

SUMMARY
The serine hydrolase α/β hydrolase domain 6 (ABHD6) has recently been implicated as a key lipase for the endocannabinoid 2-arachidonylglycerol (2-AG) in the brain. However, the biochemical and physiological function for ABHD6 outside of the central nervous system has not been established. To address this we utilized targeted antisense oligonucleotides (ASOs) to selectively knock down ABHD6 in peripheral tissues to identify in vivo substrates and to understand ABHD6's role in energy metabolism. Here we show that selective knockdown of ABHD6 in metabolic tissues protects mice from high fat diet-induced obesity, hepatic steatosis, and systemic insulin resistance. Using combined in vivo lipidomic identification and in vitro enzymology approaches we show that ABHD6 can hydrolyze several lipid substrates, positioning ABHD6 at the interface of glycerophospholipid metabolism and lipid signal transduction. Collectively, these data suggest that ABHD6 inhibitors may serve as novel therapeutics for obesity, nonalcoholic fatty liver disease, and type II diabetes.
INTRODUCTION
A major challenge for drug discovery in the post genomic era is the functional characterization of unannotated genes identified by sequencing efforts. Although many unannotated gene products belong to structurally related gene or protein families, which may provide important functional clues, membership to such families does not always accurately predict the true biochemical and physiological role of proteins. Genes encoding the α/β hydrolase fold domain (ABHD) protein family are present in all reported genomes (Nardini and Dijkstra, 1999; Hotelier et al., 2004), and conserved structural motifs shared by these proteins predict common roles in lipid metabolism and signal transduction (Lefevre et al., 2001; Fiskerstrand et al., 2010; Long et al., 2011; Simon and Cravatt, 2006; Montero-Moran et al., 2009; Blankman et al., 2007; Lord et al., 2011; Brown et al., 2010). Furthermore, mutations in several members of the ABHD protein family have been implicated in inherited inborn errors of lipid metabolism (Lefevre et al., 2001; Fiskerstrand et al., 2010). Most recently, studies in cell and animal models have revealed important roles for ABHD proteins in glycerophospholipid metabolism, lipid signal transduction, and metabolic disease (Long et al., 2011; Simon and Cravatt 2006; Montero-Moran et al., 2009; Blankman et al., 2007; Lord et al., 2011; Brown et al., 2010). However, the physiological substrates and products for these lipid metabolizing enzymes and their broader role in metabolic pathways remain largely uncharacterized. Given this, functional annotation of ABHD enzymes holds clear promise for drug discovery targeting diseases of altered lipid metabolism and lipid signaling.
ABHD5, also known as CGI-58, has been studied quite extensively due to its key role in triacylglycerol (TAG) metabolism, lipid signaling, and genetic association with the human disease Chanarin-Dorfman Syndrome (CDS) (Lefevre et al., 2001; Montero-Moran et al., 2009; Lord et al., 2011; Brown et al., 2010; Lass et al., 2006; Schweiger et al., 2009). Given ABHD5's clear role in nutrient metabolism and lipid signal transduction, we aimed to test whether the closely related enzyme ABHD6 might play a similar role in lipid signaling and metabolic disease. ABHD6 has recently been described as an enzymatic regulator of endocannabinoid (ECB) signaling in the brain (Blankman et al., 2007; Marrs et al., 2010; Marrs et al., 2011). However, ABHD6 is ubiquitously expressed, and the biochemical and physiological functions of ABHD6 outside of the central nervous system have not been studied. Furthermore, unbiased identification of ABHD6 substrates in vivo has not been reported. To address this we selectively knocked down ABHD6 in peripheral tissues, allowing us to identify novel substrates in vivo and to uncover a previously underappreciated role for ABHD6 in promoting the metabolic syndrome. These studies demonstrate that ABHD6 plays a non-redundant enzymatic role in promoting the metabolic disorders induced by high-fat feeding, and suggest that ABHD6 inhibition may be effective in preventing obesity, non-alcoholic fatty liver disease, and type II diabetes.
RESULTS
ABHD6 is Ubiquitously Expressed and Upregulated by High Fat Diet Feeding
Mouse ABHD6 is a 336 amino acid protein that shares high sequence identity with its human (94%), macaque (94%), and rat (97%) orthologues (Figure 1A). A highly conserved active site serine nucleophile is found at residue 148 (Figure 1A), which is predicted to be necessary for enzyme catalysis. ABHD6 mRNA is ubiquitously expressed (Figure 1B), with highest expression in small intestine, liver, and brown adipose tissue in mice fed standard rodent chow. Additionally, high fat diet feeding increases ABHD6 mRNA expression in the small intestine and the liver (Figure 1B). This transcriptional regulation of ABHD6 in metabolic tissue prompted us to examine whether ABHD6 may be an important mediator of high fat diet-induced metabolic disease.
Figure 1. ABHD6 is Ubiquitously Expressed and is Regulated by High Fat Diet.

(A) Alignment of human (Hu), macaque (Ma), rat (Ra), and mouse (Mu) ABHD6 orthologues showing conserved (gray) and divergent residues (white); C = consensus sequence. The underlined letters represent the consensus GXSXG “nucleophile elbow” containing the predicted serine nucleophile S148 (black box).
(B) mRNA expression analysis of ABHD6 in C57BL/6 mouse tissues following 10 weeks of chow or high fat diet (HF) feeding. Data represent the mean ± SEM (n = 4); * = P < 0.05 (vs. chow-fed group within each tissue). AU = arbitrary units; Liv = liver; He = heart; Lu = lung; BAT = brown adipose tissue; Kid = kidney; Spl = spleen; small intestine segments proximal to distal are labeled SI-1, SI-2, and SI-3; Te = testes; Br = brain; Mu = skeletal muscle.
ABHD6 Knockdown Protects Against High Fat Diet-Induced Obesity
To examine the role of ABHD6 in lipid metabolism in peripheral tissues without altering expression in the brain, we utilized antisense oligonucleotide (ASO) targeting (Crooke et al., 1997). Initially, we tested four separate ASOs targeting murine ABHD6, and found that all reduced hepatic ABHD6 mRNA by >80% after 4 weeks of administration (Figure S1A). Following 12 weeks of ASO knockdown with two of our ABHD6 ASOs (ASOα and ASOβ), we observed tissue selective knockdown of ABHD6 protein with the following rank order: liver (>90%) > white adipose tissue (>90%) > kidney (~50%) (Figure 2A and Figure S1B). In contrast, ABHD6 protein expression in the brain, spleen, and brown adipose tissue was relatively unaffected by ASO treatment (Figure S1B). Given that ABHD6 has been described as a key enzymatic regulator of endocannabinoid signaling in the brain (Blankman et al., 2007; Marrs et al., 2010; Marrs et al., 2011), we carefully examined whether ASO treatment reduced ABHD6 in brain regions relevant to metabolic disease. Importantly, ABHD6 was not altered in the hippocampus (Figure S1D), whereas livers from the same mice showed marked reductions in ABHD6 expression in the liver (Figure S1C). Furthermore, the mRNA expression of ABHD6 in the hypothalamus was likewise not altered by ASO treatment (Figure S1E).
Figure 2. ASO-Mediated Knockdown of ABHD6 Protects Against High Fat Diet-Induced Obesity.

(A) ABHD6 protein levels in the liver, epididymal white adipose tissue (WAT), and brain of mice treated with either saline, a control non-targeting ASO, or two separate ABHD6 ASOs for 12 weeks.
(B) Body weight in chow-fed or high fat diet (HFD)-fed mice; data represent the mean ± SEM (n = 6-9); * = P < 0.05 (vs. control ASO group within each time point).
(C) Gross appearance of HFD-fed mice.
(D) Gross appearance of epididymal fat pads in HFD-fed mice, and total epididymal fat pad weight in both chow-fed and HFD-fed mice; data represent the mean ± SEM (n = 6-9); * = P < 0.05 (vs. control ASO within each diet).
(E) Body fat % as measured by magnetic resonance imaging (n=4).
(F) Lean body mass as measured by magnetic resonance imaging (n=4).
(G) Snout to anus length (n=10).
(H) Cumulative food intake in ASO-treated mice (n=10).
(I) Total intestinal fat absorption in mice treated with ASOs and fed a HFD for 9 weeks (n=14-15).
(J) qPCR analyses of epididymal adipose tissue (WAT) gene expression in HFD-fed mice; data represent the mean ± SEM (n = 4); * = P < 0.05 (vs. control ASO group). ATGL, adipose triglyceride lipase; HSL, hormone-sensitive lipase, MAGL, monoacylglycerol lipase; SREBP1c, sterol response element-binding protein 1c; FAS, fatty acid synthase; ACC1, acetyl-CoA carboxylase 1; SCD1, stearoyl-CoA desaturase 1; AU = arbitrary units.
(K) Diurnal and nocturnal quantification of physical activity (total beam break counts).
(L) Diurnal and nocturnal quantification of oxygen consumption (VO2).
(M) Diurnal and nocturnal quantification of respiratory exchange ratio (RER; VCO2/VO2). For metabolic cage studies, mice were weight matched and acclimated for at least 48 h prior to measurement. Data represent the mean ± SEM (n = 4); * = P < 0.05 (vs. control ASO).
ABHD6 ASO treatment did not alter body weight (Figure 2B), adiposity (Figure 2E), or food intake (Figure 2H) in mice fed a standard rodent chow diet. However, whenchallenged with a high fat diet, ABHD6 ASO-treated mice were protected from diet-induced body weight gain (Figure 2B and 2C), which was in large part due to a reduction in adipose tissue (Figure 2D). Magnetic resonance imaging (MRI) showed that ABHD6 ASO treatment significantly reduced body fat mass (Figure 2E) without altering lean body mass (Figure 2F). Furthermore, ABHD6 ASO treatment did not result in general growth retardation, given that snout to anus lengths were unaffected (Figure 2G). The protection from high fat diet-induced obesity in ABHD6 ASO-treated mice could not be explained by reductions in food intake (Figure 2H) or by reductions in intestinal fat absorption (Figure 2I). In fact the absorption of several long chain fatty acids was actually slightly increased in ABHD6 ASO-treated mice (Table SI). Gene expression analysis in epididymal white adipose tissue revealed that ABHD6 knockdown caused increased expression of lipolyticgenes such as HSL and MAGL (Figure 2J), with very minor changes in lipogenic gene expression such as SREBP1c and SCD1 in white adipose tissue (Figure 2J). The ability of ABHD6 ASO treatment to protect against high fat diet-induced obesity could be explained in part by increases in physical activity during the dark cycle (Figure 2K) and increases in energy expenditure during both the diurnal and nocturnal phases (Figure 2L). There were no detectable alterations in the respiratory exchange ratio (RER) in control and ABHD6 ASO-treated mice fed a high fat diet (Figure 2M).
ABHD6 Knockdown Protects Against Metabolic Disorders Induced by High Fat Feeding
ABHD6 knockdown significantly reduced high fat diet-induced accumulation of total hepatic triacylglycerol (TAG) (Figure 3A) without altering total hepatic levels of diacylglycerols (DAG) or monoacylglycerols (MAG) in either dietary setting (Figure 3B). This reduction in neutral lipids was observed in almost all molecular species of TAG, with the exception of 56:6 TAG (Figure S3A). ABHD6 ASO treatment significantly blunted high fat diet-driven increases in some hepatic DAG species (32:2, 32:1, 34:3, 34:2, 34:1, and 38:6), but not in others (36:2) (Figure S3B). In parallel, ABHD6 knockdown protected mice from high fat diet-induced hyperglycemia (Figure 3C), hyperinsulinemia (Figure 3D), and improved both glucose and insulin tolerance (Figure 3H and 3I). Interestingly, reducing ABHD6 did not alter plasma TAG levels on either diet (Figure 3E), yet knockdown increased plasma non-esterified fatty acid (NEFA) levels specifically in high fat diet fed mice (Figure 3G). Additionally, ABHD6 ASO treatment did not alter VLDL triacylglycerol secretion rates (Figure 3J). However, ABHD6 knockdown protected against high fat-dietinduced hypercholesterolemia (Figure 3F), which was reflected as a significant decrease in LDL and a modest increase in HDL levels (data not shown). Importantly, hepatic short chain TAG species were significantly decreased by ABHD6 ASO treatment (50:4, 52:5,52:4, and 54:7) in chow-fed mice, where systemic insulin sensitivity and plasma triacylglycerol levels were similar to control mice (Figure S7).
Figure 3. ASO-Mediated Knockdown of ABHD6 Protects Against Metabolic Disorders Induced by High Fat Feeding.

Mice were fed a chow or high fat diet (HFD) and treated with a control ASO or an ASO targeting ABHD6 for 12 weeks.
(A) Gross appearance of livers and microscopic examination (H&E staining at 40x magnification) in HFD-fed mice.
(B) Total hepatic levels of triacylglycerols (TAG), diacylglycerols (DAG), and monoacylglycerols (MAG) in mice fed diets for 12 weeks.
(C) Plasma glucose levels in mice treated with ASOs and diets for 10-11 weeks.
(D) Plasma insulin levels in mice treated with ASOs and diets for 12 weeks.
(E) Plasma TAG levels in mice treated with ASOs and diets for 12 weeks.
(F) Plasma cholesterol levels in mice treated with ASOs and diets for 12 weeks.
(G) Plasma non-esterified fatty acids (NEFAs) in mice treated with ASOs and diets for 12 weeks.
(H) Glucose tolerance tests in mice treated with ASOs and diets for 10-11 weeks.
(I) Insulin tolerance tests in mice treated with ASOs and diets for 10-11 weeks.
(J) Hepatic VLDL-TAG secretion rates in mice treated with ASOs and high fat diet for 11 weeks.
All data represent the mean ± SEM (n = 4-6), * = P < 0.05 (vs. control ASO within each diet).
ABHD6 is a Critical Regulator of De Novo Fatty Acid Synthesis
When comparing global hepatic gene expression between high fat diet-fed control vs. ABHD6 ASO-treated mice, we found a surprisingly small number of genes that were differentially expressed by greater than 2-fold (167 upregulated genes and 138 downregulated genes; p <0.005) (Figure 4A). The most highly enriched gene ontology (-log[p-value] = 10) regulated by ABHD6 knockdown was fatty acid metabolism (Figure 4A). ABHD6 ASO treatment resulted in a 50% reduction in hepatic ABHD5 expression, which is unlikely to be from direct ASO-mediated silencing due to lack of sequence homology between the two mRNAs. Knockdown of ABHD6 also increased ATGL expression, while reducing HSL expression in the liver (Figure 4B). More consistently, we observed coordinate downregulation of genes involved in de novo fatty acid synthesis and lipogenesis (SREBP1c, FAS, ACC-1, and SCD-1) in ABHD6 ASO-treated mouse liver (Figure 4B and 4C). In agreement, the in vivo rate of de novo fatty acid synthesis wasreduced by 62% in ABHD6 ASO-treated mice (Figure 4D). Consistent with this, primary hepatocytes isolated from ABHD6 ASO-treated livers showed decreased rates of 3H-oleate esterification into triacylglycerol (Figure 4E) as well as decreased de novo lipogenesis rates as measured by the kinetic conversion of 14C-acetate into 14C-triacylglycerol (Figure 4F). It is important to note that all four ASOs targeting the knockdown of ABHD6 at different sites consistently decreased hepatic lipogenic gene expression (Figure S1B, S1C, and S1D), without altering adipose tissue (Figure 2J) or hypothalamic (Figure S1E) lipogenic gene expression. We also examined thephosphorylation state of both AMPKα and AMPKβ in mouse liver, but noted no obvious differences in activation state (Figure S2E).
Figure 4. ABHD6 is a Critical Regulator of De Novo Lipogenesis.

(A) Biologic processes overrepresented among up-regulated and down-regulated genes identified by microarray analysis from livers of ABHD6 ASO-treated mice compared with control ASO-treated mice fed a high fat diet.
(B) qPCR confirmation of hepatic genes identified by microarray analyses in HFD-fed mice; data represent the mean ± SEM (n = 4); * = P < 0.05 (vs. control ASO group). ATGL, adipose triglyceride lipase; HSL, hormone-sensitive lipase, MAGL, monoacylglycerol lipase; SREBP1c, sterol response element-binding protein 1c; FAS, fatty acid synthase; ACC1, acetyl-CoA carboxylase 1; SCD1, stearoyl-CoA desaturase 1; SREBP2, sterol response element-binding protein 2; HMG-Red, 3-hydroxy-3-methylglutaryl-CoA reductase; LDLr, low-density lipoprotein receptor; PPARα, peroxisome proliferator-activated receptor alpha; CPT-1α, carnitine palmitoyltransferase 1; AOX, acyl-CoA oxidase; AU = arbitrary units.
(C) Hepatic lipogenic protein expression in mice treated with a control non targeting ASO, or two independent ABHD6 ASOs for 12 weeks.
(D) In vivo synthesis rates of fatty acids in livers of control and ABHD6 ASO treated mice. HFD-fed male mice (6 weeks of diet and ASO) were injected intraperitoneally with 3H-labeled water, and 1 hour later livers were removed for measurement of 3H-labeled fatty acids as described in the methods section.
(E) Esterification rates in primary hepatocytes isolated from ASO treated mice. Hepatocytes were kinetically labeled with 3H-oleate to follow the conversion into 3H-triacylglycerol in the presence of lipase inhibitors to block lipolysis/re-esterification. Data represent the mean ± SEM (n = 3) from a representative experiment, which was repeated twice in pooled hepatocytes isolated from ASO-treated mice; * = P < 0.05 (vs. control ASO group).
(F) De novo lipogenesis rates in primary hepatocyte isolated from ASO treated mice. Hepatocytes were kinetically labeled with 14C-acetate to follow the conversion into 14C-triacylglycerol in the presence of lipase inhibitors to block lipolysis/re-esterification. Data represent the mean ± SEM (n = 3) from a representative experiment, which was repeated twice in pooled hepatocytes isolated from ASO-treated mice; * = P < 0.05 (vs. control ASO group).
ABHD6 is a Minor Monoacylglycerol Lipase in Mouse Liver, and Knockdown Does Not Alter Hepatic Endocannabinoid Levels or Acute Cannabinoid Receptor 1 (CB1) Signaling
Given that ABHD6 was previously described as a monoacylglycerol lipase in the brain (Blankman et al., 2007; Marrs et al., 2010; Marrs et al., 2011), we carefully examined whether ABHD6 knockdown resulted in accumulation of hepatic MAG species, or whether ABHD6 knockdown resulted in hyperactivation of the ECB system in mouse liver. ABHD6 knockdown did not alter the total hepatic levels of MAG in mice fed either chow or a high fat diet (Figure 3B), although two species of MAG containing oleate (18:1) or linoleate (18:2) did modestly increase (Figure 5A). In contrast, hepatic levels of endocannabinoid lipids (2-AG and anandamide) were not changed by ABHD6 ASO treatment (Figure 5B and 5C). There was also no apparent difference in total hepatic MAG lipase activity with ABHD6 knockdown (data not shown). Given that previous studies have demonstrated that the CB1-dependent signaling is largely desensitized when 2-AG builds up as a result of monoacylglycerol lipase (MAGL) inhibition or genetic deficiency (Schlosburg et al., 2010; Taschler, et al., 2012), we carefully examined acute CB1 signaling in ABHD6 ASO-treated mice. To examine the effect of ABHD6 knockdown on CB1 receptor desensitization, we administered the cannabinoid receptor (CB1) agonist CP-55,940 or vehicle directly into the portal vein of ASO-treated mice and followed downstream signaling. ABHD6 knockdown did not alter CP-55,940-induced ERK-MAPK activation compared to control ASO-treated mice (Figure 5D). However, basal activation of ERKMAPK was lower in ABHD6 ASO-treated mice (Figure 5D), indicating that ABHD6 may be involved in regulating other inputs into ERK-MAPK activation.
Figure 5. ABHD6 Knockdown Results in Modest Alterations in Hepatic Monoacylglycerol Levels, Yet Does Not Alter Hepatic Endocannabinoid Levels or Acute Cannabinoid Receptor 1 (CB1) Signaling in Mouse Liver.

(A) Male C57BL/6 mice were fed a chow or high fat diet (HFD) and treated with a control ASO or an ASO targeting ABHD6 for 12 weeks. The hepatic levels of monoacylglycerol (MAG) species were measured by mass spectrometry as described in the methods section. Data represent the mean ± SEM (n = 6), * = P < 0.05 (vs. control ASO within each diet).
(B and C) Male C57BL/6 mice were fed a high fat diet (HFD) and treated with a control non-targeting ASO or an ASO targeting the knockdown of ABHD6 for 12 weeks. The hepatic levels of 2-arachidonylglycerol (B) and anandamide (C) were measured by mass spectrometry as described in the methods section. Data represent the mean ± SEM (n = 6), and no significant differences were found.
(D) Male C57BL/6 mice were fed a high fat diet (HFD) and treated with a control ASO or an ASO targeting ABHD6 for 8 weeks. Following 8 weeks of ASO treatment, mice were fasted for 12 h and subsequently injected with either vehicle or CP-55,940 (0.1 mg/kg) directly into the portal vein. Exactly 5 min later, livers were excised and immediately snap frozen in liquid nitrogen. Protein extracts from the liver were analyzed by Western blotting for ABHD6, phospho-ERK (Thr202/Tyr204), total ERK MAPK, monoacylglycerol lipase (MAGL), or beta actin (β-actin); three representative animals are shown for each group.
ASO-Mediated Inhibition of ABHD6 Uncovers a Role for ABHD6 in Lysophospholipid Metabolism
Interestingly, a number of phospholipids and lysophospholipids accumulated to varying degrees in ABHD6 ASO-treated livers (Figure 6), implicating them as potential physiologically relevant substrates. ABHD6 knockdown significantly increased total hepatic levels of phosphatidylcholine (PC), lysophosphatidylcholine (LPC), phosphatidylethanolamine (PE), lysophosphatidylethanolamine (LPE), phosphatidylglycerol (PG), lysophosphatidylglycerol (LPG), phosphatidylinositol (PI), lysophosphatidylinositol (LPI), and phosphatidylserine (PS) (Figure 6B, 6C, 6D, 6E, 6F, 6H, 6I, 6J, 6K, and 6L), without altering hepatic levels of phosphatidic acid or lysophosphatidic acid species (Figure 6A, 6G, and Figure S4C). Of interest, the most prominent accumulation was observed for nearly all species of LPG (Figure 6J, Figure S6A) and PG (Figure 6D, Figure S5C) in ABHD6 knockdown mice. Inhibition of ABHD6 also promoted the accumulation of several ether-linked glycerophospholipids (plasmalogens) including all detected molecular species of plasmanylcholines (Figure S4A) and plasmenylethanolamines (Figure S4B). In parallel, nearly all species of LPE, LPG, LPI, and LPS were increased with ABHD6 knockdown regardless of diet (Figure S6). We subsequently tested whether ABHD6 can hydrolyze phospholipid and neutral lipid substrates in vitro. To accomplish this we expressed a GST-tagged ABHD6 in S. cerevisiae (Figure 6M), and the purified protein was incubated in the presence of a panel of lipid substrates (Figure 6M-Q). As previously demonstrated with ABHD6-expressing cell homogenates (Blankman et al., 2007; Marrs et al., 2010; Marrs et al., 2011; Navia-Paldanius et al., 2012) ABHD6 hydrolyzes MAG substrates including 1,(3)-rac--oleoylglycerol and 2-oleoylglycerol, and its ability to hydrolyze MAG substrates is lost when the active site serine is mutated to alanine (S148A) (Figure 6N). However, in saturation kinetic experiments recombinant MAGL exhibited a 11-fold higher specific activity (Vmax = 5359.3 μmol/h*mg) than ABHD6 (Vmax = 478.6 μmol/h*mg) (Figure 6O), and under the applied conditions the apparent Km was lower for MAGL (Km = 1.2) compared to ABHD6 (Km = 1.9), indicating that ABHD6 has a lower affinity for MAG.
Figure 6. ASO-Mediated Knockdown Annotates ABHD6 as a Physiological Lysophospholipase in Mouse Liver.

Mice were fed a chow or high fat diet and treated with a control non-targeting ASO or an ASO targeting ABHD6 for 12 weeks (panels A-L).
(A) Total hepatic phosphatidic acid (PA) levels.
(B) Total hepatic phosphatidylcholine (PC) levels.
(C) Total hepatic phosphatidylethanolamine (PE) levels.
(D) Total hepatic phosphatidylglycerol (PG) levels.
(E) Total hepatic phosphatidylinositol (PI) levels.
(F) Total hepatic phosphatidylserine (PS) levels.
(G) Hepatic levels of 16:0 lysophosphatidic acid (LPA).
(H) Hepatic levels of 18:2 lysophosphatidylcholine (LPC).
(I) Hepatic levels of 18:2 lysophosphatidylethanolamine (LPE).
(J) Hepatic levels of 18:2 lysophosphatidylglycerol (LPG).
(K) Hepatic levels of 18:0 lysophosphatidylinositol (LPI).
(L) Hepatic levels of 18:0 lysophosphatylserine (LPS). All data in panels A-L represent the mean ± SEM (n = 6), * = P < 0.05 (vs. control ASO within each diet). In panels M-Q recombinant ABHD6 or monoacylglycerol lipase (MAGL) were used to test in vitro substrate specificity of both enzymes.
(M) Coomassie stain of purified GST-tagged murine ABHD6, which was expressed in S. cerevisiae and purified by affinity chromatography; lane 1 = molecular weight ladder, lane 2 = affinity purified ABHD6.
(N) Degradation of 1(3)-oleoylglycerol [1,(3)-rac-OG; 3 mM] and 2-oleoylglycerol [2-OG; 3 mM] by wild type (WT) GST-ABHD6 and a mutant variant of ABHD6 lacking the putative active serine (S148G).
(O) Saturation kinetics of ABHD6 and monoacylglycerol lipase (MAGL) using 1(3)-monoolein as substrate. Data are presented as the mean ± S.D. and representative of at least two independent experiments.
(P) Purified GST-ABHD6 was incubated in the presence of a panel of potential glycerophospholipid substrates and the release of fatty acids was determined. Data are presented as the mean ± S.D. and are representative of at least two independent experiments. * = P < 0.05 (lysophospholipid vs. phospholipid)
(Q) Saturation kinetics of ABHD6 using 1-oleoyl lysophosphatidylglycerol (LPG) as substrate. Data are presented as the mean ± S.D. and representative of at least two independent experiments.
Next, we investigated whether affinity-purified ABHD6 hydrolyzes the other glycerophospholipid substrates identified by in vivo lipidomics (Figure 6A-6L). Recombinant ABHD6 showed considerable lipase activity towards several lysophospholipids including LPG, LPA, and LPE but not LPC (Figure 6P). In contrast, ABHD6 exhibited no lipase activity against major phospholipid classes including PG, PE, PA, PS, and PC (Figure 6P). The highest activity was observed using LPG as a substrate (Figure 6P and 6Q), and this activity was even higher in a detergent-containing (5 mM CHAPS) buffer system. Under these conditions, we determined a Vmax of 93.2 μmol/h*mg and a Km of 0.75 (Figure 6Q). Furthermore, purified ABHD6 exhibited low activity against retinyl palmitate (RP) and rac-dioleoylglycerol, whereas no activity was observed in the presence of trioleoylglycerol or cholesteryl oleate (data not shown). Notably, ABHD6's activityagainst 1,3-diacylglycerol was 5-fold higher in comparison to 1,2(2,3)-diacylglycerol (data not shown). The ability of ABHD6 to hydrolyze neutral lipid and lysophospholipid substrates was completely lost when the active site serine was mutated to alanine (Figure 6 and data not shown). It is important to note that MAGL did not hydrolyze any of the tested phospholipid substrates (data not shown) annotating MAGL, but not ABHD6, as a specific MAG hydrolase. Taken together, these observations suggest that ABHD6 can act both as a monoacylglycerol lipase and lysophospholipase exhibiting a preference for LPG among the tested lysophospholipids.
Small Molecule Inhibition of ABHD6 Protects Against High Fat Diet-Induced Glucose Intolerance and Obesity
ASO-mediated inhibition is a tissue-restricted therapeutic approach, targeting knockdown in metabolic tissues (liver, white adipose tissue, and kidney) without altering ABHD6 expression or activity in many other tissues (brain, spleen, brown adipose tissue). However, ABHD6 is ubiquitously expressed (Figure 1B), begging the question whether systemic inhibition would likewise protect against metabolic disease. Therefore, we determined whether a small molecule inhibitor of ABHD6, which would be predicted to target all tissues including the brain, also provides protection against the metabolic disorders driven by high fat diet feeding. Treatment with the small molecule inhibitor of ABHD6 (WWL-70) protected mice from high fat diet-induced body weight gain (Figure 7A), which was largely due to a reduction in adipose tissue mass (Figure 7B). WWL-70 treatment also protected mice against high fat diet-induced glucose intolerance. Although there was a trend towards a decrease, WWL-70 did not significantly reduce hepatic triacylglycerol levels in high fat diet fed mice (Figure 7D), which contrasts with what is observed with ASO-mediated inhibition (Figure 3). These results show that inhibition of ABHD6 using a systemic inhibitor improves some aspects of the metabolic syndrome, but does not protect against hepatic steatosis to the same degree that ASO-mediated inhibition does (Figure 3).
Figure 7. Small Molecule Inhibition of ABHD6 Protects Against High Fat Diet-Induced Glucose Intolerance and Obesity.
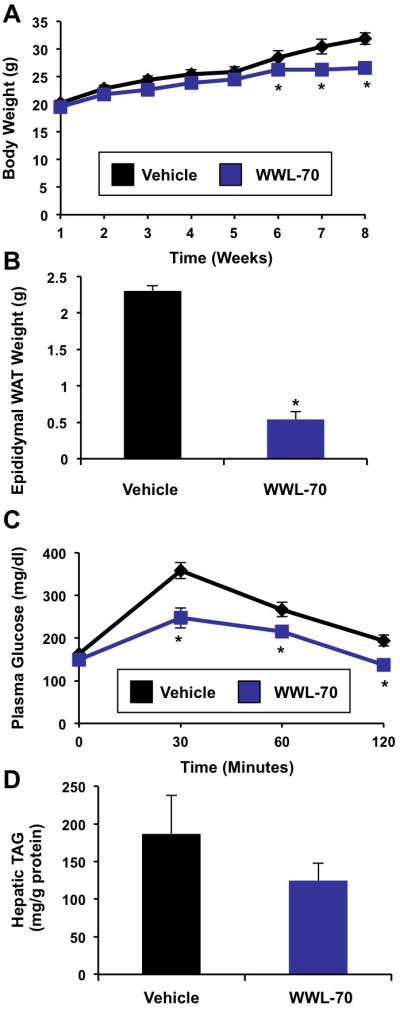
Male C57BL/6 mice were fed a high fat diet (HFD) and treated with a vehicle or 10 mg/kg of the ABHD6 inhibitor WWL-70 for 8 weeks.
(A) Body weight.
(B) Epididymal white adipose tissue (WAT) weight.
(C) Glucose tolerance tests in mice treated with inhibitor and diet for 6 weeks.
(D) Total hepatic triacylglycerol levels in mice treated with inhibitor and diet for 8 weeks. Data represent the mean ± SEM (n = 10), * = P < 0.05 (vs. vehicle).
DISCUSSION
Although other members of the ABHD protein family have been clearly linked to lipid signaling and metabolic disease (Lefevre et al., 2001; Fiskerstrand et al., 2010; Long et al., 2011; Simon and Cravatt 2006; Montero-Moran et al., 2009; Blankman et al., 2007; Lord et al., 2011; Brown et al., 2010), this is the first study to document a role for ABHD6 in promoting the metabolic disorders driven by high fat diet feeding. The major findings of this work demonstrate the following: 1) peripheral knockdown of ABHD6 protects mice from high fat diet-induced obesity, 2) ABHD6 knockdown protects mice from high fat diet-induced hepatic steatosis and associated insulin resistance, 3) ABHD6 is a critical regulator of hepatic de novo lipogenesis, and 4) ABHD6 hydrolyzes several lipid substrates in vitro, including lysophospholipids with preference for LPG. Accordingly, we show that inhibition of ABHD6 results in the accumulation of LPG and PG in vivo. Taken together, our results reveal that ABHD6 plays a role in the development of the metabolic syndrome.
ABHD6 has been shown to regulate the ECB system in the brain due to its ability to hydrolyze specific pools of 2-AG (Blankman et al., 2007; Marrs et al., 2010; Marrs et al., 2011). We also confirmed that ABHD6 exhibits MAG lipase activity in vitro (Figure 6N and 6O). Our data suggest however, that ABHD6 is a minor contributor to MAG lipolysis and ECB signaling in mouse liver. This finding is supported by the fact that total MAG levels do not change (Figure 3B), 2-AG and anandamide levels are not elevated (Figure 5B and 5C), and acute CB1-driven activation of ERK-MAPK is unaltered with ABHD6 knockdown (Figure 5D), arguing against CB1 receptor desensitization. It is important to contrast these findings with the very striking accumulation of hepatic 2-AG, marked reduction in hepatic MAG lipase activity, and CB1 receptor desensitization seen in both MAGL−/− mice and mice treated chronically with a specific MAGL inhibitor (Taschler et al., 2011; Schlosburg et al., 2010). It is also important to note that if ABHD6 was a major regulator of hepatic 2-AG levels, one would anticipate that ABHD6 knockdown would cause hyperactivation of hepatic CB1 signaling, which has been reported to promote hepatic steatosis through increasing de novo lipogenesis (Osei-Hyiaman et al., 2005, Osei-Hyiaman et al., 2008). However, we observed that ABHD6 knockdown protected mice from high fat diet induced hepatic steatosis and suppressed de novo lipogenesis (Figure 4). Collectively, these results support the notion that MAGL is the predominant MAG lipase in mouse liver. We did, however, see minor elevations in 18:1 and 18:2 MAG (Figure 5A), making it possible that ABHD6's ability to hydrolyze these lipids may affect other signaling processes regulating hepatic de novo lipogenesis.
ABHD6 contributes to 2-AG hydrolysis and ECB signaling in the brain (Blankman et al., 2007; Marrs et al., 2010; Marrs et al., 2011). However, MAGL accounts for the vast majority (>80%) of 2-AG hydrolysis in the brain, whereas ABHD6 only accounts for less than 5% of the total 2-AG hydrolase activity (Blankman et al., 2007). Our studies suggest that ABHD6's additional ability to hydrolyze a variety of lipid substrates may be important in linking lipid mediator signaling to the metabolic adaptations resulting from high fat diet feeding. In this study we utilized a targeted lipidomics approach to identify new substrates of ABHD6 in mouse liver, and verified LPG as a bona fide substrate in vitro and in vivo. LPG is considered a bioactive lipid, although its receptor remains to be identified (Makide et al., 2009; Jo et al., 2008). Lysophospholipids have previously been implicated in promoting metabolic disease via their ability to dictate membrane dynamics and initiate cell signaling, particularly in immune cells (Wymann et al., 2008; Skoura and Hla, 2009). We assume that defective LPG degradation favors the reesterification of LPG to PG, which may explain the elevated PG levels in the ABHD6-knockdown liver. PG is also an important precursor for other complex lipids including cardiolipin and bis(monoacylglycerol)phosphate (Hullin-Matsuda et al., 2007). Therefore, changes in ABHD6-driven LPG metabolism could alter many mitochondrial or lysosomal metabolic processes. Interestingly, a recent genome-wide association study (GWAS) in Pima Indians identified the lysophosphatidylglycerol acyltransferase 1 (LPGAT1) loci as being predictive of body mass index (BMI), providing additional genetic evidence that LPG metabolism may influence BMI and adiposity (Traurig et al., 2012).
Projecting forward, there are several important factors to consider for developing ABHD6 as a drug target. For example, it will be important to determine how ABHD6 expression and subcellular distribution are regulated in diverse cell and tissue contexts. Furthermore, it will be essential to evaluate whether ABHD6 has other physiological substrates in vivo, and if tissue-selective inhibitors will be necessary for safe and effective prevention of metabolic disease. Another important consideration will be whether chronic ABHD6 inhibition results in CB1 receptor desensitization in the brain or other receptor signaling abnormalities. Our data suggest that chronic ABHD6 inhibition with ASOs does not cause CB1 receptor desensitization in mouse liver (Figure 5D). However, whether CB1 receptor desensitization occurred in other tissues was not examined here. As previously documented (Bachovchin et al., 2010), we show that ABHD6 is ubiquitously expressed in mice (Figure 1B). Additional studies will be required to assess the risk versus benefits of inhibiting ABHD6 in specific tissues. Our studies provide key information in regards to ABHD6's biochemical and physiological role in certain peripheral tissues (liver, adipose, and kidney) given the tissue-selective inhibition observed with ASOs (Figure S1). However, our studies do not address ABHD6's primary role in other tissues where it is abundantly expressed (brain, small intestine, pancreas, and skeletal muscle), all of which are key sites regulatory sites for energy metabolism.
It is important to compare and contrast the results of inhibiting ABHD6 with systemic small molecule inhibitors versus a more selective approach using ASO technology. Although both ASO-mediated and WWL-70-mediated inhibition of ABHD6 protected against high fat diet-induced obesity and glucose intolerance, only ABHD6 ASO treatment improved hepatic steatosis. The mechanism underlying this difference is unclear at this point, but this most likely stems from systemic versus tissue-restricted inhibition. Interestingly, ASO-mediated inhibition of ABHD6 was not associated with alteration in food intake at any time point examined (Figure 2H). However, WWL-70 treatment caused a 20% reduction in food intake (data not shown), indicating that the mechanism by which WWL-70 protected against obesity and glucose intolerance is likely driven by hypophagia, while ASO-mediated inhibition more specifically dampened hepatic lipogenesis and increases in energy expenditure.
In addition to further studies with selective ABHD6 inhibitors, the generation of global and conditional knockout mouse models is necessary to define the tissue-specific role for ABHD6 in chronic diseases of altered lipid metabolism. In summary, our data identify ABHD6 as a key determinant in the pathogenesis of HFD-induced obesity, insulin resistance, and hepatic steatosis. Furthermore, we have uncovered novel lipid substrates of ABHD6 by coupling in vivo lipidomic and in vitro biochemical approaches. Looking forward, we believe that utilizing a similar in vivo loss-of-function approach may prove useful for mapping natural enzyme-substrate relationships for the other uncharacterized enzymes in the ABHD family. It is interesting to note that during the preparation of this manuscript that another ABHD enzyme ABHD12 was likewise identified as a dual monoacylglycerol lipase and lysophospholipase with preference towards lysophosphatidylserine species using a similar in vivo substrate identification approach (Blankman et al., 2013). Taken together, our findings suggest that ABHD6 is a key lipase involved in monoacylglycerol and lysophospholipid hydrolysis, and that ABHD6 inhibitors hold promise as therapeutics for obesity, non-alcoholic fatty liver disease, and type II diabetes.
EXPERIMENTAL PROCEDURES
Mice
For ABHD6 knockdown studies, at 6-8 weeks of age male C57BL/6N mice (Harlan) were either maintained on standard rodent chow or switched to a high fat diet for a period of 4-12 weeks, and were simultaneously injected with murine specific ABHD6 antisense oligonucleotides biweekly (25 mg/kg BW) as previously described (Lord et al., 2011; Brown et al., 2010; Brown et al., 2008a; Brown et al., 2008b). For small molecule inhibitor studies, mice were fed a high fat diet and simultaneously treated with either a vehicle or 10 mg/kg WWL-70 for 8 weeks. Intraperitoneal glucose tolerance tests and insulin tolerance tests were performed essentially as previously described (Brown et al., 2008a, Brown et al., 2010) in mice treated with diet and ASO for 10-11 weeks. Metabolic measurements were conducted in the comprehensive lab animal monitoring systems (CLAMS) from Columbus Instruments. Body composition was determined by magnetic resonance imaging (MRI). A detailed description of all mouse experiments is provided in the online supplementary methods section.
In Vivo Cannabinoid Receptor (CB1) Receptor Signaling Analyses
Mice were injected with control ASO or ABHD6 ASOβ and maintained on a HFD for a period of 8 weeks prior to experiment. After an overnight fast (9:00 p.m. - 9:00 a.m.), mice were anesthetized with isoflurane (4% for induction, 2% for maintenance), and were maintained on a 37°C heating pad to control body temperature. A minimal midline laparotomy was performed and the portal vein was visualized. Thereafter, mice received either vehicle (cremophor EL:ethanol:saline at a 1:1:18 ratio) or the CB1 agonist (CP-55,490, Cayman Chemical # 13241) at a dose of 0.1 mg/kg body weight directly into the portal vein. Exactly 5 minutes later, the liver was excised and immediately snap frozen in liquid nitrogen. Protein extracts from tissues were analyzed by Western blotting to examine CB1 signaling as described below.
In Vivo Determination of Very Low Density Lipoprotein (VLDL) Secretion, Intestinal Fat Absorption, and De Novo Fatty Acid Synthesis Rates
VLDL secretion rates were determined using the detergent block method (Li et al., 1996). Dietary fat absorption was measured using the Olestra® method (Jandacek et al., 2004), and de novo fatty acid synthesis was measured using the 3H-water method (Shimano et al., 1996). These methods are described in detail in the online supplementary methods section.
Primary Hepatocyte Studies
Hepatocytes were isolated from chow-fed ASO-treated mice using the collagenase perfusion method, and the rate of de novo lipogenesis was determined following the conversion of 14C-acetate and 3H-oleate into newly synthesized triacylglycerol in the presence of lipase inhibitors to block lipolysis/re-esterification. A detailed method is provided in the online supplementary methods.
Generation and Purification of a Polyclonal Antibody Against ABHD6
A maltose binding protein ABHD6 fusion protein construct was generated to create affinity-purified rabbit polyclonal antibodies against murine ABHD6. A detailed description of cloning, expression, and purification are included in the online supplementary methods section.
Immunoblotting
Whole tissue homogenates were made from multiple tissues in a modified RIPA buffer, and Western blotting was conducted as previously described (Brown et al., 2004).
Microarray and Quantitative Real-Time PCR Analysis of Gene Expression
Tissue RNA extraction was performed as previously described for all mRNA analyses (Lord et al., 2011; Brown et al., 2010; Brown et al., 2008a; Brown et al., 2008b). Microarray analyses were performed by the Wake Forest School of Medicine Microarray Shared Resource Core using standard operating procedures, and quantitative real time PCR (qPCR) analyses were conducted as previously described (Lord et al., 2011; Brown et al., 2010; Brown et al., 2008a; Brown et al., 2008b). A detailed description of RNA methods is available in the online supplementary methods section.
Hepatic Lipid Analyses
Extraction of liver lipids and quantification of molecular species by mass spectrometry was performed as previously described (Lord et al., 2011; Ivanova et al., 2007; Myers et al., 2011; Callender et al., 2007; Saghatelian et al., 2004).
Purification of GST-tagged murine ABHD6 for Enzymology Studies
The coding sequence of murine ABHD6 was cloned into the yeast expression vector pYEX4T-1. The resulting protein was purified using glutathione-sepharose beads, and used for enzymology studies as described in the online supplementary methods section.
Statistical Analysis
All data are expressed as the mean ± S.E.M. or S.D., and were analyzed using either a one-way or two-way analysis of variance (ANOVA) followed by Student's t tests for post hoc analysis using JMP version 5.0.12 software (SAS Institute, Cary, NC).
Supplementary Material
Highlights.
ABHD6 inhibition protects against high fat diet-induced obesity and insulin resistance
ABHD6 inhibition protects against high fat diet-induced hepatic steatosis
ABHD6 is a critical regulator of hepatic de novo lipogenesis
ABHD6 is both a monoacylglycerol lipase and a lysophospholipase
ACKNOWLEDGEMENTS
We thank Larry Rudel, Paul Dawson, and Ryan Temel (Wake Forest School of Medicine) for insightful comments and suggestions. We also sincerely thank Marc Prentki and Murthy Madiraju (Montreal Diabetes Research Center) for critical review of this work. This work was supported by the Department of Pathology at Wake Forest School of Medicine, a pilot grant from the Wake Forest School of Medicine Venture Fund, and a pilot grant awarded under the Wake Forest and Harvard Center for Botanical Lipids (P50-AT002782). These studies also received generous funding by the National Institute of General Medical Sciences LIPID MAPS (U54-GM069338 to H.A.B.), and National Institute of Diabetes and Digestive and Kidney Diseases (F32-DK084582 to J.L.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information including Extended Experimental Procedures, 7 Supplemental Figures, and 1 Supplemental Table can be found with this article online.
Other than Richard Lee, Rosanne Crooke, and Mark Graham, who are employees at ISIS pharmaceuticals, Inc. (Carlsbad, CA), all other authors report that they have no conflicts of interest.
REFERENCES
- Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc. Natl. Acad. Sci. USA. 2010;107:20941–20946. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Long JZ, Trauger SA, Siuzdak G, Cravatt BF. ABHD12 controls brain lysophophatidylserine pathways that are deregulated in a murine model of the neurodegenerative disease PHARC. Proc. Natl. Acad. Sci. USA. 2013;110:1500–1505. doi: 10.1073/pnas.1217121110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Boysen MS, Chung S, Fabiyi F, Morrison RF, Mandrup S, McIntosh MK. Conjugated linoleic acid induces human adipocyte delipidation: autocrine/paracrine regulation of MEK/ERK signaling by adipocytokines. J. Biol. Chem. 2004;279:26735–26747. doi: 10.1074/jbc.M401766200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Chung S, Sawyer JK, Degirolamo C, Alger HM, Nguyen T, Zhu X, Duong MN, Wibley AL, Shah R, et al. Inhibition of stearoyl coenzyme A desaturase 1 dissociates insulin resistance and obesity from atherosclerosis. Circulation. 2008a;118:1467–1475. doi: 10.1161/CIRCULATIONAHA.108.793182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Bell TA, 3rd, Alger HM, Sawyer JK, Smith TL, Kelley K, Shah R, Wilson MD, Davis MA, Lee RG, et al. Targeted depletion of hepatic ACAT2-driven cholesterol esterification reveals a non-biliary route for fecal neutral sterol loss. J. Biol. Chem. 2008b;283:10522–10534. doi: 10.1074/jbc.M707659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Betters JL, Lord C, Ma Y, Han X, Yang K, Alger HM, Melchior J, Sawyer JK, Shah R, et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J. Lipid Res. 2010;51:3306–3316. doi: 10.1194/jlr.M010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callender HL, Forrester JS, Ivanova P, Preininger A, Milne S, Brown HA. Quantification of diacylglycerol species from cellular extracts by electrospray ionization mass spectrometry using a linear regression algorithm. Anal. Chem. 2007;79:263–272. doi: 10.1021/ac061083q. [DOI] [PubMed] [Google Scholar]
- Crooke ST. Advances in understanding the pharmacological properties of antisense oligonucleotides. Adv. Pharmacol. 1997;40:1–49. doi: 10.1016/s1054-3589(08)60136-2. [DOI] [PubMed] [Google Scholar]
- Fiskerstrand T, H'mida-Ben Brahim D, Johansson S, M'zahem A, Haukanes BI, Drouot N, Zimmermann J, Cole AJ, Vedeler C, Bredrup C, et al. Mutations in ABHD12 cause the neurodegenerative disease PHARC: An inborn error of endocannabinoid metabolism. Am. J. Hum. Genet. 2010;87:410–417. doi: 10.1016/j.ajhg.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotelier T, Renault L, Cousin X, Negre V, Marchot P, Chatonnet A. ESTHER, the database of the alpha/beta-hydrolase fold superfamily of proteins. Nucleic Acids Res. 2004;32:D145–147. doi: 10.1093/nar/gkh141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova PT, Milne SB, Byrne MO, Xiang Y, Brown HA. Glycerophospholipid identification and quantitation by electrospray ionization mass spectrometry. Meth. Enzymol. 2007;432:21–57. doi: 10.1016/S0076-6879(07)32002-8. [DOI] [PubMed] [Google Scholar]
- Hullin-Matsuda F, Kawasaki K, Delton-Vandenbroucke I, Xu Y, Nishijima M, Lagarde M, Schlame M, Kobayashi T. De novo biosynthesis of the late endosomal lipid, bis(monoacylglycero)phosphate. J. Lipid Res. 2007;48:1997–2008. doi: 10.1194/jlr.M700154-JLR200. [DOI] [PubMed] [Google Scholar]
- Jandacek R, Heubi JE, Tso P. A novel, noninvasive method for the measurement of intestinal fat aborption. Gastroenterology. 2004;2004;127:139–144. doi: 10.1053/j.gastro.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Jo SH, Kim SD, Kim JM, Lee HY, Lee SY, Shim JW, Yun J, Im DS, Bae YS. Lysophosphatidylglycerol stimulates chemotactic migration in human natural killer cells. Biochem. Biophys. Res. Commun. 2008;372:147–151. doi: 10.1016/j.bbrc.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Lefevre F, Jobard F, Caux F, Bouadjar B, Karaduman A, Helig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach J, et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin- Dorfman syndrome. Am. J. Hum. Genet. 2001;69:1002–1012. doi: 10.1086/324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006;3:309–319. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Li X, Catalina F, Grundy SM, Patel S. Method to measure apolipoprotein B-48 and B-100 secretion rates in an individual mouse: evidence for a very rapid turnover of VLDL and preferential removal of B-48- relative to B-100- containing lipoproteins. J. Lipid Res. 1996;37:210–220. [PubMed] [Google Scholar]
- Li W, Blankman JL, Cravatt BF. A functional proteomic strategy to discover inhibitors for uncharacterized hydrolases. J. Am. Chem. Soc. 2007;129:9594–9595. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 2009;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Cisar JS, Milliken D, Niessen S, Wang C, Trauger SA, Sludzdak G, Cravatt BF. Metabolomics annotates ABHD3 as a physiologic regulator of medium-chain phospholipids. Nat. Chem. Biol. 2011;7:763–765. doi: 10.1038/nchembio.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CC, Betters JL, Ivanova PT, Milne SB, Myers DS, Madenspacher J, Thomas G, Chung S, Liu M, Davis MA, et al. CGI-58/ABHD5-derived signaling lipids regulate systemic inflammation and insulin action. Diabetes. 2012;61:355–363. doi: 10.2337/db11-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makide K, Kitamura H, Sato Y, Okutani M, Aoki J. Emerging lysophospholipid mediators. Lysophosphatidylserine, lysophosphatidylthreonine, lysophosphatidylethanolamine, and lysophosphatidylglycerol. Prostaglandins Other Lipid Mediat. 2009;89:135–139. doi: 10.1016/j.prostaglandins.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat. Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs WR, Horne EA, Ortega-Gutierrez S, Cisneros JA, Xu C, Lin YH, Muccioli GG, Lopez-Rodriguez ML, Stella N. Dual inhibition of alpha/beta-hydrolase domain 6 and fatty acid amide hydrolase increases endocannabinoid levels in neurons. J. Biol. Chem. 2011;286:28723–28728. doi: 10.1074/jbc.M110.202853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero-Moran G, Caviglia JM, McMahon D, Rothenberg A, Subramanian V, Xu Z, Lara-Gonzalez S, Storch J, Carman GM, Brasaemle DL. CGI-58/ABHD5 is a coenzyme A-dependent lysophosphatidic acid acyltransferase. J. Lipid Res. 2009;51:709–719. doi: 10.1194/jlr.M001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers DS, Ivanova PT, Milne SB, Brown HA. Quantitative analysis of glycerophospholipids by LC-MS: Acquisition, data handling and interpretation. Biochim. Biophys. Acta. 2011;1811:748–757. doi: 10.1016/j.bbalip.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagan N, Zoeller RA. Plasmalogens: biosynthesis and functions. Prog. Lipid Res. 2001;40:199–229. doi: 10.1016/s0163-7827(01)00003-0. [DOI] [PubMed] [Google Scholar]
- Nardini M, Dijkstra BW. Alpha/beta hydrolase fold enzymes: the family keeps growing. Curr. Opin. Struct. Biol. 1999;9:732–737. doi: 10.1016/s0959-440x(99)00037-8. [DOI] [PubMed] [Google Scholar]
- Navia-Paldanius D, Savinainen JR, Laitinen JT. Biochemical and pharmacological characterization of human α/β-hydrolase domain containing 6 (ABHD6) and 12 (ABHD12). J. Lipid Res. 2012;53:2413–2424. doi: 10.1194/jlr.M030411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei-Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Batkai S, Harvey-White J, Mackie K, Offertaier L, Wang L, Kunos G. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Invest. 2005;115:1298–1305. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei-Hyiaman D, Liu J, Zhou L, Godlewski G, Harvey-White J, Jeong WI, Batkai S, Marsicano G, Lutz B, Buettner C, Kunos G. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J. Clin. Invest. 2008;118:3160–3169. doi: 10.1172/JCI34827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol. Endocrinol. Metab. 2009;297:E289–E296. doi: 10.1152/ajpendo.00099.2009. [DOI] [PubMed] [Google Scholar]
- Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J. Clin. Invest. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF. Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for alpha/beta-hydrolase 4 in this pathway. J. Biol. Chem. 2006;281:26465–26472. doi: 10.1074/jbc.M604660200. [DOI] [PubMed] [Google Scholar]
- Skoura A, Hla T. Lysophospholipid receptors in vertebrate development, physiology, and pathology. J. Lipid Res. 2009;50(Suppl):S293–S298. doi: 10.1194/jlr.R800047-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschler U, Radner FP, Heier C, Schreiber R, Schweiger M, Schoiswohl G, Preiss-Landl K, Jaeger D, Reiter B, Koefeler HC, et al. Monoacylglyceride lipase deficiency in mice impairs lipolysis and attenuates diet induced insulin resistance. J. Biol. Chem. 2011;286:17467–17477. doi: 10.1074/jbc.M110.215434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traurig MT, Orczewska JI, Ortiz DJ, Blan L, Marinelerena AM, Kobes S, Malhotra A, Hanson RL, Mason CC, Knowler WC, et al. Evidence for a role of LPGAT1 in influencing BMI and percent body fat in Native Americans. Obesity (Silver Spring) 2013;21:193–202. doi: 10.1002/oby.20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann P, Schneiter R. Lipid signaling in disease. Nature Rev. Mol. Cell. Biol. 2008;9:162–176. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.