plants: a case study in the crop species, maize. Plant Cell Environ. 2009;32:1211–1229. doi: 10.1111/j.1365-3040.2009.01978.x. [DOI] [PubMed] [Google Scholar]
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37:914–939. doi: 10.1111/j.1365-313X.2004.02016.x. [DOI] [PubMed] [Google Scholar]
- Meins F Jr, Si-Ammour A, Blevins T. RNA silencing systems and their relevance to plant development. Annu Rev Cell Dev Biol. 2005;21:297–318. doi: 10.1146/annurev.cellbio.21.122303.114706. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10:1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom K-H, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant microRNA database. http://bioinformatics.cau.edu.cn/PMRD/
- Rodriguez MC, Petersen M, Mundy J. Mitogen-activated protein kinase signaling in plants. Annu Rev Plant Biol. 2010;61:621–649. doi: 10.1146/annurev-arplant-042809-112252. [DOI] [PubMed] [Google Scholar]
- Bartels S, Besteiro MA, Lang D, Ulm R. Emerging functions for plant MAP kinase phosphatases. Trends Plant Sci. 2010;15:322–329. doi: 10.1016/j.tplants.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Fuchs S, Grill E, Meskiene I, Schweighofer A. Type 2C protein phosphatases in plants. FEBS J. 2013;280:681–693. doi: 10.1111/j.1742-4658.2012.08670.x. [DOI] [PubMed] [Google Scholar]
- Schweighofer A, Hirt H, Meskiene I. Plant PP2C phosphatases: emerging functions in stress signaling. Trends Plant Sci. 2004;9:236–243. doi: 10.1016/j.tplants.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Xue T, Wang D, Zhang S, Ehlting J, Ni F, Jakab S, Zheng C, Zhong Y. Genome-wide and expression analysis of protein phosphatase 2C in rice and Arabidopsis. BMC Genomics. 2008;9:550. doi: 10.1186/1471-2164-9-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerk D, Templeton G, Moorhead GB. Evolutionary radiation pattern of novel protein phosphatases revealed by analysis of protein data from the completely sequenced genomes of humans, green algae, and higher plants. Plant Physiol. 2008;146:351–367. doi: 10.1104/pp.107.111393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel LP, Nicole MC, Sritubtim S, Morency MJ, Ellis M, Ehlting J, Beaudoin N, Barbazuk B, Klessig D, Lee J. et al. Ancient signals: comparative genomics of plant MAPK and MAPKK gene families. Trends Plant Sci. 2006;11:192–198. doi: 10.1016/j.tplants.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Tunnacliffe A, Hincha DK, Leprince O, Macherel D. In: Sleeping Beauties - Dormancy and Resistance in Harsh Environments. Lubzens E, Cerda J, Clark M, editor. Berlin: Springer; 2010. LEA proteins: versatility of form and function; pp. 91–108. [Google Scholar]
- Tunnacliffe A, Wise MJ. The continuing conundrum of LEA proteins. Naturwissenschaften. 2007;94:791–812. doi: 10.1007/s00114-007-0254-y. [DOI] [PubMed] [Google Scholar]
- Hundertmark M, Hincha DK. LEA (Late Embryogenesis Abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genomics. 2008;9:118. doi: 10.1186/1471-2164-9-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- imaGenes Pre-Selection Strategy. http://www.lifesciences.sourcebioscience.com/genomic-services/gene-expression-/microarray-platforms.aspx.
- Agilent. http://www.agilent.com.
- Tan PK, Downey TJ, Spitznagel EL Jr, Xu P, Fu D, Dimitrov DS, Lempicki RA, Raaka BM, Cam MC. Evaluation of gene expression measurements from commercial microarray platforms. Nucl Acids Res. 2003;31:5676–5684. doi: 10.1093/nar/gkg763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahurak M, Parmigiani G, Yu W, Scharpf RB, Berman D, Schaeffer E, Shabbeer S, Cope L. Pre-processing Agilent microarray data. BMC Bioinforma. 2007;8:142. doi: 10.1186/1471-2105-8-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thellungiella MapMan ontology/mapping file. http://mapman.gabipd.org/web/guest/mapmanstore.
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300. [Google Scholar]
- Fowler S, Thomashow MF. Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell. 2002;14:1675–1690. doi: 10.1105/tpc.003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah MA, Heyer AG, Hincha DK. A global survey of gene regulation during cold acclimation in Arabidopsis thaliana. PLoS Genet. 2005;1:e26. doi: 10.1371/journal.pgen.0010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreps JA, Wu Y, Chang HS, Zhu T, Wang X, Harper JF. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002;130:2129–2141. doi: 10.1104/pp.008532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, Narusaka M, Abe H, Kasuga M, Yamaguchi-Shinozaki K, Carninci P, Hayashizaki Y, Shinozaki K. Monitoring the expression pattern of 1300 Arabidopsis genes under drought and cold stresses by using a full-length cDNA microarray. Plant Cell. 2001;13:61–72. doi: 10.1105/tpc.13.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, Oono Y, Kamiya A, Nakajima M, Enju A, Sakurai T. et al. Monitoring of the expression profiles of 7000 Arabidopsis genes under drought, cold, and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002;31:279–292. doi: 10.1046/j.1365-313X.2002.01359.x. [DOI] [PubMed] [Google Scholar]
- Foyer CH, Noctor G. Redox regulation in photosynthetic organisms: signaling, acclimation, and practical implications. Antioxid Redox Signal. 2009;11:861–905. doi: 10.1089/ars.2008.2177. [DOI] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16:1667–1678. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, De Bodt S, Raes J, Casneuf T, Van Montagu M, Kuiper M, Van de Peer Y. Modeling gene and genome duplications in eukaryotes. Proc Natl Acad Sci USA. 2005;102:5454–5459. doi: 10.1073/pnas.0501102102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell. 2004;16:1679–1691. doi: 10.1105/tpc.021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DH, Dassanayake M, Bohnert HJ, Cheeseman JM. Life at the extreme: lessons from the genome. Genome Biol. 2012;13:241. doi: 10.1186/gb4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis spotted microarray. http://ag.arizona.edu/microarray.
- Gehlenborg N, O’Donoghue SI, Baliga NS, Goesmann A, Hibbs MA, Kitano H, Kohlbacher O, Neuweger H, Schneider R, Tenenbaum D. et al. Visualization of omics data for systems biology. Nat Meth. 2010;7:S56–S68. doi: 10.1038/nmeth.1436. [DOI] [PubMed] [Google Scholar]
- Usadel B, Nagel A, Thimm O, Redestig H, Blaesing OE, Palacios-Rojas N, Selbig J, Hannemann J, Piques MC, Steinhauser D. et al. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of coresponding genes, and comparison with known responses. Plant Physiol. 2005;138:1195–1204. doi: 10.1104/pp.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M, Nunes-Nesi A, Krüger P, Nagel A, Hannemann J, Giorgi FM, Childs L, Osorio S, Walther D, Selbig J. et al. Robin: an intuitive wizard application for R-based expression microarray quality assessment and analysis. Plant Physiol. 2010;153:642–651. doi: 10.1104/pp.109.152553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah MA, Wiese D, Freund S, Fiehn O, Heyer AG, Hincha DK. Natural genetic variation of freezing tolerance in Arabidopsis. Plant Physiol. 2006;142:98–112. doi: 10.1104/pp.106.081141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde P, Hincha DK, Heyer AG. Heterosis in the freezing tolerance of crosses between two Arabidopsis thaliana accessions (Columbia-0 and C24) that show differences in non-acclimated and acclimated freezing tolerance. Plant J. 2004;38:790–799. doi: 10.1111/j.1365-313X.2004.02080.x. [DOI] [PubMed] [Google Scholar]
- Zhulidov PA, Bogdanova EA, Shcheglov AS, Vagner LL, Khaspekov GL, Kozhemyako VB, Matz MV, Meleshkevitch E, Moroz LL, Lukyanov SA. et al. Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucl Acids Res. 2004;32:e37. doi: 10.1093/nar/gnh031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Min XJ, Butler G, Storms R, Tsang A. OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucl Acids Res. 2005;33:W677–W680. doi: 10.1093/nar/gki394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAIR. http://www.arabidopsis.org.
- Rhee SY, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, Garcia-Hernandez M, Huala E, Lander G, Montoya M. et al. The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucl Acids Res. 2003;31:224–228. doi: 10.1093/nar/gkg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Schneider M, Bairoch A, Wu CH, Apweiler R. Plant protein annotation in the UniProt knowledgebase. Plant Physiol. 2005;138:59–66. doi: 10.1104/pp.104.058933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z. et al. CDD: a Conserved Domain Database for protein classification. Nucl Acids Res. 2005;33:D192–D196. doi: 10.1093/nar/gni191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R, Fedorova N, Jackson J, Jacobs A, Kiryutin B, Koonin E, Krylov D, Mazumder R, Mekhedov S, Nikolskaya A. et al. The COG database: an updated version includes eukaryotes. BMC Bioinforma. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- Schäffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucl Acids Res. 2001;29:2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yu J, Li D, Zhang Z, Liu F, Zhou X, Wang T, Ling Y, Su Z. PMRD: plant microRNA database. Nucl Acids Res. 2010;38:D806–D813. doi: 10.1093/nar/gkp818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thellungiella salsuginea genome sequence. http://www.phytozome.org/search.php.
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinforma. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012;28:1166–1167. doi: 10.1093/bioinformatics/bts091. [DOI] [PubMed] [Google Scholar]
- Unrooted dendrograms. http://align.genome.jp.
- PROTPARAM. http://www.expasy.org/tools/protparam.html.
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP, and related tools. Nat Prot. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Lohse M, Bolger AM, Nagel A, Fernie AR, Lunn JE, Stitt M, Usadel B. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucl Acids Res. 2012;40:W622–W627. doi: 10.1093/nar/gks540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefisto software. http://www.usadellab.org/cms/index.php?page=mefisto.
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAPK Group. Mitogen-activated protein kinase cascades in plants: a new nomenclature. Trends Plant Sci. 2002;7:301–308. doi: 10.1016/S1360-1385(02)02302-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A list of non-ambiguous single nucleotide variations identified between Shandong gene models [[12]] and Yukon unigenes using VarScan [[37]].
Arabidopsis MAPK and MAPKK genes and the corresponding T. salsuginea contigs. The indicated groups refer to the Arabidopsis MAPK and MAPKK protein nomenclature [103].
Arabidopsis PP2C genes and the corresponding T. salsuginea contigs. The indicated clusters refer to the Arabidopsis PP2C protein nomenclature [52] and the phylogenetic tree shown in Additional file 5.
Arabidopsis PTP and DSP genes and the corresponding T. salsuginea contigs.
Unrooted phylogenetic tree of all Arabidopsis and T. salsuginea PP2C proteins shown in Additional file3.
Unrooted phylogenetic tree of all Arabidopsis and T. salsuginea PTP and DSP proteins shown in Additional file4.
Unrooted phylogenetic tree of all Arabidopsis and T. salsuginea MAPK and MAPKK proteins shown in Additional file2. The indicated groups refer to the Arabidopsis MAPK and MAPKK protein nomenclature [103].
List of all genes encoding MAPK, MAPKK, PP2C, PTP and DSP proteins in Arabidospis and of all putative genes encoding orthologous proteins in Thellungiella salsuginea. The table contains the gene identifiers, the number of encoded amino acids for each gene and the derived protein sequences.
List of all genes encoding LEA proteins in Arabidospis and of all putative genes encoding orthologous LEA proteins in T. salsuginea. The table contains the gene identifiers, the number of encoded amino acids for each gene and the derived protein sequences.
Unrooted dendogram of all Arabidopsis and T. salsuginea LEA proteins. The sequence numbers and their corresponding protein sequences are listed in Additional file 9.
Assignment of putative T. salsuginea LEA proteins to different protein family domains based on multiple sequence alignment and phylogeny analyses of putative T. salsuginea and known Arabidopsis LEA proteins.
Characteristics of T. salsuginea LEA proteins.
Reproducibility of signal intensities of all hybridized probes among biological replicates of non-acclimated (NA) and cold acclimated leaf samples (ACC).
Correlation of fold change of different probes designed from the same contigs. Log2-transformed fold change of mean signal intensities for 2,237 probe pairs from the same contigs for the three biological replicates.
Number of probes identified from the reciprocal hybridization of RNA samples derived from non-acclimated or cold acclimated leaves to Arabidopsis and T. salsuginea arrays.
Statistical analysis showing over- and under-representation of MapMan functional groups among significantly cold regulated genes.
List of putative duplicated Yukon unigenes with respect to Shandong gene models. The annotations provided are derived from Mercator and Mapman software (Yukon unigenes) and from the Phytozome annotation (Shandong gene models).