

Abstract
The primary purpose of this study was to evaluate the intestinal permeability (Peff) of N-formyl-methionyl-leucyl-phenylalanine (fMet-Leu-Phe), a bacterially derived chemotactic tripeptide, in the duodenum, jejunum, ileum, and colon of wild-type and PepT1 knockout mice. A secondary purpose was to determine if the presence of intestinal PepT1 translated into fMet-Leu-Phe directed neutrophil migration in these animals. Using an in situ single pass perfusion technique, the Peff of [3H]fMet-Leu-Phe was substantially reduced in the duodenum, jejunum, and ileum of PepT1 knockout mice as compared to wild-type animals. In contrast, the Peff of [3H]fMet-Leu-Phe in colon was unchanged between genotypes and about 5% of that in small intestine. Jejunal uptake of [3H]fMet-Leu-Phe was specific for PepT1 and saturable with an intrinsic K0.5 of 1.6 mM. The peptide/histidine transporters PhT1 and PhT2 were not involved in [3H]fMet-Leu-Phe uptake. Myeloperoxidase activity (a measure of neutrophil migration) was significantly increased following 4 h perfusions of 10 μM fMet-Leu-Phe in the jejunum of wild-type mice and was abolished by 50 mM glycylglycine; no change was observed in the jejunum of PepT1 knockout mice. Likewise, fMet-Leu-Phe perfusions had no effect on myeloperoxidase activity in the colon of either genotype. In conclusion, these findings demonstrated that PepT1 had a major influence on the permeability of fMet-Leu-Phe in duodenum, jejunum, and ileum in wild-type mice and on inflammatory response in intestinal regions that expressed PepT1.
Keywords: PepT1, fMet-Leu-Phe, regional permeability, myeloperoxidase activity
INTRODUCTION
Since PepT1, a proton-coupled oligopeptide transporter, had been cloned from the rabbit intestine,1 subsequent studies suggested that the end products of digested protein were absorbed predominantly by PepT1 in the form of di/tripeptides rather than free amino acids.2 PepT1 was shown to be expressed throughout the small intestine (duodenum, jejunum, and ileum) with little or no expression in the colon.3 Immunofluorescent staining demonstrated that PepT1 was expressed at the apical side of the enterocyte with decreasing expression from the tip to the crypt of the villus.3 In theory, PepT1 can transport 400 dipeptides and 8000 tripeptides, but it has been suggested that not all possible di/tripeptides are transported by this protein.4 In addition to endogenous small peptides, PepT1 can transport several peptide-like drugs such as some β-lactam antibiotics,5 angiotensin-converting enzyme inhibitors,6 the anticancer drug bestatin,7 and the antiviral prodrug valacyclovir.8 Because of its broad substrate specificity, PepT1 has been proposed as a drug delivery target to increase the intestinal permeability of poorly absorbed compounds. A successful example, valacyclovir, has been shown to exhibit a 3-to 5-fold increase in oral bioavailability as compared to its parent compound acyclovir.9
PepT1 regulation, under a variety of experimental and clinical conditions, has been recently reviewed by Smith et al.10 Pharmacological agents, hormones, diet, and disease have been shown to modify the expression of PepT1 mRNA or protein levels. Most interestingly, PepT1 was upregulated in the colon, where normally no PepT1 is expressed, under certain intestinal disease states. For example, PepT1 was detected at the apical membrane of colon in patients with inflammatory bowel disease (IBD) as shown by immunohistochemical staining.11 The upregulation of colonic PepT1 was also observed in patients with short bowel syndrome (SBS).12 Therefore, a pathological role was suggested for PepT1 based on its ability to transport bacterially produced chemotactic peptides and its aberrant expression in colon.13
One type of naturally occurring product of bacteria was the N-formyl peptides. In fact, the tripeptide N-formyl-methionyl-leucyl-phenylalanine (fMet-Leu-Phe) was shown to be the major peptide neutrophil chemotactic factor produced by E. coli.14 PepT1 as well as its homologue PepT2 have been shown to transport some bacterially derived compounds such as fMet-Leu-Phe,15 muramyl dipeptide (MDP),16 and γ-D-glutamyl-meso-diaminopimelic acid (γ-iE-DAP).17 The functional consequence of taking up bacterially derived compounds has also been demonstrated in cell culture and perfusion studies. Thus, luminal fMet-Leu-Phe resulted in the direct movement of neutrophils across Caco-2-BBE cells and specific inhibition of hPepT1-mediated fMet-Leu-Phe transport decreased neutrophil migration by about 50%.15 Moreover, the hPepT1-mediated epithelial transport of fMet-Leu-Phe increased MHC Class I surface expression in HT29-CI.19A cells expressing GFP-hPepT1 but not in untransfected control cells.11 In situ 4 h jejunal perfusions of fMet-Leu-Phe in rats also demonstrated an increase of myeloperoxidase (MPO) activity, a biomarker of neutrophil migration, presumably because of the presence of PepT1 in this intestinal region.18 Finally, in an animal model of SBS, 4 h colonic infusions of fMet-Leu-Phe resulted in both increased PepT1 mRNA expression and MPO activity two weeks following the 80% small intestinal resection, effects that were reversed by dipeptide inhibition.19 Overall, these studies support the hypothesis that colonic PepT1 is involved in the pathological progression of intestinal inflammation in addition to being a nutritional and drug transporter.13
Crohn’s disease, a major form of IBD that usually affects the ileum and colon, may attack any part of the digestive tract. Yet, little if any information is available on the regional permeability of bacterially derived chemotactic peptides such as fMet-Leu-Phe in the small and large intestines. In addition, the relative contribution of intestinal PepT1 (Slc15a1) in this process is unknown as is the contribution of other potential peptide transporters of the Slc15 family such as PepT2 (Slc15a2),20 PhT1 (Slc15a4), and PhT2 (Slc15a3).21,22 The in situ singlepass intestinal perfusion technique has proven to be a useful tool in examining the effective permeability of a model dipeptide, glycylsarcosine (GlySar), in wild-type and PepT1 null mice.23,24 Therefore, the specific aims of the present study were (1) to characterize the intestinal permeability of fMet-Leu-Phe in the duodenum, jejunum, ileum, and colon of mice and (2) to determine if the presence of intestinal PepT1 translates into fMet-Leu-Phe directed neutrophil migration in these animals. Specifically, the regional permeability, concentration dependency, substrate specificity, and dynamic activity of fMet-Leu-Phe were studied in PepT1-competent and-deficient mice.
EXPERIMENTAL SECTION
Materials
[3H-Phe]fMet-Leu-Phe (15 Ci/mmol) was purchased from AmBios Laboratories (Newington, CT), [14C]inulin (2.38 mCi/g) from Moravek Biochemicals and Radiochemicals (Brea, CA), and glycylproline (GlyPro) from Bachem (Torrance, CA). All other chemicals were acquired from Sigma-Aldrich (St. Louis, MO).
Animals
Mouse experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the U.S. National Institutes of Health. Gender-matched wild-type and PepT1 knockout mice (8–10 week old) were used in all the experiments. The mice were kept under 12 h light and dark cycles and fed a standard diet and water ad libitum (Unit for Laboratory Animal Medicine, University of Michigan, Ann Arbor, MI).
Metabolism of fMet-Leu-Phe in Intestinal Segments and Exposure in Portal Vein
One centimeter pieces of intestinal segments (duodenum, jejunum, ileum, and colon) were taken from wild-type mice and the luminal surface exposed longitudinally. Each intestinal segment was incubated with 0.5 mL phosphate buffered saline (pH 7.4) containing 0.8 mM fMet-Leu-Phe for 5 min at 37 °C. The incubation medium was then placed on ice and 10 μL of 10% trifluoroacetic acid (TFA) was added to stop the reaction. The mixture was centrifuged at 12 000g for 5 min and an aliquot of supernatant was analyzed by high-performance liquid chromatography (HPLC) consisting of a 600S controller, 616 pump, 2487 dual λ absorbance detector, and Empower 2 data acquisition software (Waters, Milford, MA). HPLC separations were carried out on a reversed-phase C18 column, 250 × 4.6 mm (Symmetric, Waters), under ambient conditions, with ultraviolet detection at 210 nm. The mobile phase consisted of 100% acetonitrile plus 0.1% TFA (buffer A) and 100% H2O plus 0.1% TFA (buffer B). A gradient elution was run over 30 min, starting from 15% buffer A to 30% buffer A, at a flow rate of 1.0 mL/min. Under these conditions, the retention times were 5.0 min for Phe, 6.5 min for fMet, 13.0 min for Leu-Phe, 14.5 min for fMet-Leu, and 24.0 min for fMet-Leu-Phe. All compounds were identified using known standards.
Portal venous blood was obtained during the 80–90 min time period of jejunal perfusion in wild-type mice with 2 μCi/ml (0.1 μM) of [3H-Phe]fMet-Leu-Phe. The blood sample was centrifuged at 3300g for 3 min after which a 100 μL aliquot of plasma was deproteinated with 200 μL of acetonitrile. The supernatant was evaporated to dryness by a Savant SVC 200H SpeedVac Concentrator and then reconstituted in 20 μL of water. The reconstituted sample was analyzed on an HPLC system, which consisted of a Waters 515 pump, a Packard 500TR radiochemical detector (PerkinElmer Life and Analytical Sciences, Boston, MA), and a reversed-phase C18 column, 250 × 4.6 mm (Discovery, Supelco, Bellefonte, PA). The mobile phase consisted of 30% acetonitrile plus 0.1% TFA, pumped isocratically at 1.0 mL/min under ambient conditions. Under these conditions, the retention times were 3.5 min for [3H-Phe] and 14.5 min for [3H-Phe]fMet-Leu-Phe. Known radiolabeled standards were used to identify compounds of interest.
In Situ Single-Pass Jejunal Perfusion of fMet-Leu-Phe
Jejunal perfusions were performed in wild-type and PepT1 knockout mice (8–10 week old) as described previously.23,24 In brief, animals were fasted overnight and then anesthetized with sodium pentobarbital (40–60 mg/kg ip). An 8 cm segment was isolated (i.e., ∼2 cm distal to the ligament of Treitz), and glass cannulas (2.0 mm outer diameter), attached to Tygon Laboratory tubing, were inserted at both ends of the intestinal segment and secured in place with silk sutures. Following cannulation, the isolated intestinal segment was rinsed with isotonic saline solution, and then covered with saline-wetted gauze and parafilm to prevent dehydration. After the surgical procedure, the mice were transferred to a temperature-controlled chamber (31 °C) to maintain body temperature during the experiment. The inlet cannula was connected to a 30 mL syringe containing perfusate and placed on a pump (Harvard Apparatus, Syringe Infusion Pump 22, South Natick, MA).
The perfusate contained 135 mM NaCl, 5 mM KCl, 10 mM MES (pH 6.5), 0.01% [14C]inulin, and 2 μCi/ml (0.1 μM) of [3H-Phe]fMet-Leu-Phe, in the presence of 100 mM phenylalanine, at a flow rate of 0.1 mL/min. The exiting perfusate was collected every 10 min for 90 min. A 100 μL aliquot of this perfusate was added to a vial containing scintillation fluid (CytoScint, MP Biomedicals, Solon, OH) and measured on a dual-channel liquid scintillation counter (Beckman LS 6000 SC, Beckman Coulter, Inc., Fullerton, CA). Water flux was corrected by the nonpermeable marker [14C]inulin. During the concentration-dependent studies, [3H-Phe]fMet-Leu-Phe was studied over 0.1 μM to 7.5 mM and during the specificity studies, potential inhibitors were present in perfusate at 50 mM.
Regional Intestinal Permeability of fMet-Leu-Phe
Each intestinal segment (i.e., duodenum, jejunum, ileum, and colon) was studied separately in wild-type and PepT1 knockout mice, as described previously for jejunal perfusion. For duodenal perfusions, a 2 cm segment was studied (i.e., ~0.25 cm distal to the pyloric sphincter), for jejunal perfusions, an 8 cm segment was studied (i.e., ~2 cm distal to the ligament of Treitz), for ileal perfusions, a 6 cm segment was studied (i.e., ~1 cm proximal to the cecum), and for colonic perfusions, a 3 cm segment was studied (i.e., ~0.5 cm distal to the cecum).
Effect of fMet-Leu-Phe on Myeloperoxidase (MPO) Activity
Wild-type and PepT1 knockout mice (8–10 week old) were perfused as described above in the jejunum and colon with minor modifications. In particular, the Krebs–Ringer buffer was perfused at a flow rate of 0.067 mL/min in jejunum and 0.1 mL/min in colon for 30 min, after which 10 μM of fMet-Leu-Phe was added to the buffer and perfused for another 4 h. For the inhibition study, 50 mM of glycylglycine (GlyGly) was added to the perfusate with fMet-Leu-Phe. Following the 4 h perfusion, MPO activity was determined by a spectrophotometric method based on the oxidation of o-dianisidine, as previously described.18 Briefly, intestinal segments (50–100 mg) were homogenized (PowerGen Model 125 Homogenizer, Fisher Scientific, Pittsburgh, PA) on ice for 30 s in 50 mM potassium phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide (1 mL per 50–100 mg intestine). The homogenates underwent three freeze–thaw procedures (−80 °C/37 °C) and were then centrifuged at 12 800g for 15 min at 4 °C. The supernatant (0.1 mL) was added to 2.9 mL of 50 mM phosphate buffer (pH 6.0) containing 0.167 mg/mL (0.53 mM) of o-dianisidine dihydrochloride and 0.0005% (0.15 mM) of hydrogen peroxide. Changes in absorbance were measured at 460 nm over 15 s intervals for 2 min on a UV/Vis spectrophotometer (BU-530 Beckman Coulter, Fullerton, CA). The MPO activity was reported as IU/g wet tissue where one IU was defined as the quantity of enzyme able to convert 1 μmol of hydrogen peroxide to water in 1 min at room temperature.
Hydrolysis Kinetics of fMet-Leu-Phe in Jejunal Homogenates
The mucous layers of jejunum from wild-type and PepT1 knockout mice were scraped off with a glass slide and homogenized on ice in 0.5 mL of ice-cold PBS for 2 min. The homogenates were centrifuged at 12 000g for 15 min, and the supernatant measured for total protein using a Pierce protein assay kit (Thermo Scientific, Rockford, IL). A 25 μL aliquot of 0.5 mg/mL protein was then incubated with different concentrations of [3H-Phe]fMet-Leu-Phe (0.4–100 μM) for 1 min at 37 °C. The reaction mixture was stopped by the addition of 50 μL of 10% TFA and centrifuged at 15 000g for 15 min. A 20 μL aliquot of supernatant was then analyzed by HPLC/radiochemical detection as described above. The rate of hydrolysis was calculated by the loss of fMet-Leu-Phe in the supernatant.
Data Analysis
Effective permeability (Peff) was determined by the steady-state loss of fMet-Leu-Phe from perfusate according to a complete radial mixing (parallel tube) model:25,26
where Q is the flow rate (mL/min), R is the intestinal radius (cm), L is the length of intestine (cm), Cout is the outlet concentration (water flux-corrected), and Cin is the inlet concentration. In our perfusion studies, steady-state was reached by 30 min. The intrinsic transport parameters (Tmax, K05) were determined by analyzing the steady-state flux (J) of fMet-Leu-Phe, referenced to estimated intestinal wall concentrations (Cw), after factoring out resistance of the unstirred water layer:
The transformation of inlet to intestinal wall concentrations was based on the equation27
where Paq is the estimated unstirred aqueous permeability as determined by
in which the aqueous diffusion coefficient (D = 3.18 × 10−4 cm2/min) was calculated by the Hayduk–Laudie expression.28 Gz is the Graetz number (0.0399), and A is a unitless constant (1.225) estimated by A = 2.5Gz + 1.125.
Kinetic parameters (Vmax, Km) from the fMet-Leu-Phe hydrolysis study were estimated by
where V is the hydrolysis rate of fMet-Leu-Phe (nmol/mg/min), and C is the substrate concentration (fMet-Leu-Phe, in μM).
Statistical Analysis
Data were expressed as mean ± SE. A two-tailed Student’s t test was used to compare differences between two groups. For multiple comparisons, one-way analysis of variance (ANOVA) was used followed by Dunnett’s or Tukey’s post hoc test (Prism v5.0, GraphPad Software, Inc., La Jolla, CA). A probability of p ≤ 0.05 was considered significant. Nonlinear regression analyses were also performed using GraphPad Prism in which the goodness of fit was determined by the coefficient of determination (r2), the standard error of parameter estimates, and by visual inspection of the residuals.
RESULTS
Metabolism of fMet-Leu-Phe in Intestinal Segments and Exposure in Portal Vein
Studies were performed initially to evaluate the stability of 0.8 mM fMet-Leu-Phe in different segments of the small and large intestines. As shown in Figure 1 (panels A–D), fMet-Leu-Phe was hydrolyzed to Phe, fMet, and fMet-Leu, but not to Leu-Phe (retention time of 13.0 min, if present), in these segments. Moreover, substantial hydrolysis occurred in the duodenum and jejunum with only minor hydrolysis in the ileum and colon. Following jejunal perfusions of 0.1 μM [3H-Phe]fMet-Leu-Phe for 90 min, only phenylalanine (shown as Phe* in Figure 1E) was detectable in the blood of portal vein. Thus, once fMet-Leu-Phe was transported across the apical membrane of enterocytes, only degradation products entered the portal vein prior to reaching the liver and systemic circulation.
Figure 1.
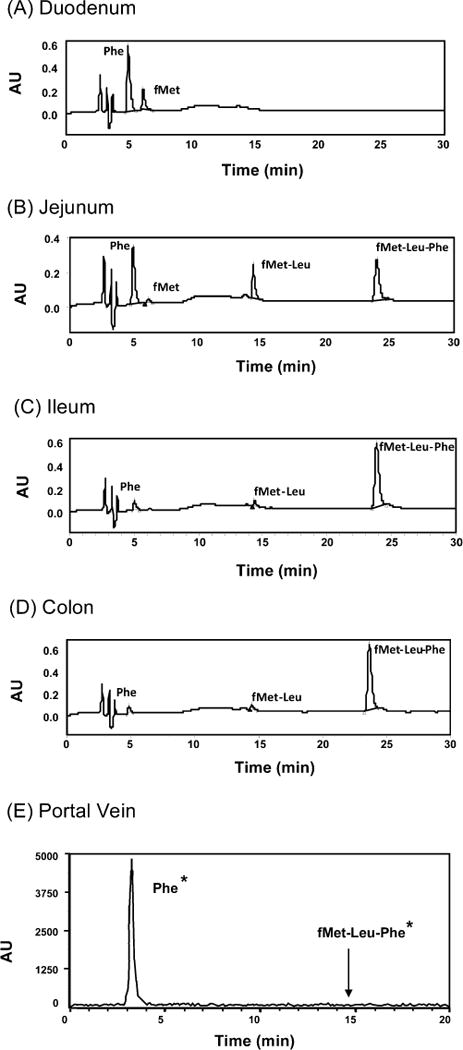
HPLC analyses of phenylalanine (Phe), N-formyl-methionine (fMet), N-formyl-methionyl-leucine (fMet-Leu), and N-formyl-methionyl-leucyl-phenylalanine (fMet-Leu-Phe) after 5 min incubations of 0.8 mM fMet-Leu-Phe (37 °C) in intestinal segments of duodenum (panel A), jejunum (panel B), ileum (panel C), and colon (panel D) of wild-type mice. All compounds were identified by UV detection using known standards; the retention times were 5.0 min for Phe, 6.5 min for fMet, 14.5 min for fMet-Leu, and 24.0 min for fMet-Leu-Phe. The portal vein was also sampled during a 90 min jejunal perfusion of 0.1 μM [3H]fMet-Leu-Phe (label on Phe*) (panel E). Using different chromatographic conditions (HPLC/radiochemical detection), retention times were 3.5 min for Phe* and 14.5 min for fMet-Leu-Phe* (no signal observed).
In Situ Single-Pass Jejunal Perfusion of fMet-Leu-Phe
Because of its degradation pattern in intestine (i.e., Phe but not Leu-Phe being formed), in situ perfusions of [3H-Phe]fMet-Leu-Phe were performed in the presence of high concentrations of unlabeled Phe (in order to prevent the uptake of radiolabeled Phe), thereby ensuring a correct interpretation of the permeability results. As shown in Figure 2, the Peff values of fMet-Leu-Phe were significantly reduced when 50, 100, or 150 mM of Phe was present in the perfusate. Thus, subsequent studies were performed with 100 mM Phe even though no statistical differences were observed between the three Phe treatments.
Figure 2.
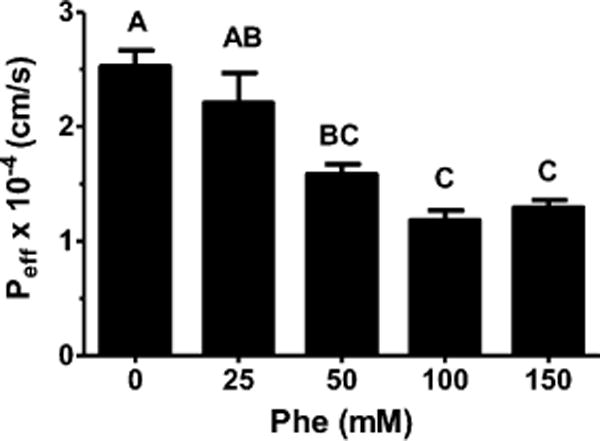
Effective permeability (Peff) of 0.1 μM [3H]fMet-Leu-Phe during jejunal perfusions of wild-type mice. Studies were performed in pH 6.5 buffer with varying concentrations of unlabeled Phe (n = 3, mean ± SE). Groups with different letters were statistically different as determined by one-way ANOVA and Tukey’s test.
To examine whether the permeability of fMet-Leu-Phe was saturable, the concentration-dependent uptake of [3H-Phe]-fMet-Leu-Phe was evaluated during jejunal perfusions in wildtype mice (total substrate ranged from 0.1 μM to 7.5 mM). As shown in Figure 3, fMet-Leu-Phe exhibited a nonlinear uptake in which the data were best fitted to a single Langmuir term. As such, the intrinsic transport parameters were estimated as Tmax = 0.73 ± 0.07 nmol/cm2/s and K0.5 = 1.6 ± 0.4 mM (r2 = 0.964).
Figure 3.
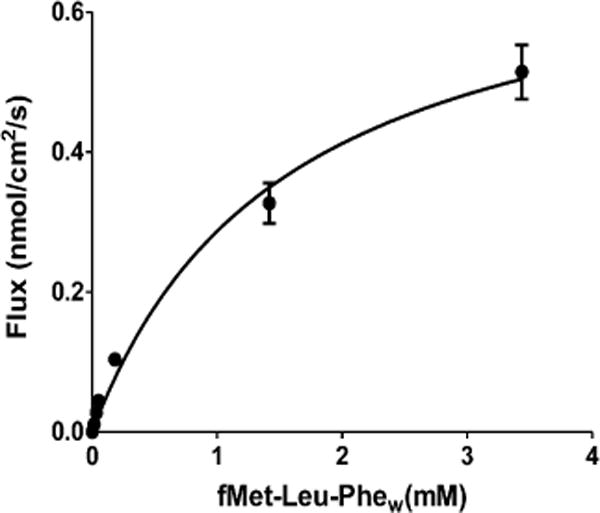
Concentration-dependent uptake of [3H]fMet-Leu-Phe (0.1 μM to 7.5 mM total substrate in perfusate) during jejunal perfusions of wild-type mice. Studies were performed in pH 6.5 buffer with 100 mM unlabeled Phe (n = 4, mean ± SE). fMet-Leu-Phew represents mean estimated concentrations of tripeptide at the intestinal wall.
The specificity of 0.1 μM [3H-Phe]fMet-Leu-Phe uptake was examined during jejunal perfusions in wild-type and PeptT1 knockout mice by adding 50 mM of the PepT1 substrate glycylproline (GlyPro) or the PhT1/PhT2 substrate L-histidine. Figure 4 demonstrates that GlyPro significantly reduced the permeability of fMet-Leu-Phe in wild-type mice but not in PepT1 knockout animals. The inhibitory effect was weak, however, in which fMet-Leu-Phe uptake was reduced by only 33%. On the basis of a competitive inhibition model, the Ki of GlyPro was estimated at 100 mM. In contrast, L-histidine had no effect on the permeability of fMet-Leu-Phe in either genotype. These results show that fMet-Leu-Phe transport was specific for PepT1 but not PhT1/PhT2.
Figure 4.
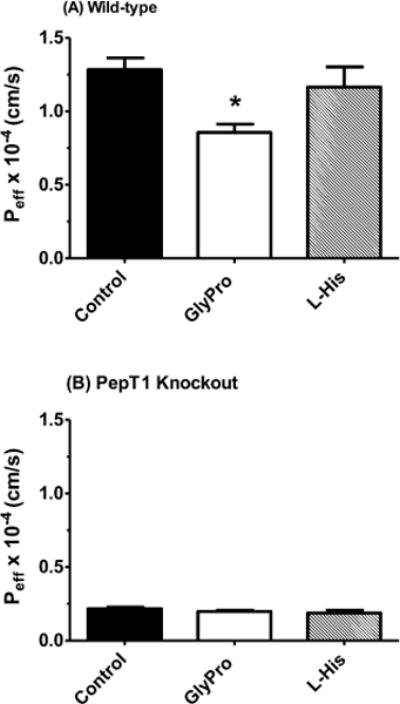
Effect of potential inhibitors (50 mM) on the Peff of 0.1 μM [3H]fMet-Leu-Phe during jejunal perfusions of wild-type (panel A) and PepT1 knockout (panel B) mice. Studies were performed in pH 6.5 buffer with 100 mM unlabeled Phe (n = 4, mean ± SE). Statistical analyses were performed by one-way ANOVA and Dunnett’s test. *p ≤ 0.05 as compared to control.
Regional Intestinal Permeability of fMet-Leu-Phe
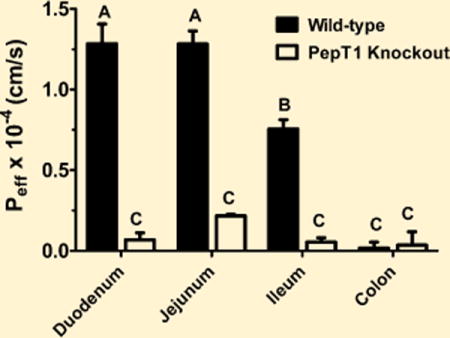
The intestinal permeability of 0.1 μM [3H-Phe]fMet-Leu-Phe was determined in different regions of the small and large intestines, as shown in Figure 5. We observed that the effective permeability of fMet-Leu-Phe was considerably lower in the duodenum, jejunum, and ileum of PepT1 knockout mice as compared to wild-type animals. Moreover, the permeability of fMet-Leu-Phe was very low in the colon (i.e., ~5% of that in the small intestine) and did not differ between the two genotypes. It was also observed that the permeability of fMet-Leu-Phe in wild-type mice was duodenum ≈ jejunum > ileum > colon, whereas the permeability in PepT1 knockout mice was not different among the four intestinal segments.
Figure 5.
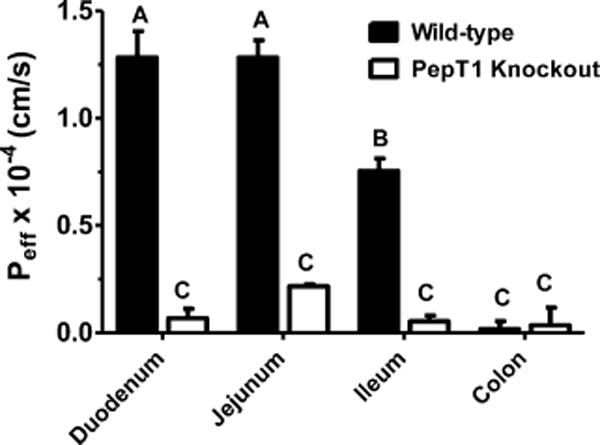
Effective permeability (Peff) of 0.1 μM [3H]fMet-Leu-Phe during intestinal perfusions of wild-type and PepT1 knockout mice. Studies were performed in pH 6.5 buffer with 100 mM unlabeled Phe (n = 6, mean ± SE). Groups with different letters were statistically different as determined by one-way ANOVA and Tukey’s test.
Effect of fMet-Leu-Phe on Myeloperoxidase (MPO) Activity
The role of PepT1 in facilitating bacterial-induced neutrophil migration was evaluated following 4 h perfusions of 10 μM fMet-Leu-Phe in the intestines of mice. Figure 6A shows that, in wild-type animals, exposure of the jejunum to this tripeptide resulted in significant increases in MPO activity (about 2-fold), which returned to control values when fMet-Leu-Phe was coperfused with saturating (inhibiting) concentrations of the dipeptide GlyGly. In contrast, fMet-Leu-Phe had no effect on MPO activity in PepT1 knockout mice, which were similar to that of wild-type animals in the absence of tripeptide. Likewise, given the absence of PepT1 protein in colon, fMet-Leu-Phe perfusions of this region were without change in MPO activity for both wild-type and PepT1 null mice (Figure 6B). Although the MPO activity was 3-fold higher in wild-type as compared to PepT1 knockout mice, all MPO values in colon were much lower than that in jejunum.
Figure 6.
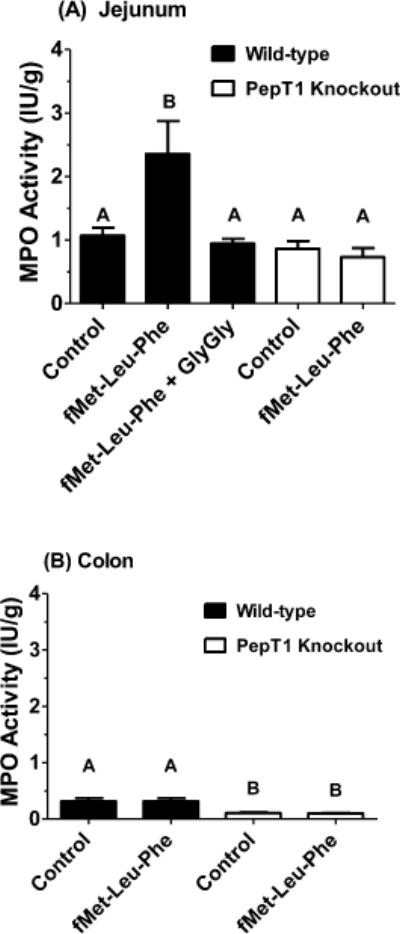
Myeloperoxidase (MPO) activity, a measure of neutrophil migration, was measured in the jejunum (panel A) and colon (panel B) of wild-type and PepT1 knockout mice following 4 h in situ perfusions of 10 μM fMet-Leu-Phe ± 50 mM GlyGly (n = 5, mean ± SE). Groups with different letters were statistically different as determined by one-way ANOVA and Tukey’s test.
Hydrolysis Kinetics of fMet-Leu-Phe in Jejunal Homogenates
To rule out any potential differences in hydrolytic activity between genotypes, the hydrolysis of [3H-Phe]fMet-Leu-Phe was examined in jejunal homogenates (total substrate ranged from 0.4–100 μM). Figure 7 demonstrates that the hydrolysis rates were nonlinear and the curves virtually superimposable between wild-type and PepT1 knockout mice. Using a single Michaelis–Menten term to fit the results, the estimated parameters showed no difference between genotypes in which Vmax = 3.2 ± 0.2 nmol/mg/min and Km = 7.0 ± 2.4 μM for wild-type mice (r2 = 0.860) and Vmax = 3.3 ± 0.1 nmol/mg/min and Km = 6.9 ± 1.2 μM (r2 = 0.959) for PepT1 knockout animals.
Figure 7.
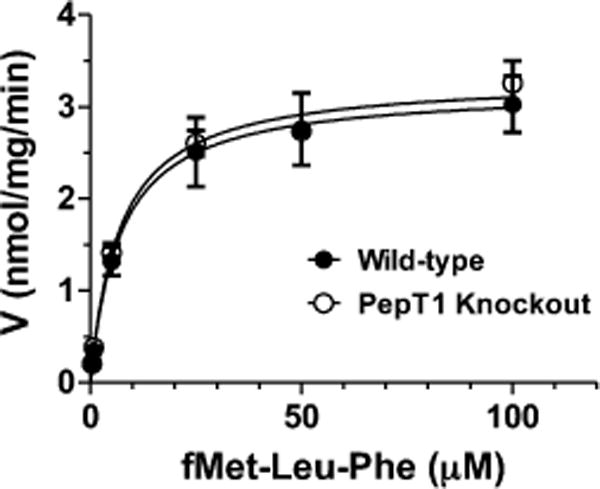
Hydrolysis kinetics of [3H]fMet-Leu-Phe (0.4–100 μM total substrate) in the jejunal homogenates of wild-type and PepT1 knockout mice (n = 4, mean ± SE).
DISCUSSION
The proposed studies have provided several unique findings in regard to the intestinal disposition and dynamics of fMet-Leu-Phe in wild-type and PepT1 knockout mice. Specifically, we found that (1) fMet-Leu-Phe exhibited substantial hydrolysis in duodenum and jejunum but only minor hydrolysis in ileum and colon. In addition, no intact tripeptide was found in the portal vein during in situ jejunal perfusions; (2) fMet-Leu-Phe uptake was specific for PepT1 and saturable with a K0.5 value of 1.6 mM. The peptide-histidine transporters PhT1/PhT2 were not involved with fMet-Leu-Phe transport across the enterocytes; (3) the permeability of fMet-Leu-Phe was substantially larger in duodenal, jejunal, and ileal segments of wild-type mice as compared to PepT1 knockout animals (∼10% residual values, on average). No difference was observed between genotypes in the permeability of fMet-Leu-Phe in colon, which was <6% of that observed in small intestine. These results are consistent with previously reported immunoblot analyses in wild-type mice in which PepT1 protein was abundantly expressed in small intestine with no protein expression in colon;24 (4) jejunal perfusions of fMet-Leu-Phe in wild-type mice caused an increase in neutrophil migration, which was completely blocked by coperfusion with saturating concentrations of the PepT1 substrate GlyGly. In the absence of PepT1, such as in PepT1 null mice or colonic perfusions in either genotype, fMet-Leu-Phe was without effect; (5) there was no difference in the hydrolysis kinetics of fMet-Leu-Phe in jejunum of wild-type and PepT1 knockout mice.
Previous studies have shown that fMet-Leu-Phe was accumulated in Caco2-BBE monolayers and Xenopus oocytes expressing hPepT115 and that the tripeptide had a K0.5 value of 2.9 mM in KG-1, a human monocytic cell line.29 The K0.5 value of 1.6 mM in wild-type mice, as determined during in situ jejunal perfusions, was comparable to the value in KG-1 cells and was similar to other PepT1 substrates reported in the literature for this low-affinity transporter.30,31 Our in situ perfusion results differed, however, from a study in rats where the authors32 reported that, during ileal loop incubations, fMet-Leu-Phe was not absorbed because of restricted mucosal permeability and carboxypeptidase degradation. To clarify this apparent discrepancy, we subsequently performed jejunal perfusion studies with GlySar in which carboxypeptidase inhibitors (i.e., 25 mM benzylsuccinic acid or 10 mM EDTA) were added to the perfusate, as had the previous authors to their ileal preparation. For these experiments, we found that, in wild-type mice, the permeability of GlySar, a nonhydrolyzable substrate of PepT1, was significantly reduced from 1.7 × 10−4 cm/s (control) to 0.21 × 10−4 cm/s in the presence of benzylsuccinic acid or to 0.69 × 10−4 cm/s in the presence of EDTA (n = 3 for each group; p < 0.001). Thus, the addition of carboxypeptidase inhibitors such as benzylsuccinic acid or EDTA can compromise the transport activity of PepT1 and, as a result, lead to the erroneous conclusion that the gut mucosa is impermeable to fMet-Leu-Phe.
Intestinal perfusions of the bacterial-derived fMet-Leu-Phe in wild-type and Pept1 knockout mice demonstrated that PepT1-mediated uptake of this chemotactic peptide in enterocytes was a key step in the dynamics of increasing neutrophil migration. Our findings in Figure 6A clearly showed that, during jejunal perfusions of fMet-Leu-Phe, the MPO activity was increased only in mice that were PepT1-competent or functionally active (i.e., in the absence of inhibiting GlyGly). Likewise, since little or no PepT1 was expressed in mouse colon, fMet-Leu-Phe perfusions of this region were without effect in either genotype (Figure 6B). It is highly unlikely that Phe or fMet-Leu would have any effect on MPO activity since no difference in neutrophil migration is observed in the jejunum of control vs fMet-Leu-Phe + GlyGly studies in wild-type mice or the jejunum of control vs fMet-Leu-Phe studies in KO mice (Figure 6A). The same is true in colon where no differences are observed in MPO activity between the colon of control vs fMet-Leu-Phe studies in both genotypes (Figure 6B). Further, it has been demonstrated that amino-terminal formyl peptides, of which fMet-Leu-Phe is the major component of E. coli culture filtrates, are the peptide mediators responsible for the potent chemotaxis of leukocytes.14,33 Interestingly, there was a small but significant difference between wild-type and PepT1 knockout mice in their baseline values for MPO activity in colon (Figure 6B). The reason for this difference is unknown but may be due, in part, to experimental variation since the measured activity was very low in colon as compared to jejunum. Alternatively, this difference may be the result of sparse but undetectable levels of PepT1 expression in the colon of wild-type mice. More experiments will be needed to clarify this unexpected result.
To rule out the possibility that the lack of effect of fMet-Leu-Phe jejunal perfusions on MPO activity in PepT1 knockout mice was the result of differences in enzymatic activity between genotypes, the hydrolysis kinetics of fMet-Leu-Phe were evaluated in PepT1-competent and -deficient mice. As described previously, no differences were found between genotypes in the hydrolysis kinetic parameters (Vmax,Km) of fMet-Leu-Phe when incubated in homogenates prepared from jejunum. Because the enzyme(s) responsible for fMet-Leu-Phe hydrolysis are uncertain, we studied this process using a nonspecific approach. Notwithstanding this uncertainty, one of the enzymes capable of hydrolyzing fMet-Leu-Phe was a carboxypeptidase, inhibitable by benzylsuccinic acid.34 Carboxypeptidase M belongs to the family of metallocarboxypeptidases, a class of enzymes that catalyze the hydrolysis of the C-terminal peptide bond in peptides and proteins. Depending upon the specific isoform, substrate, and experimental system, the Km of these carboxypeptidases can range from 6–1573 μM.35 The Km of 7 μM for fMet-Leu-Phe in our study is consistent with these literature values. Moreover, with fMet-Leu but not Leu-Phe being formed during intestinal degradation (see Figure 1), it appears that the initial direction of hydrolysis was from the carboxyl terminus of the tripeptide.
IBD, which comprises Crohn’s disease and ulcerative colitis, is a chronic disorder characterized by nonspecific inflammation and intestinal tissue damage.36 The pathophysiology of IBD is incompletely understood but disease susceptibility includes a complex interaction of host genetics, gut microbiota, environmental factors, and a dysregulated mucosal immune system. Mounting evidence suggests an important role for PepT1, including its aberrant expression in colon, in linking the intestinal uptake of chemotactic bacterial-derived peptides and an aggravated inflammatory response.10,37 Thus, internalization of these peptides leads to activation of NF-κB and MAPK pathways within the enterocyte which, in turn, triggers proinflammatory cytokine/chemokine production and the infiltration of neutrophils into inflammatory regions.37 However, it has also been shown that β-actin/hPepT1 transgenic mice infected with Citrobacter rodentium, which can induce PepT1 expression in mouse colon, had reduced bacterial colonization, production of proinflammatory cytokines, and neutrophil infiltration of the colon as compared to wild-type animals.38 Thus, colonic PepT1 may serve a dual purpose in exacerbating intestinal inflammation via its transport of chemotactic peptides produced by commensal bacteria, while also mitigating intestinal inflammation by interfering with those lipid rafts used as docking sites for murine attaching and effacing pathogens. The molecular mechanism underlying the aberrant expression of PepT1 in colon is unknown but reveals a novel role for PepT1 in both host susceptibility or host defense to IBD. It is noteworthy that a bidirectional association was found between a functional SLC15A1 single nucleotide polymorphism and Crohn’s disease in two cohorts of Swedish (increased risk) and Finnish (increased protection) patients.39 This discussion illustrates, in part, the complex relationship of colonic PepT1 expression and regulation and its influence on intestinal inflammation. To further explore the role and relevance of intestinal PepT1 in IBD, it may prove useful to perform more sophisticated studies in which transgenic PepT1 null mice expressing the human PepT1 gene are evaluated under a variety of experimental conditions such as inducible colitis and germ-free and gnotobiotic conditions.
Acknowledgments
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [R01GM035498] (to D.E.S.) and Michigan Gastrointestinal Peptide Research Center (5 P30 DK34933).
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Fei YJ, Kanai Y, Nussberger S, Ganapathy V, Leibach FH, Romero MF, Singh SK, Boron WF, Hediger MA. Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature. 1994;368:563–566. doi: 10.1038/368563a0. [DOI] [PubMed] [Google Scholar]
- 2.Ganapathy V, Gupta N, Martindale RG. Protein Digestion and Absorption. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. 4. Elsevier; Amsterdam, The Netherlands: 2006. pp. 1667–1692. [Google Scholar]
- 3.Ogihara H, Saito H, Shin BC, Terado T, Takenoshita S, Nagamachi Y, Inui K, Takata K. Immuno-localization of H+/peptide cotransporter in rat digestive tract. Biochem Biophys Res Commun. 1996;220:848–852. doi: 10.1006/bbrc.1996.0493. [DOI] [PubMed] [Google Scholar]
- 4.Vig BS, Stouch TR, Timoszyk JK, Quan Y, Wall DA, Smith RL, Faria TN. Human PepT1 pharmacophore distinguishes between dipeptide transport and binding. J Med Chem. 2006;49:3636–3644. doi: 10.1021/jm0511029. [DOI] [PubMed] [Google Scholar]
- 5.Okano T, Inui K, Takano M, Hori R. H+ gradient-dependent transport of aminocephalosporins in rat intestinal brush-border membrane vesicles: role of dipeptide transport system. Biochem Pharmacol. 1986;35:1781–1786. doi: 10.1016/0006-2952(86)90292-3. [DOI] [PubMed] [Google Scholar]
- 6.Thwaites DT, Cavet M, Hirst BH, Simmons NL. Angiotensin-converting enzyme (ACE) inhibitor transport in human intestinal epithelial (Caco-2) cells. Br J Pharmacol. 1995;114:981–986. doi: 10.1111/j.1476-5381.1995.tb13301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomita Y, Katsura T, Okano T, Inui K, Hori R. Transport mechanisms of bestatin in rabbit intestinal brush-border membranes: role of H+/dipeptide cotransport system. J Pharmacol Exp Ther. 1990;252:859–862. [PubMed] [Google Scholar]
- 8.Ganapathy ME, Huang W, Wang H, Ganapathy V, Leibach FH. Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem Biophys Res Commun. 1998;246:470–475. doi: 10.1006/bbrc.1998.8628. [DOI] [PubMed] [Google Scholar]
- 9.Soul-Lawton J, Seaber E, On N, Wootton R, Rolan P, Posner J. Absolute bioavailability and metabolic disposition of valaciclovir, the L-valyl ester of acyclovir, following oral administration to humans. Antimicrob Agents Chemother. 1995;39:2759–2764. doi: 10.1128/aac.39.12.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith DE, Clémencon B, Hediger MA. Proton-coupled oligopeptide transporter family SLC15: physiological, pharmacological and pathological implications. Mol Aspects Med. 2013 doi: 10.1016/j.mam.2012.11.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Merlin D, Si-Tahar M, Sitaraman SV, Eastburn K, Williams I, Liu X, Hediger MA, Madara JL. Colonic epithelial hPepT1 expression occurs in inflammatory bowel disease: transport of bacterial peptides influences expression of MHC class 1 molecules. Gastroenterology. 2001;120:1666–1679. doi: 10.1053/gast.2001.24845. [DOI] [PubMed] [Google Scholar]
- 12.Ziegler TR, Fernandez-Estivariz C, Gu LH, Bazargan N, Umeakunne K, Wallace TM, Diaz EE, Rosado KE, Pascal RR, Galloway JR, Wilcox JN, Leader LM. Distribution of the H+/peptide transporter PepT1 in human intestine: up-regulated expression in the colonic mucosa of patients with short-bowel syndrome. Am J Clin Nutr. 2002;75:922–930. doi: 10.1093/ajcn/75.5.922. [DOI] [PubMed] [Google Scholar]
- 13.Charrier L, Merlin D. The oligopeptide transporter hPepT1: gateway to the innate immune response. Lab Invest. 2006;86:538–546. doi: 10.1038/labinvest.3700423. [DOI] [PubMed] [Google Scholar]
- 14.Marasco WA, Phan SH, Krutzsch H, Showell HJ, Feltner DE, Nairn R, Becker EL, Ward PA. Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. J Biol Chem. 1984;259:5430–5439. [PubMed] [Google Scholar]
- 15.Merlin D, Steel A, Gewirtz AT, Si-Tahar M, Hediger MA, Madara JL. hPepT1-mediated epithelial transport of bacteria-derived chemotactic peptides enhances neutrophil-epithelial interactions. J Clin Invest. 1998;102:2011–2018. doi: 10.1172/JCI4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vavricka SR, Musch MW, Chang JE, Nakagawa Y, Phanvijhitsiri K, Waypa TS, Merlin D, Schneewind O, Chang EB. hPepT1 transports muramyl dipeptide, activating NF-kappaB and stimulating IL-8 secretion in human colonic Caco2/bbe cells. Gastroenterology. 2004;127:1401–1409. doi: 10.1053/j.gastro.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 17.Swaan PW, Bensman T, Bahadduri PM, Hall MW, Sarkar A, Bao S, Khantwal CM, Ekins S, Knoell DL. Bacterial peptide recognition and immune activation facilitated by human peptide transporter PEPT2. Am J Respir Cell Mol Biol. 2008;39:536–542. doi: 10.1165/rcmb.2008-0059OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buyse M, Tsocas A, Walker F, Merlin D, Bado A. PepT1-mediated fMLP transport induces intestinal inflammation in vivo. Am J Physiol Cell Physiol. 2002;283:C1795–C1800. doi: 10.1152/ajpcell.00186.2002. [DOI] [PubMed] [Google Scholar]
- 19.Shi B, Song D, Xue H, Li N, Li J. PepT1 mediates colon damage by transporting fMLP in rats with bowel resection. J Surg Res. 2006;136:38–44. doi: 10.1016/j.jss.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 20.Ruhl A, Hoppe S, Frey I, Daniel H, Schemann M. Functional expression of the peptide transporter PEPT2 in the mammalian enteric nervous system. J Comp Neurol. 2005;490:1–11. doi: 10.1002/cne.20617. [DOI] [PubMed] [Google Scholar]
- 21.Herrera-Ruiz D, Wang Q, Gudmundsson OS, Cook TJ, Smith RL, Faria TN, Knipp GT. Spatial expression patterns of peptide transporters in the human and rat gastrointestinal tracts, Caco-2 in vitro cell culture model, and multiple human tissues. AAPS PharmSci. 2001;3(1):E9. doi: 10.1208/ps030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhardwaj RK, Herrera-Ruiz D, Eltoukhy N, Saad M, Knipp GT. The functional evaluation of human peptide/histidine transporter 1 (hPHT1) in transiently transfected COS-7 cells. Eur J Pharm Sci. 2006;27:533–542. doi: 10.1016/j.ejps.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Hu Y, Smith DE, Ma K, Jappar D, Thomas W, Hillgren KM. Targeted disruption of peptide transporter Pept1 gene in mice significantly reduces dipeptide absorption in intestine. Mol Pharmaceutics. 2008;5:1122–1130. doi: 10.1021/mp8001655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jappar D, Wu SP, Hu Y, Smith DE. Significance and regional dependency of peptide transporter (PEPT) 1 in the intestinal permeability of glycylsarcosine: in situ single-pass perfusion studies in wild-type and Pept1 knockout mice. Drug Metab Dispos. 2010;38:1740–1746. doi: 10.1124/dmd.110.034025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komiya I, Park JY, Kamani A, Ho NFH, Higuchi WI. Quantitative mechanistic studies in simultaneous fluid flow and intestinal absorption using steroids as model solutes. Int J Pharm. 1980;4:249–262. [Google Scholar]
- 26.Kou JH, Fleisher D, Amidon GL. Calculation of the aqueous diffusion layer resistance for absorption in a tube: application to intestinal membrane permeability determination. Pharm Res. 1991;8:298–305. doi: 10.1023/a:1015829128646. [DOI] [PubMed] [Google Scholar]
- 27.Johnson DA, Amidon GL. Determination of intrinsic membrane transport parameters from perfused intestine experiments: a boundary layer approach to estimating the aqueous and unbiased membrane permeabilities. J Theor Biol. 1988;131:93–106. doi: 10.1016/s0022-5193(88)80123-1. [DOI] [PubMed] [Google Scholar]
- 28.Reid RC, Prausnitz JM, Sherwood TK. The Properties of Gases and Liquids. 3. McGraw-Hill Book Company; New York: 1977. p. 573. [Google Scholar]
- 29.Charrier L, Driss A, Yan Y, Nduati V, Klapproth J-M, Sitaraman SV, Merlin D. hPepT1 mediates bacterial tripeptide fMLP uptake in human monocytes. Lab Invest. 2006;86:490–503. doi: 10.1038/labinvest.3700413. [DOI] [PubMed] [Google Scholar]
- 30.Rubio-Aliaga I, Daniel H. Peptide transporters and their roles in physiological processes and drug disposition. Xenobiotica. 2008;38:1022–1042. doi: 10.1080/00498250701875254. [DOI] [PubMed] [Google Scholar]
- 31.Brandsch M, Knutter I, Bosse-Doenecke E. Pharmaceutical and pharmacological importance of peptide transporters. J Pharm Pharmacol. 2008;60:543–585. doi: 10.1211/jpp.60.5.0002. [DOI] [PubMed] [Google Scholar]
- 32.Woodhouse AF, Anderson RP, Myers DB, Brtoom MF, Hobson CH, Chadwick VS. Intestinal absorption, metabolism and effects of bacterial chemotactic peptides in rat intestine. J Gastroenterol Hepatol. 1987;2:35–43. [Google Scholar]
- 33.Chadwick VS, Mellor DM, Myers DB, Selden AC, Keshavarzian A, Broom MF, Hobson CH. Production of peptides inducing chemotaxis and lysosomal enzyme release in human neutrophils by intestinal bacteria in vitro and in vivo. Scand J Gastroenterol. 1988;23:121–128. doi: 10.3109/00365528809093861. [DOI] [PubMed] [Google Scholar]
- 34.Chadwick VS, Schlup MMT, Cooper BT, Broom MF. Enzymes degrading bacterial chemotactic F-met peptides in human ileal and colonic mucosa. J Gastroenterol Hepatol. 1990;5:375–381. doi: 10.1111/j.1440-1746.1990.tb01412.x. [DOI] [PubMed] [Google Scholar]
- 35.Deiteren K, Surpateanu G, Gilany K, Willemse JL, Hendriks DF, Augustyns K, Laroche Y, Scharpe S, Lambeir AM. The role of the S1 binding site of carboxypeptidase M in substrate specificity and turn-over. Biochim Biophys Acta. 2007;1774:267–277. doi: 10.1016/j.bbapap.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 36.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 37.Ingersoll SA, Ayyadurai S, Charania MA, Laroui H, Yan Y, Merlin D. The role and pathophysiological relevance of membrane transporter PepT1 in intestinal inflammation and inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2012;302:G484–G492. doi: 10.1152/ajpgi.00477.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen HT, Dalmasso G, Powell KR, Yan Y, Bhatt S, Kalman D, Sitaraman SV, Merlin D. Pathogenic bacteria induce colonic PepT1 expression: an implication in host defense response. Gastroenterology. 2009;137:1435–1447. e1431–1432. doi: 10.1053/j.gastro.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zucchelli M, Torkvist L, Bresso F, Halfvarson J, Hellquist A, Anedda F, Assadi G, Lindgren GB, Svanfeldt M, Janson M, Noble CL, Pettersson S, Lappalainen M, Paavola-Sakki P, Halme L, Farkkila M, Turunen U, Satsangi J, Kontula K, Lofberg R, Kere J, D’Amato M. 2009. PepT1 oligopeptide transporter (SLC15A1) gene polymorphism in inflammatory bowel disease. Inflammatory Bowel Dis. 2009;15:1562–1569. doi: 10.1002/ibd.20963. [DOI] [PubMed] [Google Scholar]