

Abstract
Matrix metalloproteinases (MMPs) are members of the neutral proteinase family. They were previously thought to be anti-fibrotic because of their ability to degrade and remodel of extracellular matrix. However, recent studies have shown that MMPs are implicated in initiation and progression of kidney fibrosis through tubular cell epithelial–mesenchymal transition (EMT) as well as activation of resident fibroblasts, endothelial-mesenchymal transition (EndoMT) and pericyte-myofibroblast transdifferentiation. Interstitial macrophage infiltration has also been shown to correlate with the severity of kidney fibrosis in various chronic kidney diseases. MMPs secreted by macrophages, especially MMP-9, has been shown by us to be profibrotic by induction of tubular cells EMT. EMT is mainly induced by transforming growth factor-β (TGF-β). However, MMP-9 was found by us and others to be up-regulated by TGF-β1 in kidney tubular epithelial cells and secreted by activated macrophages, resulting in EMT and ultimately kidney fibrosis. Therefore, MMP-9 may serve as a potential therapeutic target to prevent kidney fibrosis in chronic kidney disease. This review, by a particular focus on EMT, seeks to provide a comprehensive understanding of MMPs, especially MMP-9, in kidney fibrosis.
Keywords: Matrix metalloproteinase, Chronic kidney disease, Kidney fibrosis, Epithelial–mesenchymal transition Transforming growth factor-β
Core tip: Matrix metalloproteinases (MMPs) were previously known to be anti-fibrotic for their ability to degrade and remodel extracellular matrix proteins. Recent studies including our own have shown that MMPs are implicated in initiation and progression of kidney fibrosis. MMP-9 of both tubular and macrophage origins were found to be able to induce epithelial-mesenchymal transition of tubular cells, an important mechanism causing kidney fibrosis. This review, by focus on MMP-9 and epithelial–mesenchymal transition, seeks to provide a comprehensive understanding for the roles of MMPs in kidney fibrosis.
INTRODUCTION
Kidney fibrosis is the final common pathway of parenchymal destruction for diverse chronic kidney diseases (CKD) including those resulting from glomerulonephritis, diabetes and hypertension[1-3]. It is characterized by substantial accumulation and activation of interstitial myofibroblasts, and excessive deposition and accumulation of extracellular matrix by myofibroblasts[4]. Myofibroblasts plays a pivotal role in the development of CKD and kidney fibrosis. Several cellular events, including tubular cell epithelial-mesenchymal transition (EMT), endothelial-mesenchymal transtion[5-7] and fibroblast activation, have been recognized as major sources of myofibroblast in kidney fibrosis. Although EMT has been challenged recently as a source of interstitial myofibroblasts[8], it remains generally accepted that EMT does contribute to kidney fibrosis[9-12].
Matrix metalloproteinases (MMPs) are a family of neutral proteinases,well known for their degradation and remodeling of extracellular matrix proteins[13]. However, the biological functions of MMPs are much more complex and diverse than previously assumed. MMPs also play a role in cell migration, cell-cell and cell-matrix adhesion and in release and activation of extracellular matrix-bond growth factors and cytokines. Some of these functions have been shown to play a role in the initiation and/or the progression of CKD and kidney fibrosis[14]. MMP-9 is known to be capable of cleaving osteopontin, a potent macrophage chemoattractant; activating transforming growth factor-β (TGF-β), a well known inducer of fibrosis; and inducing tubular cell EMT, an important source of myofibroblasts in renal diseases[15,16]. In this review, we will discuss roles of MMPs, especially MMP-9, in the development of CKD and kidney fibrosis.
MMPS AND INFLAMMATION IN CKD
CKD is characterized by pathological changes of glomerulosclerosis, tubular atrophy and tubulointerstitial fibrosis resulting from chronic injuries and inflammation of kidney[3]. Hypertension and diabetes are also major causes of CKD. MMPs are known to play important roles during inflammation. Besides the known roles for MMPs in extracellular matrix remodeling, they were found to be critical in recruitment and chemotaxis of inflammatory cells[17]. Elevated serum levels MMP-2 and -9 have been found in CKD patients[18] while increased levels of MMPs may serve as stress markers in these patients[19]. Previous studies have shown that macrophages play a critical role in progression of kidney fibrosis of all forms of chronic kidney diseases[20-22]. As one of the major sources for MMPs and profibrotic cytokines, macrophage has been associated with excessive accumulation of extracellular matrix proteins and myofibroblasts in CKD patients as well as experimental models[15,23]. Moreover, macrophages also release TGF-β, leading to upregulation of MMP-9 by tubular epithelial cells. MMP-9 was fond by us to mediate EMT of the tubular epithelial cells downstream of TGF-β[15,16]. A previous report has shown that serum levels of MMP-2 were correlated with proteinuria, intima media thickness and reduced kidney function in patients with CKD[24]. Plasma concentration of MMP-9 was found to be increased in the early stage of diabetic kidney disease[14]. In a rat model of chronic glomerulonephritis, expression of MMP-2 and TGF-β were shown to be significantly up-regulated[25]. In hypertension and hypertensive end-stage kidney disease (ESKD), up-regulation of MMP-9, MMP-2 and MMP-10 were observed[26]. These evidences all demonstrated upregulation of MMPs in the progression of CKD.
ROLE OF MMPS IN KIDNEY FIBROSIS
Matrix metalloproteinases, in particular MMP-2 and MMP-9 are known to play an important role in kidney fibrosis through the induction of tubular cell EMT. Results from our studies and that of Cheng et al[27] have demonstrated that MMP-2 and MMP-9 can directly induce the entire course of renal tubular cell EMT in vitro[15,27]. MMP-2 and MMP-9, which specifically cleave type IV collagen and laminin[28], major constituents of tubular basement membrane, contribute to tubular cell EMT via the disruption of tubular cell membrane integrity. This process has been recognized as a complementary step required for complete induction of tubular cell EMT, where it enables the newly transformed mesenchymal cells to migrate and invade the interstitial space and contributes to the development of fibrosis through extracellular matrix deposition[29]. In fact, induction of tubular cell EMT in vitro[30] and in vivo[31] has been shown to be associated with increased expression of MMP-2 and MMP-9. The importance of MMPs in tubular basement membrane disruption has been demonstrated by Yang et al[31] where indirect reduction of MMP-9 activity in tissue-type plasminogen activator (t-PA) deficient mice was associated with preservation of tubular basement membrane integrity, and a reduction in tubular cell EMT and kidney fibrosis in obstructive nephropathy. Recent studies from the same group showed consistent results in MMP-9 knockout mice with obstructive nephropathy[32]. However, evidence for cross basement membrane migration of mesenchymal-transited tubular epithelial cells is still lacking from current studies. Therefore, the claim of a complete EMT in kidney fibrosis as defined in cancer cells has been questioned. For this reason, a limited (type 2) EMT has been designated in the context of fibrosis[9].
EPITHELIAL-MESENCHYMAL TRANSITION IN KIDNEY FIBROSIS
EMT of renal tubular epithelial cells has been shown to be an important mechanism for development of kidney fibrosis in CKD[33,34]. It can be induced by TGF-β represents a functional transition of polarized epithelial cells into mesenchymal myofibroblast cells which are responsible for extracellular matrix deposition that leads ultimately to kidney fibrosis[35-37]. EMT is characterized by loss of epithelial markers such as E-cadherin and cytokeratin, and by nuclear translocation of β-catenin accompanied by de novo expression of mesenchymal markers typically α-smooth muscle actin (α-SMA), vimentin and fibroblast specific protein 1[38]. It is found in the development of human kidney disease and fibrosis as indicated by the findings from renal biopsy specimens that tubular epithelial cells underwent phenotypic changes including de novo SMA expression and loss of cytokeratin[29,39]. In murine model of unilateral ureteral obstruction (UUO), EMT was observed to actively participate in kidney fibrosis[36]. Other studies have shown that the number of tubular epithelial cells with EMT features is associated with decreased serum creatinine and the degree of interstitial damage in human renal biopsies of different renal diseases[29,40]. Furthermore, the progression of myofibroblast accumulation was reversed in the absence of EMT[29,31]. Whereas EMT has been extensively investigated as a major mechanism for kidney fibrosis, mesenchymal transition of kidney endothelial cells[41], activation of resident fibroblasts and pericytes[8] have also been demonstrated to contribute to myofibroblast population in kidney fibrosis. Iwano et al[33] showed conclusively using proximal tubule lineage-tagged mice that up to 36% of tubulointerstitial myofibroblasts were originated by tubular EMT. Kidney endothelial cells by endothelial lineage tracing have been shown to contribute to 30%-50% of myofibroblasts through EndoMT[34].
MMPS MEDIATE TGF-β-INDUCED EMT IN KIDNEY FIBROSIS
TGF-β plays a key role in development of kidney fibrosis. Its role in tubular cell EMT induction has been studied extensively[42]. As a sole factor, TGF-β is capable of inducing the entire course of tubular cell EMT and has also been recognized as its most potent inducer[36]. Moreover, other cytokines appear to play an indirect role dependent on TGF-β induction[43], or function synergistically with TGF-β to cause tubular cell EMT[30]. Our studies and that of Cheng et al[27] have indicated that TGF-β-induced tubular cell EMT was abrogated by inhibition of MMP activity, demonstrating the involvement of MMP in TGF-β-induced tubular cell EMT[27]. It has been reported that TGF-β is capable of inducing MMP-2 and MMP-9 expression by rat tubular epithelial cells (NRK52e)[30]. Using a different murine tubule cell line, one of our previous studies found that MMP-9 expression and activity was only induced after TGF-β treatment. TGF-β-induced tubular cell EMT was abrogated by inhibition of MMP-9 activity, suggesting that MMP-9 may play a downstream role in TGF-β induced tubular cell EMT[15]. This result is confirmed by a recent study, where we observed a significant reduction in TGF-β-induced tubular cell EMT in MMP-9 knockout mice compared to wild-type mice, despite a compensatory upregulation of MMP-3 and MMP-7[44]. Our previous study revealed that TGF-β1 induced proteolytic shedding by MMP of tubular epithelial E-cadherin. The MMP-mediated disruption of E-cadherin caused nuclear translocation of β-catenin, transcriptional induction of Slug and repression of E-cadherin transcription, and consequent tubular cell EMT[16].
CONTRIBUTION OF MACROPHAGE MMP-9 IN EMT
Macrophages are well known to play substantial roles in many aspects of human and animal models of fibrotic kidney diseases[20]. In various experimental models, ablation of macrophages has been shown to markedly attenuate kidney fibrosis. As a rich source of pro-fibrotic growth factors and proteolytic enzymes[45], macrophages play a major role in determining the outcome of CKD and kidney fibrosis. Secretion of pro-fibrotic growth factors may increase the myofibroblast population by activation of interstitial fibroblast or through the induction of tubular cell EMT. Lange-Sperandio et al[46] showed that blockade of leukocyte recruitment, including that of macrophages, by a chemokine receptor antagonist reduced tubular cell EMT and renal fibrosis, suggesting a role for macrophages in tubular cell EMT induction. Our study showed that macrophages induced tubular cell EMT via the secretion of MMPs, especially MMP-9. This result is further supported by our recent study, where a significant reduction in macrophage-induced tubular cell EMT was observed from MMP-9 knockout macrophages as compared to wild-type macrophages[44]. Moreover, in situ hybridization showed macrophages as a major source of MMP-9 in murine UUO kidney, a well established model of kidney fibrosis. We also demonstrated co-localization of macrophage MMP-9 with myofibroblasts in UUO kidney, suggesting its involvement in tubular EMT[44].
TARGETING MMP-9
The contribution of MMP-9 to kidney fibrosis has been demonstrated previously in several studies, either by indirect inhibition through the use of t-PA deficient mice (as t-PA is an inducer of MMP-9) or the use of MMP-9 knockout mice[31]. However the utility of MMP knockout models is questionable due to compensatory up-regulation of other MMPs[47,48], and because of conflicting results in cancer studies[49], where MMPs were highly involved in cancer progression.
To define the role of MMP-9 in vivo, direct inhibition of MMP-9 activity is preferable. Knowing the expression pattern and cellular origin of an MMP and whether it plays a protective or destructive role is essential in the development of an effective MMP-based therapeutic strategy. In fact, a study of Zeisberg et al[48] demonstrated that early inhibition of MMP activity, specifically that of MMP-2, 3 and 9 is protective against Alport disease in mice deficient in α3 (IV) chain of type IV collagen, whilst late-stage inhibition of MMP activity led to acceleration of disease progression associated with interstitial fibrosis and early death.
Results from our recent study demonstrated a biphasic expression of MMP-9 during early- and late-, but not mid-stage in the course of UUO[44]. Interestingly, we showed tubular cells to be the predominant source of MMP-9 during early-stage, whereas tubular cells, macrophages and myofibroblasts produced MMP-9 during late-stage UUO. Early and late-stage inhibition of MMP-9 by MMP-9 neutralizing antibody or MMP-2/9 inhibitor resulted in a reduction in: (1) MMP-9 cleaved osteopontin[44] which is known to play a role in macrophage recruitment[50]; (2) infiltration of macrophages; (3) tubular cell EMT as indicated by a reduced translocation of β-catenin and α-SMA expression in tubular epithelial cells; and (4) in kidney fibrosis[44]. Taken together, findings from our previous and recent studies have suggested a potential mechanism underlying the contribution of MMP-9 to kidney fibrosis (Figure 1).
Figure 1.
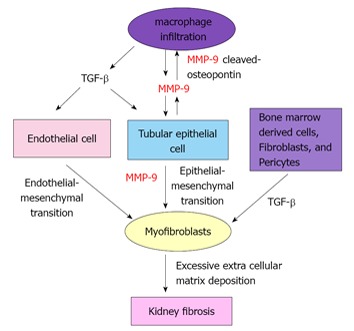
Mechanisms by which matrix metalloproteinases-9 contributes to kidney fibrosis in chronic kidney diseases. MMP-9: matrix metalloproteinases-9.
CONCLUSION
Kidney fibrosis represents a failed wound healing in progressive chronic kidney diseases. It is characterized by interstitial infiltration with mononuclear inflammatory cells, substantial accumulation and activation of interstitial myofibroblasts and consequent progressive deposition and accumulation of extracellular matrix, mainly by myofibroblasts. MMPs are proteolytic enzymes involved in degradation of extracellular matrix and basement membrane and play important roles in the progression of CKD and interstitial fibrosis. Macrophages and myofibroblasts are two major effector cells in CKD and kidney fibrosis. We and others found that MMP-induced EMT contributes to generation of myofibroblasts, and MMP-9 secreted by activated macrophages is at least partially responsible for the profibrotic role of interstitial macrophages. Disruption of E-cadherin by MMPs directly mediates tubular cell EMT downstream of TGF-β1.
Traditionally, MMPs have been considered to be anti-fibrotic factors due to their proteolytic degradation of extracellular matrix. Reduced MMP proteolytic activity or an increase of tissue inhibitors of MMP (TIMPs) was thought to account for extracellular matrix accumulation and fibrosis[14,19]. Discovery of their in vivo physiological non-extracellular matrix substrates in recent years has revealed diverse biological functions of MMPs. Against expectations, MMP-2 and MMP-9 have been shown by us and others to be profibrotic through induction of renal tubular cell EMT. Moreover, failure of MMP inhibitors in anti-cancer clinical trials has revealed that the biological function of MMPs is not simply the destruction of extracellular matrix, as originally assumed, but has emphasized that MMPs also target diverse non-extracellular matrix substrates, such as cell surface molecules, cytokines, growth factors and adhesion molecules. MMPs are dysregulated and are involved in virtually every aspect of inflammation and tissue repair. Our studies suggest that specific inhibition rather than promotion of proteolytic actions of MMP, in particular MMP-9, may offer a novel therapeutic approach to prevent renal fibrosis.
Footnotes
P- Reviewer Friedman EA S- Editor Wen LL L- Editor A E- Editor Lu YJ
References
- 1.Eddy AA. Can renal fibrosis be reversed? Pediatr Nephrol. 2005;20:1369–1375. doi: 10.1007/s00467-005-1995-5. [DOI] [PubMed] [Google Scholar]
- 2.Meguid El Nahas A, Bello AK. Chronic kidney disease: the global challenge. Lancet. 2005;365:331–340. doi: 10.1016/S0140-6736(05)17789-7. [DOI] [PubMed] [Google Scholar]
- 3.Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15:290–301. doi: 10.1007/s004670000461. [DOI] [PubMed] [Google Scholar]
- 4.Bicer A, Guclu B, Ozkan A, Kurtkaya O, Koc DY, Necmettin Pamir M, Kilic T. Expressions of angiogenesis associated matrix metalloproteinases and extracellular matrix proteins in cerebral vascular malformations. J Clin Neurosci. 2010;17:232–236. doi: 10.1016/j.jocn.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Hertig A. [Epithelial-mesenchymal transition of the renal graft] Nephrol Ther. 2008;4 Suppl 1:S25–S28. doi: 10.1016/S1769-7255(08)73648-4. [DOI] [PubMed] [Google Scholar]
- 6.Guarino M, Tosoni A, Nebuloni M. Direct contribution of epithelium to organ fibrosis: epithelial-mesenchymal transition. Hum Pathol. 2009;40:1365–1376. doi: 10.1016/j.humpath.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 7.Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179:1074–1080. doi: 10.1016/j.ajpath.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeisberg M, Duffield JS. Resolved: EMT produces fibroblasts in the kidney. J Am Soc Nephrol. 2010;21:1247–1253. doi: 10.1681/ASN.2010060616. [DOI] [PubMed] [Google Scholar]
- 10.Zeisberg M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21:1819–1834. doi: 10.1681/ASN.2010080793. [DOI] [PubMed] [Google Scholar]
- 11.Fragiadaki M, Mason RM. Epithelial-mesenchymal transition in renal fibrosis - evidence for and against. Int J Exp Pathol. 2011;92:143–150. doi: 10.1111/j.1365-2613.2011.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hertig A, Gangadhar T, Kalluri R. Renal studies provide an insight into cardiac extracellular matrix remodeling during health and disease. J Mol Cell Cardiol. 2010;48:497–503. doi: 10.1016/j.yjmcc.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hijova E. Matrix metalloproteinases: their biological functions and clinical implications. Bratisl Lek Listy. 2005;106:127–132. [PubMed] [Google Scholar]
- 14.Catania JM, Chen G, Parrish AR. Role of matrix metalloproteinases in renal pathophysiologies. Am J Physiol Renal Physiol. 2007;292:F905–F911. doi: 10.1152/ajprenal.00421.2006. [DOI] [PubMed] [Google Scholar]
- 15.Tan TK, Zheng G, Hsu TT, Wang Y, Lee VW, Tian X, Wang Y, Cao Q, Wang Y, Harris DC. Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. Am J Pathol. 2010;176:1256–1270. doi: 10.2353/ajpath.2010.090188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng G, Lyons JG, Tan TK, Wang Y, Hsu TT, Min D, Succar L, Rangan GK, Hu M, Henderson BR, et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. Am J Pathol. 2009;175:580–591. doi: 10.2353/ajpath.2009.080983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 18.Pawlak K, Mysliwiec M, Pawlak D. Peripheral blood level alterations of MMP-2 and MMP-9 in patients with chronic kidney disease on conservative treatment and on hemodialysis. Clin Biochem. 2011;44:838–843. doi: 10.1016/j.clinbiochem.2011.03.143. [DOI] [PubMed] [Google Scholar]
- 19.Musiał K, Zwolińska D. Matrix metalloproteinases (MMP-2,9) and their tissue inhibitors (TIMP-1,2) as novel markers of stress response and atherogenesis in children with chronic kidney disease (CKD) on conservative treatment. Cell Stress Chaperones. 2011;16:97–103. doi: 10.1007/s12192-010-0214-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duffield JS. Macrophages and immunologic inflammation of the kidney. Semin Nephrol. 2010;30:234–254. doi: 10.1016/j.semnephrol.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manabe I. Chronic inflammation links cardiovascular, metabolic and renal diseases. Circ J. 2011;75:2739–2748. doi: 10.1253/circj.cj-11-1184. [DOI] [PubMed] [Google Scholar]
- 22.Rodríguez-Iturbe B, Pons H, Herrera-Acosta J, Johnson RJ. Role of immunocompetent cells in nonimmune renal diseases. Kidney Int. 2001;59:1626–1640. doi: 10.1046/j.1523-1755.2001.0590051626.x. [DOI] [PubMed] [Google Scholar]
- 23.Ebihara I, Nakamura T, Tomino Y, Shimada N, Koide H. Metalloproteinase-9 mRNA expression in monocytes from patients with chronic renal failure. Am J Nephrol. 1998;18:305–310. doi: 10.1159/000013355. [DOI] [PubMed] [Google Scholar]
- 24.Nagano M, Fukami K, Yamagishi S, Ueda S, Kaida Y, Matsumoto T, Yoshimura J, Hazama T, Takamiya Y, Kusumoto T, et al. Circulating matrix metalloproteinase-2 is an independent correlate of proteinuria in patients with chronic kidney disease. Am J Nephrol. 2009;29:109–115. doi: 10.1159/000151439. [DOI] [PubMed] [Google Scholar]
- 25.Harendza S, Schneider A, Helmchen U, Stahl RA. Extracellular matrix deposition and cell proliferation in a model of chronic glomerulonephritis in the rat. Nephrol Dial Transplant. 1999;14:2873–2879. doi: 10.1093/ndt/14.12.2873. [DOI] [PubMed] [Google Scholar]
- 26.Friese RS, Rao F, Khandrika S, Thomas B, Ziegler MG, Schmid-Schönbein GW, O’Connor DT. Matrix metalloproteinases: discrete elevations in essential hypertension and hypertensive end-stage renal disease. Clin Exp Hypertens. 2009;31:521–533. doi: 10.3109/10641960802668730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng S, Lovett DH. Gelatinase A (MMP-2) is necessary and sufficient for renal tubular cell epithelial-mesenchymal transformation. Am J Pathol. 2003;162:1937–1949. doi: 10.1016/S0002-9440(10)64327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenz O, Elliot SJ, Stetler-Stevenson WG. Matrix metalloproteinases in renal development and disease. J Am Soc Nephrol. 2000;11:574–581. doi: 10.1681/ASN.V113574. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15:1–12. doi: 10.1097/01.asn.0000106015.29070.e7. [DOI] [PubMed] [Google Scholar]
- 30.Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Müller GA, Neilson EG. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002;61:1714–1728. doi: 10.1046/j.1523-1755.2002.00333.x. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Shultz RW, Mars WM, Wegner RE, Li Y, Dai C, Nejak K, Liu Y. Disruption of tissue-type plasminogen activator gene in mice reduces renal interstitial fibrosis in obstructive nephropathy. J Clin Invest. 2002;110:1525–1538. doi: 10.1172/JCI16219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Zhou Y, Tan R, Xiong M, He W, Fang L, Wen P, Jiang L, Yang J. Mice lacking the matrix metalloproteinase-9 gene reduce renal interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol. 2010;299:F973–F982. doi: 10.1152/ajprenal.00216.2010. [DOI] [PubMed] [Google Scholar]
- 33.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol. 2001;159:1465–1475. doi: 10.1016/S0002-9440(10)62533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeisberg M, Maeshima Y, Mosterman B, Kalluri R. Renal fibrosis. Extracellular matrix microenvironment regulates migratory behavior of activated tubular epithelial cells. Am J Pathol. 2002;160:2001–2008. doi: 10.1016/S0002-9440(10)61150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thiery JP, Chua K, Sim WJ, Huang R. [Epithelial mesenchymal transition during development in fibrosis and in the progression of carcinoma] Bull Cancer. 2010;97:1285–1295. doi: 10.1684/bdc.2010.1206. [DOI] [PubMed] [Google Scholar]
- 39.Jinde K, Nikolic-Paterson DJ, Huang XR, Sakai H, Kurokawa K, Atkins RC, Lan HY. Tubular phenotypic change in progressive tubulointerstitial fibrosis in human glomerulonephritis. Am J Kidney Dis. 2001;38:761–769. doi: 10.1053/ajkd.2001.27693. [DOI] [PubMed] [Google Scholar]
- 40.Rastaldi MP, Ferrario F, Giardino L, Dell’Antonio G, Grillo C, Grillo P, Strutz F, Müller GA, Colasanti G, D’Amico G. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002;62:137–146. doi: 10.1046/j.1523-1755.2002.00430.x. [DOI] [PubMed] [Google Scholar]
- 41.Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y, Yamamoto H, Bertram JF. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59:2612–2624. doi: 10.2337/db09-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lan HY. Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr Opin Nephrol Hypertens. 2003;12:25–29. doi: 10.1097/00041552-200301000-00005. [DOI] [PubMed] [Google Scholar]
- 43.Fan JM, Huang XR, Ng YY, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Interleukin-1 induces tubular epithelial-myofibroblast transdifferentiation through a transforming growth factor-beta1-dependent mechanism in vitro. Am J Kidney Dis. 2001;37:820–831. doi: 10.1016/s0272-6386(01)80132-3. [DOI] [PubMed] [Google Scholar]
- 44.Tan TK, Zheng G, Hsu TT, Lee SR, Zhang J, Zhao Y, Tian X, Wang Y, Wang YM, Cao Q, et al. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Lab Invest. 2013;93:434–449. doi: 10.1038/labinvest.2013.3. [DOI] [PubMed] [Google Scholar]
- 45.Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79:319–326. doi: 10.1172/JCI112815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lange-Sperandio B, Trautmann A, Eickelberg O, Jayachandran A, Oberle S, Schmidutz F, Rodenbeck B, Hömme M, Horuk R, Schaefer F. Leukocytes induce epithelial to mesenchymal transition after unilateral ureteral obstruction in neonatal mice. Am J Pathol. 2007;171:861–871. doi: 10.2353/ajpath.2007.061199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lim DH, Cho JY, Miller M, McElwain K, McElwain S, Broide DH. Reduced peribronchial fibrosis in allergen-challenged MMP-9-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2006;291:L265–L271. doi: 10.1152/ajplung.00305.2005. [DOI] [PubMed] [Google Scholar]
- 48.Zeisberg M, Khurana M, Rao VH, Cosgrove D, Rougier JP, Werner MC, Shield CF, Werb Z, Kalluri R. Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med. 2006;3:e100. doi: 10.1371/journal.pmed.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krüger A. Functional genetic mouse models: promising tools for investigation of the proteolytic internet. Biol Chem. 2009;390:91–97. doi: 10.1515/BC.2009.015. [DOI] [PubMed] [Google Scholar]
- 50.O’Regan AW, Hayden JM, Body S, Liaw L, Mulligan N, Goetschkes M, Berman JS. Abnormal pulmonary granuloma formation in osteopontin-deficient mice. Am J Respir Crit Care Med. 2001;164:2243–2247. doi: 10.1164/ajrccm.164.12.2104139. [DOI] [PubMed] [Google Scholar]