

Abstract
Pash is a computer program for efficient, parallel, all-against-all comparison of very long DNA sequences. Pash implements Positional Hashing, a novel parallelizable method for sequence comparison based on k-mer representation of sequences. The Positional Hashing method breaks the comparison problem in a unique way that avoids the quadratic penalty encountered with other sensitive methods and confers inherent low-level parallelism. Furthermore, Positional Hashing allows one to readily and predictably trade between sensitivity and speed. In a simulated comparison task, anchoring computationally mutated reads onto a genome, the sensitivity of Pash was equal to or greater than that of BLAST and BLAT, with Pash outperforming these programs as the reads became shorter and less similar to the genome. Using modest computing resources, we employed Pash for two large-scale sequence comparison tasks: comparison of three mammalian genomes, and anchoring millions of chimpanzee whole-genome shotgun sequencing reads onto the human genome. The results of these comparisons by Pash agree with those computed by other methods that use more than an order of magnitude more computing resources. These results confirm the sensitivity of Positional Hashing.
One expected benefit of genome sequencing is the identification of functional DNA elements through comparative methods. For example, comparison of the sequenced genome of Saccharomyces cerevisiae to the genomes of three related yeast species resulted in substantial revision to the catalog of open reading frames (ORFs) and noncoding conserved sequence motifs that had been discovered by examining the sequence of S. cerevisiae alone (Kellis et al. 2003). Strikingly, comparison of the mouse and human genomes revealed that approximately 5% of these genomes are under purifying selection (Waterston et al. 2002). Rat/human or rat/mouse genome comparisons yield similar statistics (Rat Genome Sequencing Consortium 2004), yet only about a third of this conserved sequence is accounted for by known genes, indicating that a large set of functional elements remain uncharacterized.
Identification of functional elements by genome comparison depends heavily on the quality of sequence alignments. Standard Dynamic Programming algorithms (Needleman and Wunsch 1970; Smith and Waterman 1981) are highly sensitive DNA comparison methods but are too computationally expensive to directly apply on the scale of multiple mammalian genomes. Even recent faster implementations of dynamic programming such as LAGAN (Brudno et al. 2003) that perform well on a megabase scale are not applicable on a genome scale without prior information (“anchors”) that direct comparison to orthologous regions.
Faster comparison is achieved by “seed-and-extend” methods such as BLAST, BLAT, BLASTZ, SSAHA, PatternHunter, and FASTA (Pearson and Lipman 1988; Altschul et al. 1997; Ning et al. 2001; Kent 2002; Ma et al. 2002; Schwartz et al. 2003). In a seed-and-extend method, one or more exactly matching k-mers (“seeds” or “hot-spots”) provide initial evidence of possible similarity. The seeds are then extended into sequence alignments. The extension step is more accurate than the seeding step, but it is computationally expensive, so these methods quickly abandon most candidate similarities because they do not immediately yield alignments that are likely to be statistically significant.
Seed-and-extend methods work well when a relatively short query is used to search a large database, but they were not initially designed for genome-scale comparison. Sensitive genome-scale comparison is achieved by dividing the sequence databases into hundreds or thousands of multimegabase subsets and performing all possible pairwise comparisons between them (Altschul et al. 1997; Ning et al. 2001; Ma et al. 2002; Schwartz et al. 2003). This contributes to the quadratic running time of these comparison methods, because the number of such comparison jobs is proportional to the square of the number of subsets. This makes large-scale comparisons computationally expensive, practically restricting them to labs that have access to large computing clusters. A further limitation of current implementations of seed-and-extend methods is that they provide few options to trade sensitivity for speed.
To address the limitations of seed-and-extend methods, we developed the Positional Hashing method and implemented it in the Pash (Positional Hashing) program. Although other sensitive comparison methods ultimately perform nucleotide alignments, the Positional Hashing method represents sequences as collections of short k-mers rather than as individual bases, throughout the comparison process. Local clusters of matching k-mers are collated together to identify sequence similarity. Whereas other methods achieve parallelism by requiring users to divide the sequences into many subsequences and perform all pairwise comparisons between them (thus incurring a quadratic penalty), Positional Hashing achieves seamless parallelism in linear time by assigning computing nodes to compare subsets of diagonals.
We compared the speed and sensitivity of Pash to BLAST and BLAT in sequence-anchoring tasks. The tasks included simulated anchoring of sequence fragments of various lengths across various evolutionary distances onto a genome of a related species, and the anchoring of chimpanzee reads onto the human genome. Pash was also applied to detect orthologous regions between the genomes of human, mouse, and rat. Our tests confirmed that this novel parallelizable method of comparing sequences on the basis of their k-mer representation is as sensitive as methods that perform nucleotide alignments, although performing much faster and conferring superior ability to trade sensitivity for speed.
METHODS
Dividing the Comparison Problem Across Diagonals
Any sequence similarity occurs along a particular diagonal of the comparison matrix (Fig. 1A, diagonal lines). In contrast to other sensitive comparison methods, Positional Hashing divides the comparison problem into the subproblems of finding similarities within subsets of diagonals, each subset consisting of diagonals L basepairs apart (Fig. 1B). These subproblems are each independently solvable on a separate node of a computer cluster. To further localize detection of similarities, diagonals are divided into diagonal segments, also of length L (Fig. 1C, dashed lines).
Figure 1.

Division of the comparison problem along diagonals. (A) Sequence similarities are contained within diagonals of the comparison matrix. (B) A subset of these diagonals, separated by a fixed offset L, is simultaneously considered by each computing node and is referred to as “diagonal” (modulo L). (C) The diagonals are conceptually divided into segments of length L, delineated by broken horizontal lines. All k-mer matches in a specific sampled position (short, bold, line segments) along each diagonal segment are simultaneously detected in a single positional hash table. This is repeated for all sampled positions along diagonal segments. The k-mer matches that occur within the same diagonal segment are collated together and assigned a significance score.
The alignment diagonals that start at the same position modulo a fixed distance L (typically around 500 bp) are jointly referred to as a “diagonal” and are denoted by D(d), d = 0,..., L – 1. The two compared sequences, S and T, are conceptually divided into the following nonoverlapping subsequences of length L: Si = S[i * L + 1,..., (i + 1)*L] where i = 0,...,|S|/L – 1 and Ti′ = T[i′ * L + 1,..., (i′ + 1) * L], where i′ = 0,..., |T|/L – 1. Positional hash tables 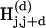 , where j = 0,..., L – k, which correspond to the diagonal D(d) contain the indices i and i′ of k-mers Si[j + 1,..., j + k] and Ti′[d + j + 1,..., d + j + k] for all i and i′. Identical k-mers are translated into the same hash key, and their corresponding indices are consequently collected in the same hash table bin. The k-mers hashed in an individual table are illustrated in Figure 1C (short bold line segments).
, where j = 0,..., L – k, which correspond to the diagonal D(d) contain the indices i and i′ of k-mers Si[j + 1,..., j + k] and Ti′[d + j + 1,..., d + j + k] for all i and i′. Identical k-mers are translated into the same hash key, and their corresponding indices are consequently collected in the same hash table bin. The k-mers hashed in an individual table are illustrated in Figure 1C (short bold line segments).
Positional hash tables are created in the first step of the Pash algorithm (Fig. 2, step 1). In the subsequent inversion step (Fig. 2, step 2), pairs of matching k-mers in S and T are detected. Specifically, a k-mer match along diagonal D(d) between subsequences Si and Ti′ in position j is detected whenever indices i and i′ co-occur in the same bin of hash table 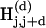 .
.
Figure 2.

Flowchart of sequence comparison using Pash. Short, exactly matching k-mers are detected during Hashing and Inversion. Lists of proximal matching k-mers are collated to detect larger matches, which are assigned a score. An optional reciprocal match filter is then employed to increase specificity.
In the collation step (Fig. 2, step 3), the lists of matching k-mers are grouped by subsequence pair (Si, Ti′). Subsequence pairs with one or more k-mer matches are assigned a significance score (see Significance of Similarities).
The hash tables 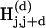 , where j = 0... L – k, corresponding to diagonal D(d), are processed on the same node of a computer cluster. Each of the L diagonals D(d), d = 0,..., L – 1 may be processed independently and in parallel across up to L computing nodes. If only one computer is available, the diagonals may be processed sequentially.
, where j = 0... L – k, corresponding to diagonal D(d), are processed on the same node of a computer cluster. Each of the L diagonals D(d), d = 0,..., L – 1 may be processed independently and in parallel across up to L computing nodes. If only one computer is available, the diagonals may be processed sequentially.
Analysis of Running Time
In the worst case, an all-against-all comparison of two sequences of length M and N requires time proportional to M * N because the size of the output can be proportional to M * N. The worst case may occur, for example, when sequences are made up entirely of the same kind of repetitive element. In many practical applications, such as detection of orthologous anchors between two mammalian genomes, the size of the output is roughly linearly proportional to M + N. In the following text, we show that under reasonable assumptions, Pash runs in O(M + N) wall time on a cluster consisting of L nodes, for inputs that result in outputs of size O(M + N).
Each of approximately L2 positional hash tables is populated (Fig. 2, step 1) with (M + N)/L sampled k-mers (e.g., Fig. 1C, short, bold, line segments). To avoid the need for subsequent sorting, k-mers are inserted into the hash table in the order of their position in the input sequences. The hash bin occupied by each k-mer is recorded in a list to preserve this ordering information. In the subsequent inversion step (Fig. 2, step 2), matching k-mers are detected by visiting the hash table bins in the same order and recording the positions of matching k-mers. The list of matching k-mers is thus produced in order, sorted by position in the input sequences.
If k-mers of sufficient length are used, the number of k-mer matches detected per single hash table is O((M + N)/L). For example, the results in this paper were obtained with k = 13 and genome sizes of approximately 3 Gbp each. Each 13-mer is expected to occur by chance approximately 45 times in genomes of this size, resulting in about 2000 randomly occurring k-mer matches. For genomes that are four times larger (12 Gbp), for the time spent on detecting random matches to increase only linearly, one would need to use 14-mers. In general, to keep the number of random matches constant, the parameter k would need to be logarithmically proportional to genome size. Beause the lists of matching k-mers are sorted, collation to find groups of matching k-mers (Fig. 2, step 3) involves single traversal of each list.
Thus, under the key assumptions of linear-sized output and sufficient k-mer size, the time required to hash, invert, and collate is O((M + N)/L) per hash table. A total of approximately L2 hash tables are employed in a complete comparison, resulting in the total running time of O((M + N) * L). In practice, even for genomes of the largest complexity, L can be set to few hundred basepairs. If computation is spread across as many nodes in the computer cluster, one diagonal per node, the wall time reduces to O(M + N).
Sampling Patterns
For simplicity, our discussion above assumed that the positional hash tables are populated with contiguous k-mers. In practice, to increase sensitivity, k-mers are formed according to a discontiguous sampling pattern that minimizes the number of overlapping bases between k-mers in consecutive positions (Califano and Rigoutsos 1993; Buhler and Tompa 2001; Ma et al. 2002; Buhler et al. 2003). This approach minimizes mutual dependence between overlapping k-mers. For a given frequency of randomly distributed mismatches, this approach does not change the total expected number of k-mer matches, but it improves sensitivity by achieving a more even spatial distribution.
The length of a k-mer and the sampling pattern used are adjustable to fit the total size of the sequences under comparison and the evolutionary distance between them. For comparison of mammalian genomes, we used a sampling pattern of length 19 bases containing 13 sampled bases, as this represents a good tradeoff between RAM usage (longer k-mers use a larger hash table, requiring greater RAM), sensitivity (shorter k-mers increase mismatch tolerance), and coincidental matches (shorter k-mers are more likely to match by chance, increasing the time spent on isolated, insignificant matches).
Significance of Similarities
For each sequence match found, Pash reports both the number of matching bases detected and a bit score that indicates significance, calculated using the Algorithmic Significance method (Milosavljevic and Jurka 1993; Milosavljevic 1995). In brief, we measure sequence similarity as the number of bits saved in a minimal encoding of the target sequence X = Ti′ given that the source (“query”) sequence Si is known. The Algorithmic Significance method provides significance values for any sampling pattern (see Sampling Patterns) and sampling density based on the number of bits saved.
Let d be the number of bits saved or the “bit score” of the match. Then,
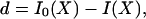 |
where I0(X) denotes the minimal number of bits required to encode the target sequence by itself, and I(X) denotes the number of bits required to encode the target using similarity to the source.
Let P0be the null hypothesis assigning probability p0(X) to target sequence X. Then I0(X) = –log2p0(X). We make the simplifying assumption that sequences are completely random; thus, I0(X) = 2 × n bits, where n is the length of the target sequence (i.e., each base is encoded using 2 bits).
We adapt standard encoding techniques to efficiently use similarity between the source and target to compute I(X). We choose one of two different encoding methods on a per case basis, indicating which method was chosen with a 1-bit flag. The two encoding methods, which we refer to as the k-mer method and the Base method, record matching k-mer and base positions, respectively.
For the k-mer encoding, let w be the number of matching k-mers, and let W be the maximum possible number of k-mers that can be contained in a match. W is dictated by the diagonal segment length and is constant throughout a given comparison. Following the aforementioned 1-bit flag, the k-mer encoding records the number of matching k-mers, which requires log2W bits. For a given value of w, there are
 |
possible lists of matching k-mers. The particular set of matching k-mers is encoded as the set's position in a lexicographically sorted list of all possible sets of w-matching k-mers, which requires
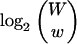 |
bits. The k-mer encoding IW(X) thus requires
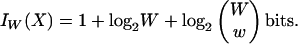 |
The Base encoding is analogous to the k-mer encoding, but it records the base positions rather than the k-mer positions. We similarly define b to be the number of bases contained in a match and B to be the maximum possible number of bases that can be contained in a match. Analogous to the k-mer encoding, the Base encoding requires
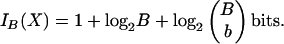 |
The encoding chosen is then IMIN(X) = min(IB(X), IW(X)). In general, matches that contain few k-mers are encoded most efficiently by the k-mer method. When there are many matching k-mers, the k-mers tend to overlap with one another extensively, and each additional k-mer contributes few additional bases; thus, it becomes more efficient to use the Base method.
Whether the k-mer or Base encoding is chosen for a given match, the bases not involved in the match must also be encoded. These remaining bases are encoded using the same model that is used for the null hypothesis (see earlier). The number of bits in the complete encoding is I(X) = IMIN(X) + 2 × (n – b). Combining and simplifying the above expressions, the bit score of a match found by Pash is
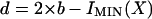 |
For a bit score d, the probability p of finding such a match by chance alone is
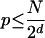 |
where N = |T| |S|/L2 is the total number of comparisons done.
Adjustable Speed/Sensitivity Tradeoff
One of the strengths of the Positional Hashing method is the ability to readily and predictably trade sensitivity for speed. If the sequences are known to be highly similar and few mismatches are expected, the density of k-mers can be decreased so that each base is sampled in fewer k-mers. For example, by halving the number of k-mers, we can halve the time required for a comparison. Conversely, this approach permits dense sampling of each base by a multitude of overlapping sampling patterns, further increasing the ability to detect similarities at large evolutionary distances.
In our current implementation, the parameter that controls the trade-off between speed and sensitivity is the “k-mer offset gap,” which is the number of bases between the start of consecutive sampled positions along a diagonal segment (Fig. 1C). The anchoring simulation, genome comparison, and chimp read-anchoring discussed below were conducted with high sensitivity (k-mer offset gap = 2), medium sensitivity (k-mer offset gap = 6), and low sensitivity (k-mer offset gap = 18), respectively.
Tolerance of Repetitive Sequences
Pash does not require the usual preprocessing step in which sequences are “masked” to remove repeat sequences from consideration. Rather, Pash uses k-mer frequency information (measured by hash table bin size) to ignore k-mers that are overrepresented (above a user-specified threshold) because of their presence in a repeated sequence. The k-mers that are shared among repeat and nonrepeat sequences, however, are ignored in both contexts. As a consequence, the use of unmasked, repeat-containing sequences has some effect on sensitivity.
Reciprocal Best Matches
To separate homologous sequences from true orthologous sequences, and to remove some residual repeat sequences that were not excluded by ignoring overused k-mers, the similarities found may be postprocessed by applying Pash's “reciprocal best match” filter. This filter ensures that for each pairwise similarity reported, each of the two sequences must appear on the other's list of top matches. This filter implicitly uses the full set of sequences as positive controls to increase the specificity of anchoring and to reduce the number of false positive matches.
One specific application of Pash is the detection of orthologous anchors and blocks of conserved synteny between two genomes. In this case, anchors may be declared only when each sequence is the other's top match. This may not be appropriate for other applications; for example, if one is interested in studying lineage-specific duplications. For such applications, the reciprocal match criterion is relaxed to allow up to a fixed number of matches for every location in each sequence. Similarly, when anchoring sequencing reads onto genomic sequence, the reciprocal match filter can be adjusted to keep the best match for each read, but map multiple reads to the same genomic location.
Merging Collinear Anchors and Visualization
In the case of comparison of genome assemblies, reciprocal best matches can further be merged into blocks of conserved synteny. Blocks are inferred from the existence of multiple proximal orthologous anchors. A postprocessing script merges anchors that are within a specified radius in both genomes. Depending on the application, the user can specify whether to tolerate local rearrangements or to require that the anchors occur in the same order and orientation in both genomes. This merging program is part of the Virtual Genome Painting program package (http://www.genboree.org), employed here for display of genome comparison results (Fig. 3).
Figure 3.

Rat/Mouse/Human comparison. Pash was used for pairwise comparison of the genome of rat with those of mouse and human. Chromosomes are shown with p-arm down. Each chromosome of the rat genome is represented as a pane (identified along the x-axis) containing two columns that have been “painted” using the Virtual Genome Painting method (http://www.genboree.org) according to similarity to chromosomes of mouse and human. Position within the rat chromosomes is indicated along the y-axis in megabases (Mb). Columns are colored according to similarity to human chromosomes (left columns, “H”), and mouse chromosomes (right columns, “M”). The same color code is used for similarity to both human and mouse, as indicated by the legend. For example, the first 25 Mb of rat chromosome 1 is similar to sequences from human chromosome 6 (indicated by pink coloring in the left column) and mouse chromosome 10 (indicated by yellow coloring in the right column). An interactive version of this figure is available at the Genboree site (http://www.genboree.org).
BLAT and BLAST
For comparison experiments, we used BLAT Client/Server version 23 (http://www.soe.ucsc.edu/~kent/src/blatSrc23.zip) and National Center for Biotechnology Information BLAST (Altschul et al. 1997). BLAT comparison parameters were adjusted to allow low-similarity matches that would be filtered by default, using the options minScore = 20 and minIdentity = 0 (A. Polakov, pers. comm.). To reduce the number of heat-sensitive proteins grouped together by BLAT, we set maxIntron to the length of the sequences being mapped. For BLAST, we used an expectation value of 1e-15 for the anchoring simulation and of 1e-50 for the chimp read mapping. We used the default values for all other BLAST and BLAT parameters.
The nodes in our computing cluster did not have sufficient RAM to run BLAT with the entire human genome, so we divided the human genome into two roughly equal halves and compared each read against both halves on separate nodes. BLAST was run using a different computing node for each chromosome.
Program Availability
Code and licenses for Pash, Positional Hashing, the Reciprocal Match postprocessing filter, and the anchor-merging script are available free of charge for academic use. Current access and licensing information is posted at http://www.brl.bcm.tmc.edu.
RESULTS
Genome Comparison
We compared the latest assembly of the rat genome (HGSC v3.1, http://www.hgsc.bcm.tmc.edu/projects/rat/) to the genomes of mouse and human (UCSC hg15 and UCSC mm3, respectively, http://genome.ucsc.edu/downloads.html), using Pash in medium-sensitivity mode (see Adjustable Speed/Sensitivity Tradeoff). Each pairwise comparison was completed in 4 days using six CPUs, for a total of 24 CPU days per comparison. The computers used had 750 MHz Pentium III processors running Linux. Peak RAM usage was less than 500 MB. We merged the Pash anchors that were within a radius of up to 400 kb (the radius was adjusted to fit the evolutionary distance between species) to facilitate display of the comparison results using the Virtual Genome Painting program (http://www.genboree.org).
At 1 Mbp resolution, the resulting picture of global similarities (Fig. 3) is virtually identical to that found by the alignment program BLASTZ, which requires 481 CPU-days to align two mammalian genomes using similar machines (Schwartz et al. 2003). For the human–rat comparison, we examined the similarity between the Pash anchors and the blocks of conserved synteny defined by the BLASTZ alignments (“syntenyRat” data tables, http://genome.ucsc.edu) in more detail. Consistent with the high specificity of Pash anchoring, 83% of the Pash anchors are contained within the blocks of conserved synteny found by BLASTZ. We are currently investigating whether any of the remaining 17% of Pash anchors indicate similarities that were not found by BLASTZ. Similarly, we determined that over 99% of bases within conserved syntenic regions defined by the BLASTZ alignments contain multiple Pash anchors, indicating that Pash correctly identified the vast majority of conserved synteny between human and rat genomes.
Anchoring Simulation
We evaluated the sensitivity and specificity of Pash relative to BLAT and BLAST using a simulated anchoring task in which “reads” containing computationally introduced mutations were anchored onto a mammalian genome. For this test, we created a library of sequences by mutating randomly chosen sequence fragments from the finished human genome (UCSC hg15, http://genome.ucsc.edu/downloads.html). The library of mutated fragments contained all combinations of four different degrees of sequence identity (95%, 85%, 75%, and 65%) and four different lengths (1000, 500, 200, and 100 bp). Each of the 16 sets contained 10,000 fragments, for a total of 160,000 fragments. Each base had the indicated percent chance to be the site of a simulated mutational event (e.g., “65% identity” implies that each base had a 35% chance to either change to a new base or be the site of a single base insertion or deletion event). Each base underwent at most one mutational event; thus, reversion to the original sequence was not possible. Most mutational events were a change to a randomly chosen different base, but 1% of events were indels (half of such events were deletions and half were insertion of a random base). The same experiments were also conducted using a 3% indel rate with very similar results (data not shown). Successful mapping was counted if any of the 10 highest scoring matches returned by a program overlapped with the original location.
We determined the frequency with which the fragments could be mapped to their original location by Pash, BLAT, and BLAST. As expected, all three programs were highly successful at 95% sequence identity (Fig. 4A). At lower sequence identity and with shorter fragment lengths, Pash becomes increasingly more sensitive than BLAT and BLAST (Fig. 4B–D). We believe that the lower-than-expected sensitivity of BLAST is the result of similarity regions sometimes being broken into multiple individual matches with relatively low scores that are not included in the set of the top 10 hits, thus precluding correct anchoring. These results indicate that when the sequences are highly similar (>95%) the choice of anchoring method should be dictated by whatever is fastest and most convenient. When the sequences are less similar (<85%), Pash performs more accurate anchoring than BLAST or BLAT.
Figure 4.

Simulated anchoring. Randomly selected sequence fragments of varying lengths (1000, 500, 200, or 100 bp) from the human genome were randomly mutated to be 95% (A), 85% (B), 75% (C), or 65% (D) identical to their original sequence. The fraction of fragments that were correctly mapped to their original location by Pash, BLAT, and BLAST indicate the sensitivity of these methods at varying levels of sequence identity.
Anchoring Chimpanzee Reads
The genome of the chimpanzee Pan troglodytes is estimated to be over 95% identical to the human genome (Chen and Li 2001; Britten 2002; Ebersberger et al. 2002) and is an attractive target for comparative assembly (Milosavljevic 1999). Anchoring the full set of chimpanzee sequencing reads onto the human genome is the first step in this process. We used a set of approximately 2.7 million whole-genome shotgun reads from the chimpanzee (http://www.ncbi.nih.gov) to determine whether Pash is sufficiently fast and sensitive for this task. These reads were selected from the set of reads of lengths between 860 and 999 bp in the National Center for Biotechnology Information trace archive but were not otherwise masked or filtered for quality.
Given the expected high similarity between the human and chimpanzee genomes, we employed Pash in a high-speed/low-sensitivity mode (see Adjustable Speed/Sensitivity Tradeoff). Because of a large CPU time requirement, we chose a random subset of 225,000 and 15,000 of these reads to estimate the speed of BLAT and BLAST, respectively. The rate of read mapping by Pash was approximately six times higher than that of BLAT and over 400 times higher than the read mapping rate of BLAST (Table 1).
Table 1.
Read Anchoring Speed and Accuracy of Pash, BLAT, and BLAST
| Program | Reads mapped/minute | Fraction in agreement with BLAST |
|---|---|---|
| Pash | 235.5 | 99.2% |
| BLAT | 39.6 | 99.8% |
| BLAST | 0.6 | N/A |
Whole-genome shotgun sequencing reads from the chimpanzee genome-sequencing project were independently mapped onto the human genome by Pash, BLAT, and BLAST. BLAST mappings to a single unambiguous location were used as a reference for comparison. Because chimpanzees are closely related to humans, Pash was employed in a high-speed/low-sensitivity mode (see Adjustable Speed/Sensitivity Tradeoff for details).
N/A means not applicable.
To confirm agreement between these methods on this anchoring task, we considered the subset of reads that were mapped by BLAST with high confidence (expectation value more significant than 1e-50, and at least 65% base identity between the read and the mapped region) and low ambiguity (the second-best match having read identity not more than 85% of that of the best match). Both Pash and BLAT agree with BLAST for over 99% of these reads, defined by one of the 10 highest-scoring mappings overlapping with the BLAST mapping (Table 1).
DISCUSSION
In contrast to seed-and-extend methods, which examine individual basepairs in the extension step, the Positional Hashing method collates together locally matching k-mers to identify sequence similarity. Throughout the comparison process, sequences are represented as short k-mers rather than individual bases. The k-mer representation confers exceptional speed and simplicity to the comparison algorithm. Moreover, both speed and specificity are controllable through selection of k-mer length and k-mer sampling density. In contrast to most other sensitive comparison methods (Pearson and Lipman 1988; Altschul et al. 1997; Ning et al. 2001; Ma et al. 2002; Schwartz et al. 2003), Positional Hashing achieves low-level parallelism without incurring a quadratic time penalty. Computation can be efficiently spread across hundreds of nodes in a computer cluster with an approximately linear reduction in wall time.
Our current, incompletely optimized implementation of the Pash program requires modest hardware resources to perform sequence anchoring even on the scale of mammalian genomes. Compared to seed-and-extend methods, Pash can achieve substantial time savings when creating orthologous maps between genomes, anchoring expressed sequence tags (ESTs) onto genomic sequence, anchoring reads of one organism onto an assembled genome of another for the purpose of comparative sequence assembly, or when constructing read overlap graphs for sequence assembly.
For applications that require basepair-level alignments, Pash may be used as an anchoring module whose results are postprocessed by a program like LAGAN, AVID, or BLASTZ (Bray et al. 2003; Brudno et al. 2003; Schwartz et al. 2003). Because the use of Pash anchors breaks the all-against-all comparison of entire genomes into much smaller pairwise comparisons of specific regions that are all highly likely to yield significant alignments, the use of Pash anchors saves significant computing time.
Pash was designed to address the problem of comparing very long sequences or large collections of short sequence fragments. Its linear running time is advantageous when the lengths of compared sequences M and N are similar (e.g., when comparing two mammalian genomes), but when one of them, say M, is much larger than the other, N, the time required for comparison is dictated by the size of M. This makes Pash ill-suited to the task of comparing a single short query with a long target.
Planned improvements to the Positional Hashing method include simultaneously considering adjacent diagonals to improve sensitivity in the presence of insertions and deletions, and collation across diagonal segment boundaries to improve sensitivity. The Positional Hashing method is applicable beyond simple sequence comparison. Other possible applications include pattern discovery through self-comparison, discovery of repeated patterns, sequence assembly, and evolutionary reconstructions.
Acknowledgments
We are grateful to the Baylor Human Genome Sequencing Center for use of computing resources; the Rat Genome Sequencing Consortium for providing valuable feedback; Manuel L. Gonzalez-Garay, Andrei Volkov, Yu A. Feng, and Jesse Reklaw for work on image generation; R. Alan Harris for testing and data generation; Qikai Xu and Ruben Valas for work on an early Pash prototype; Miklós Csürös for comments about this manuscript; and Elke Norwig-Eastaugh for administrative support. K.K. was partially supported by a training fellowship from the W.M. Keck Foundation to the Gulf Coast Consortia through the Keck Center for Computational and Structural Biology. This research was funded by the Baylor College of Medicine and Howard Hughes Medical Institute faculty startup grants to A.M.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1963804.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, N., Dubchak, I., and Pachter, L. 2003. AVID: A global alignment program. Genome Res. 13: 97-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten, R.J. 2002. Divergence between samples of chimpanzee and human DNA sequences is 5%, counting indels. Proc. Natl. Acad. Sci. 99: 13633-13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno, M., Do, C.B., Cooper, G.M., Kim, M.F., Davydov, E., NISC Comparative Sequencing Program, Green, E.D., Sidow, A., and Batzoglou, S. 2003. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13: 721-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler, J., Keich, U., and Sun, Y. 2003. Designing seeds for similarity search in genomic DNA. Proceedings of the RECOMB Conference, Berlin, Germany. The Association for Computing Machinery (ACM).
- Buhler, J. and Tompa, M. 2001. Finding motifs using random projections. Proceedings of the RECOMB Conference, Montreal, Canada. The Association for Computing Machinery (ACM).
- Califano, A. and Rigoutsos, I. 1993. FLASH: a fast look-up algorithm for string homology. IEEE Computer Society Conference on Computer Vision and Pattern Recognition, New York. [PubMed]
- Chen, F.C. and Li, W.H. 2001. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 68: 444-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersberger, I., Metzler, D., Schwarz, C., and Paabo, S. 2002. Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet. 70: 1490-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis, M., Patterson, N., Endrizzi, M., Birren, B., and Lander, E.S. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423: 241-254. [DOI] [PubMed] [Google Scholar]
- Kent, W.J. 2002. BLAT—the BLAST-like alignment tool. Genome Res. 12: 656-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, B., Tromp, J., and Li, M. 2002. PatternHunter: Faster and more sensitive homology search. Bioinformatics 18: 440-445. [DOI] [PubMed] [Google Scholar]
- Milosavljevic, A. 1995. Discovering dependencies via algorithmic mutual information: A case study in DNA sequence comparisons. Machine Learning 21: 35-50. [Google Scholar]
- Milosavljevic, A. 1999. DNA sequence similarity recognition by hybridization to short oligomers. U.S. Patent 6001562.
- Milosavljevic, A. and Jurka J. 1993. Discovering simple DNA sequences by the algorithmic significance method. Comput. Appl. Biosci. 9: 407-411. [DOI] [PubMed] [Google Scholar]
- Needleman, S.B. and Wunsch, C.D. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48: 443-453. [DOI] [PubMed] [Google Scholar]
- Ning, Z., Cox, A.J., and Mullikin, J.C. 2001. SSAHA: A fast search method for large DNA databases. Genome Res. 11: 1725-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, W.R. and Lipman, D.J. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. 85: 2444-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rat Genome Sequencing Project Consortium. 2004. Genome sequence of the Brown Norway Rat yields insights into mammalian evolution. Nature (in press). [DOI] [PubMed]
- Schwartz, S., Kent, W.J. Smit, A., Zhang, Z., Baertsch, R., Hardison, R.C., Haussler, D., and Miller, W. 2003. Human–mouse alignments with BLASTZ. Genome Res. 13: 103-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147: 195-197. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.brl.bcm.tmc.edu/; Pash program download.
- http://www.genboree.org; Interactive version of Figure 3 and the Virtual Genome Painting program.
- http://www.soe.ucsc.edu/~kent/src/blatSrc23.zip; BLAT version 23 download.
- http://www.hgsc.bcm.tmc.edu/projects/rat/; Rat Genome Project.
- http://genome.ucsc.edu/downloads.html; University of California, Santa Cruz, genome assembly download.
- http://genome.ucsc.edu/; University of California, Santa Cruz, genome browser.
- http://www.ncbi.nih.gov/; National Center for Biotechnology Information home page.