

Abstract
Cells of the adaptive immune system (T and B cells) causing autoimmunity require activation signals that are normally provided by the innate immune system. Innate signaling receptors are obvious candidates for participation in the induction of autoimmunity, and the nature of these receptors suggests that microbes could be the triggers. Recent publications describing the development of autoimmunity in sterile conditions and in animals deficient in innate signaling question the requirement of these receptors for initiation of autoimmunity. In addition, the role of the non-pathogenic (commnesal) microbiota as a regulator of autoimmunity has come into the spotlight. In this review we discuss recent reports that deal with the link between innate signaling receptors and ‘adaptive’ autoimmunity.
Introduction
The studies of autoimmune diseases concordance in identical twins, emigrants from areas with high or low incidence of a particular autoimmune disease to areas with respectively low or high incidence, and genetically related populations living in dissimilar conditions, all find that environment influences the diseases (reviewed in [1-3]. For example, the results of the identical twin studies in type 1 diabetes (T1D) find that the concordance rate is about 40-50%, which is higher than in non-identical twins (around 7%), but is much less than 100%. Taken at the face value the figure suggests both strong genetic and environmental influences. It also suggests that either 50% of identical twins were unlucky to be infected with T1D-provoking pathogens, or that 50% of twins were lucky to be exposed to environmental cues that stopped the autoimmunity. That the second interpretation is plausible was confirmed by the studies of T1D in genetically susceptible mice (that are also genetically identical). It was found that the incidence of diabetes was dependent on the quality of animal care (good care=less pathogens=high incidence) [4].
Microbial involvement in autoimmunity suggested that receptors that have been evolutionary selected to detect conserved molecular patterns associated with pathogens (PAMPS, according to Janeway [5]) could be involved. These innate pattern-recognition receptors (PRRs) can see microbial products that are absent from eukaryotic host (glycolipids, lipopeptides, special proteins, modified nucleic acids), or not available to them in the host under normal circumstances because of the rapid clearance or sequestration from the receptors. The receptors of the adaptive system (T cell receptors and antibodies) lack the ability to tell apart self from non-self but are sensitive to self-antigens during development. Immunity to pathogens is based on sequential reactivity of the innate and adaptive immune systems (Fig. 1A a). The goal of this review is to discuss recent data linking PRRs with ‘adaptive’ autoimmunity.

A. a. Janeway’s model of communication between innate and adaptive immune responses. Activation of the innate system produces three different signals that stimulate T cells: active antigen processing and presentation (I), costimulation (II) and a milieu of cytokines (III) that provides for the ‘adequacy’ of the immune response based on recognition of a pathogen such as an extracellular or an intracellular bacterium, virus, or a fungus, or a worm. In the absence of signal II, T cells engaging cognate antigens are supposed to die. That is likely to be one of the key mechanisms that suppress autoimmunity.
b. Costimulatory signals elicited by pathogens through PRRs can also activate self-reactive T cells that have escaped negative selection. That process requires that the antigen presenting cell (APC) also express the self-peptide/MHC complex. That may be achieved by cross-presentation of peptides from infected cells engulfed by the APC. In this scenario antigenic similarity between a pathogen and self is not required. In fact, such similarity, known as ‘molecular mimicry’ has been documented only in limited number of systems. The bystander activation of autoimmune T cells still requires genetic predisposition to autoimmunity and/or a failure of other protective mechanisms. It is normally controlled by mechanisms that limit self-antigen processing and presentation in response to detection of the PRR agonists by the APCs[16]
c and d. Since autoimmunity in diseases such as T1D develops in the absence of microbes and TLR signaling the genetic predisposition to autoimmunity could be explained by two scenarios with faulty negative selection being a prerequisite to both of them. Either expression of costimulatory signals is genetically elevated in susceptible individuals (c), or the T cells have lower threshold for activation that can utilize background costimulatory signals (d). Obviously, these two scenarios are not mutually exclusive. Although TLR signals are not required, they can still be crucial for acceleration of the disease induction by pathogens (as in b).
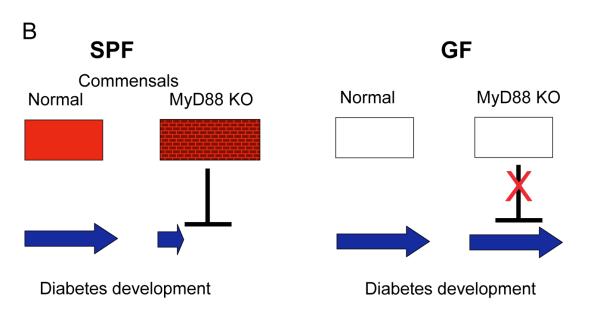
B. Influence of PRRs and commensal microbiota of autoimmunity. In T1D the lack of MyD88 signaling removes control over commensal microbiota changing its composition. Microbes that are now thriving induce tolerance of T cells in regional lymph nodes and protect animals from T1D. In GF mice lacking microbiota the protective mechanisms cannot work and they proceed to develop T1D independently of TLRs. No endogenous ligands for TLRs are involved in disease induction. Protective microbes are likely tolerizing the animal’s immune system towards themselves and affecting autoimmune T cells by a bystander mechanism.
Germ-free animals: a test tube for sterile autoimmunity
The question of whether microbes are required to induce an autoimmune disease can be addressed (not so easily) by derivation of autoimmunity-prone animals into germ-free (GF, or axenic, sterile) animals. Such animals can be analyzed for the signs of the disease and also can be colonized with a defined set of microbes (making them ‘gnotobiotic’ animals). GF animals have been used to test the development of various autoimmune and autoinflammatory conditions. It is clear that T1D develops in GF rodents[6-8]. GF Non-obese diabetic (NOD) mice demonstrated a high incidence of the disease in both females and males (whereas it is skewed towards females in specific-pathogen free, SPF, conditions). However, GF mice did not develop T1D as a cohort, but showed quite a stretch of the disease onset over time. Two important conclusions can be drawn from this experiment:
that microbes were not required for the induction of T1D, and
that a prominent stochastic component is involved in T1D development in the absence of microbiota. This stochastic component is likely a reflection of the random nature of the rearrangement of the T cell receptor (TCR) genomic segments leading to production of autoimmune T cells with a certain probability and explaining in part the low disease concordance in twin studies. These data and the well-established genetic linkage of organ-specific autoimmune diseases with the Major Histocompatibility Complex (MHC) strongly suggest that faulty negative selection is the primary and the most important component in the pathogenesis of autoimmune diseases. In support of this view, development of autoimmunity in GF mice deficient in transcription factor AIRE [9], that controls negative selection at the early stages of development[10], was an excellent demonstration of the lack of necessity for microbial involvement in the onset of disease.
The processes of negative selection are hardly complete even in the carriers of MHC alleles without known association with autoimmunity or in animals sufficient for AIRE. That is best demonstrated by rampant autoimmunity in adult animals in which regulatory T cells (Tregs) were ablated by a toxin [11]. Therefore, in addition to active tolerance induced by Tregs multiple additional barriers exist to curb autoimmunity: immune privilege, induction of inhibitory receptors [12-14], limitation of self-antigen processing and presentation by the PRR agonists [15,16], and contraction of expanded T cell clones by either self-limiting mechanisms [17-19] or by elimination of activated APCs [20,21]. Importantly, the latter mechanism requires stimulation of PRRs to induce the sensitivity of APCs to Fas-mediated elimination [21].
Other autoimmune models were tested in GF conditions and some were microbe-independent (autoimmune gastritis in mice lacking activation-induced cytidine deaminase, AID[22]), whereas others (ankylosing enthesopathy [23] or arthritis caused by deletion of the IL-1 receptor antagonist [24]) were ablated in GF animals. A hypomorphic mutation in the SHP1 encoding gene led to the ‘motheaten’-like phenotype in the SPF but not in the GF environment [25]. Notably, the predominant pathology in motheaten mice is caused by activation of innate inflammatory effector cells (macrophages and neutrophils) and is also observed in Rag-deficient animals [26]. In this case, the autoinflammatory component turned out to be dependent on the microbiota.
Important conclusions from the studies of GF animals are that a) ‘adaptive’ autoimmunity can develop in the absence of microbes, and b) if it does not, than endogenous ligands for PRRs are not important without microbial stimuli. Which brings us to the key question of whether development of ‘adaptive’ autoimmunity requires innate signaling through PRRs.
PRRs and autoimmunity
A major set of signaling PRRs is the Toll-like receptors (TLRs), which have been shown beyond any reasonable doubt to work in a way predicted by Charlie Janeway: they activate innate cells to provide signals ‘one’ and ‘two’ to the cells of the adaptive immune system[27-29].
Three questions can be formulated that are important for the understanding of the role of the TLRs (and other PRRs) in autoimmunity:
Are TLRs required to induce an autoimmune disease?
If they are, do they use exogenous or endogenous ligands?
Can TLRs modulate the course of the disease, and by what mechanisms?
The answers are likely to be disease-specific, but it is clear that at least two diseases (T1D in NOD mice and poly-specific autoimmunity caused by the disruption of aire gene) were independent of TLR signaling[8,9].
In the systems where TLRs appear to be dispensable for induction of autoimmunity, the question of endogenous vs. exogenous ligands is not valid. However, the results of many studies claiming the opposite cannot be simply discarded based on this conclusion, they just need to be re-interpreted. For example, a study showing TLR2 involvement in T1D development [30] could in fact suggest that TLR2 is critical for the homeostasis of the commensal microbiota (see below). For experimental models in which autoimmunity does not develop in GF conditions, the answer to question ‘b’ is that the TLR ligands must be of exogenous origin. That may be only partly true if TLRs are used in a positive amplification loop rather than for initiation of the disease.
The answer to question ‘c’ is a definite ‘yes’, as it has been shown in many systems. The influence of TLRs on T1D that can be both protective and inductive has been recently reviewed [31-33] including induction of T1D in BBDR rats by viral infections. TLR-dependence of the disease in BB-DR rats was shown by multiple indirect approaches since the lack of relevant knock-outs in rats makes it hard to establish directly. In mice with transgenic expression of an LCMV-derived antigen and a TCR that recognizes that antigen, the triggering of autoimmunity required either infection with the virus (LCMV) or TLR engagement [34]. Interestingly, the anti-LCMV response is dependent on TLR but not other PRR’s signaling [35]. Among many other viruses implicated in T1D development, Coxsakie viruses are considered main culprits both in humans (where evidence is obviously circumstantial) and in mice (reviewed in detail in [36]). The role of TLRs in the process appeared to be hard to address because TLR3-deficient mice (but not MyD88-deficient mice) were highly sensitive to infection with Coxsakie B4 virus [37]. This is one of the few known instances where susceptibility to a pathogen is solely dependent on a single PRR, but it also makes the point that the importance of TLR signaling for protection against infections outweighs their importance for development of autoimmunity.
Are other PRRs involved in autoimmunity? The known signaling PRRs are divided into membrane/endosomal receptors (TLRs and dectins), and cytosolic receptors: nucleotide-binding oligomerization domain containg (NOD-like) receptors (NLRs), retinoic acid-inducible gene I (RIG-I) like receptors (RLRs), and intracellular DNA sensor(s) inducing type I interferons, IDS).
Dectins (C-type lectins signaling through a Syk kinase-dependent pathway) are involved in the anti-fungal responses and promote Th17 differentiation [38,39]. In a recent study[40], the yeast cell wall derivative zymosan was found to work as an adjuvant promoting induction of Experimental Autoimmune Encephalomyelitis (EAE) under conditions where TLR2 signaling was ablated. Interestingly, TLR2 induced suppression of the disease when mice were immunized with the antigen (MOG) with zymosan but not when immunized with MOG+CFA. Thus, exposure to different TLR agonists can decide the course of an autoimmune disease.
Mutations in NLRs, which are part of caspase-1 activating inflammasomes, seem to lead primarily to autoinflammatory disorders[41], suggesting that in general signaling through these receptors does not lead to induction of costimulation and triggering of adaptive immune responses. In addition, inflammasomes require transcription of pro-IL1, which is triggered by TLR agonists, making it hard to dissect the role of these receptors in autoimmunity [29,42]. NOD-like receptors have been associated by genetic screens with IBD and have been found to to activate adaptive immunity [43] and stimulate Th17 development in humans [44].
The role for RLRs in autoimmunity is not yet determined, although they are capable of sensing both foreign and self-RNA species. Quite interestingly, recent screens for genes involved in T1D [45,46] pointed at IFIH1 (MDA-5, an RLR) as a player in T1D development. Although there is no direct evidence yet on how it could work, observed polymorphism of the gene suggested that protective alleles were loss-of-function mutants. Thus IFIH1 can be involved in T1D as a trigger of costimulation for the activation of autoimmunity in response to a viral infection.
Sensing of DNA by multiple receptors leads to production of type I interferons which can lead for activation of costimulatory signals and autoimmunity (recently reviewed in [47]. Because DNA sensors can serve as receptors for self-DNA, several mechanisms prevent such recognition. However, mutations in secreted and intracellular exo- and endo-nucleases lead to production of type I interferon and systemic or organ-specific autoimmunity. The reaction can be amplified by intracellular delivery of DNA bound by antimicrobial peptide LL37 (psoriasis) anti-DNA antibodies (lupus models) or by a DNA-binding molecule HMGB1[42]. In systemic lupus-like diseases, TLR7 and TLR9 are likely to be important for the amplification loop of the autoimmune process: in Y-associated autoimmune (Yaa) mouse carrying a duplication of a portion of X chromosome (including tlr7 gene), autoimmunity was still detectable when TLR7 expression dose was returned to 1X [48]. However, higher levels of expression of TLR7 are sufficient to induce autoimmunity even in resistant animals [48]. Thus, evolutionary conserved fine-tuning of the expression of receptors that can use self-molecules as agonists is an important anti-autoimmune mechanism.
Non-pathogenic microbes and autoimmunity
Whereas encounters with pathogens are random, eukaryotic organisms are in a constant mutualistic relationship with commensal microbiota which is essential for the fitness of the eukaryotic host. Besides nutritional and lymphoid organogenic functions, commensals are needed for protection against pathogens and support of the integrity of the intestinal epithelium. Our recent study[8] has highlighted the role of commensals in autoimmunity. NOD mice lacking the MyD88 adaptor were resistant to T1D development when housed in SPF conditions. However, GF conditions reversed the trend and made the knock-out mice susceptible to T1D. Colonization of GF MyD88-negative mice with a defined bacterial consortium led to a significant decrease in T1D development. At the same time, transplantation of the microbiota of SPF MyD88-negative mice to GF wild-type mice led to a decrease in T1D-associated pathology in the latter. This result and the deep sequencing of 16S RNA genes from ceacal microbiota of MyD88-sufficient and -deficient mice confirmed the hypothesis that specific changes in microbiota composition can affect the development of autoimmunity. Fig. 1B shows the model explaining the hypothesis. In the case of T1D, commensal bacteria that are normally controlled by a MyD88-dependent mechanism(s) render diabetogenic T cells tolerant. The microbial signals and receptors that are involved in the process are currently unknown. It is important to appreciate that many effects of innate receptor signaling on autoimmunity could be indirect. The aforementioned finding of T1D dependence on TLR2 is a likely example of that, with TLR2 (and TLR9 [49]) being responsible for control over the intestinal microbiota. We have recently found that double TLR4/MyD88 negative (a double knock-out that disables all TLRs except TLR3) GF NOD mice were still highly susceptible to T1D (Volchkov P., unpublished data), a result that in combination with the lack of TLR3 participation in T1D development [8,49] shows that TLRs are totally dispensable for T1D development when commensals are absent. TLRs are likely involved in control over intestinal microbiota through causing secretion of immunoglobulins, activation of neutrophils and macrophages, and secretion of antimicrobial peptides. Because besides inflammatory signals, TLRs deliver commensal-dependant tissue repair signals[50], it is possible that viral enteritis (or other types of stress on microbiota such as antibiotics) affects the commensal microbiota reducing its tolerogenic function and enhancing delivery of PRR agonists across the epithelial barrier. Moreover, other PRRs (e.g. NOD1) [51] and the microbiota itself [52,53] could be involved in commensal-dependent development of gut-associated lymphoid organs, adding to the complexity of the host-commensal relationship.
Commensals can also be involved in induction of autoimmunity by other unknown mechanisms. Joint diseases frequently accompany intestinal inflammation, and in gnotobiotic mice with ankylosing enthesopathy colonization with typical intestinal commensal bacteria were critical for disease progression [54]. Similarly, commensals were required for arthritis development in IL1R antagonist-negative mice [24], suggesting that microbial agonists of PRRs can be delivered systemically, or that they induce inflammatory mediators acting systemically.
An important issue is the role of diet in development of autoimmunity. Besides provision of nutrients that can affect immune reactivity directly (e.g. vitamins A and D) [55], diet can affect microbial communities in the gut. For example, an obesity-promoting diet affects microbiota and sets its metabolism to sustain obesity[56,57]. Diet can control many physiological and pathophysiological processes including PRR signaling [58]. Microbiota-induced metabolic changes can be detected in the blood by ‘metabolomics’ analysis [59]. Thus, the longitudinal studies of T1D may contribute in a significant way to the understanding of the role of the microbiota and diet in disease development : metabolomics analysis of sera from children before and after conversion to diabetes has revealed metabolic changes that precede the appearance of autoantibodies[60].
Metabolism, diet and microbiota make a functional triangle where every side affects two others also affecting the host’s health including autoimmunity.
Conclusions and future directions
Two major points need to be made.
First, the lack of a requirement for TLR signaling for development of autoimmunity in some systems suggests that in addition to faulty negative selection, a glitch in either T cell requirements for costimulation (low threshold for T cell activation) or a glitch in sensitivity of T cells to the background costimulatory signals can result in autoimmunity. These background signals must be important for the homeostasis of the immune system and are present in sterile conditions [61]. Genetic linkage of autoimmunity to genes controlling T cell activation [ptpn22 phosphatase [45], IL2RA [62], UBASH3A (human homolog of mouse STS2 [63])], costimulation (CTLA-4 [64]), lymphocyte-lymphocyte interactions (SLAM family members[65]), and transcription factors regulating multiple immunity-related genes (BACH2 [63]) support this conclusion. Some of these genes are predictably universal for all types of autoimmunity and some are disease-specific [66].
Second, involvement of PRRs in development of autoimmunity can be indirect and work through regulation of the host-commensal microbiota interactions. Clearly, comprehensive deep microbial DNA sequencing studies are needed to clarify the effects of inactivation of various antimicrobial mechanisms on commensal composition and metabolism in animal models, as well as to test microbiota dynamics in longitudinal studies of human autoimmunity. The use of GF mice deficient in PRR signaling, although technically challenging, would be helpful in elucidating the role of innate signaling receptors in other models of autoimmunity.
Acknowledgments
This work was supported by JDRF grants 2005-204 and 2007-353, by NIH grant DK063452 and by NIH/NIDDK Digestive Disease Research Core Center grant DK42086.
The author is grateful to Tatyana Golovkina, Ruslan Medzhitov and Joseph Pickard for comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
• of special interest
•• of outstanding interest
- 1.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347:911–920. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 2.Bach JF. Infections and autoimmune diseases. J Autoimmun. 2005;25(Suppl):74–80. doi: 10.1016/j.jaut.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 3.Zipris D. Epidemiology of type 1 diabetes and what animal models teach us about the role of viruses in disease mechanisms. Clin Immunol. 2009;131:11–23. doi: 10.1016/j.clim.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 4.Pozzilli P, Signore A, Williams AJ, Beales PE. NOD mouse colonies around the world--recent facts and figures. Immunol Today. 1993;14:193–196. doi: 10.1016/0167-5699(93)90160-M. [DOI] [PubMed] [Google Scholar]
- 5.Janeway CA., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Rossini AA, Williams RM, Mordes JP, Appel MC, Like AA. Spontaneous diabetes in the gnotobiotic BB/W rat. Diabetes. 1979;28:1031–1032. doi: 10.2337/diab.28.11.1031. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki TYT, Fujimura T, Kawamura E, Shimizu M, Yamashit R a, Nomoto K. In: Rygaard JBN, Graem N, Spang-Thomsen M, editors. Karger; Basel: 1985. [Google Scholar]
- 8 ••.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. The paper showed that T1D develops independently from TLR signaling and that TLRs control commensal microbiota in specific fashion. The changes in commensals were shown to affect the disease development.
- 9 ••.Gray DH, Gavanescu I, Benoist C, Mathis D. Danger-free autoimmune disease in Aire-deficient mice. Proc Natl Acad Sci U S A. 2007;104:18193–18198. doi: 10.1073/pnas.0709160104. This paper demonstrated independence of autoimmnuty cause by AIRE deletion from both microbes and TLR signaling.
- 10.Guerau-de-Arellano M, Martinic M, Benoist C, Mathis D. Neonatal tolerance revisited: a perinatal window for Aire control of autoimmunity. J Exp Med. 2009;206:1245–1252. doi: 10.1084/jem.20090300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11 •.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. This work showed that negative selection is incomplete even in regular, non-autoimmunity-prone animals.
- 12.Crawford A, Wherry EJ. The diversity of costimulatory and inhibitory receptor pathways and the regulation of antiviral T cell responses. Curr Opin Immunol. 2009;21:179–186. doi: 10.1016/j.coi.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuchroo VK, Dardalhon V, Xiao S, Anderson AC. New roles for TIM family members in immune regulation. Nat Rev Immunol. 2008;8:577–580. doi: 10.1038/nri2366. [DOI] [PubMed] [Google Scholar]
- 14.Arnett HA, Escobar SS, Viney JL. Regulation of costimulation in the era of butyrophilins. Cytokine. 2009;46:370–375. doi: 10.1016/j.cyto.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 16.Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7:1029–1035. doi: 10.1038/ni1006-1029. [DOI] [PubMed] [Google Scholar]
- 17.Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity. 2008;28:218–230. doi: 10.1016/j.immuni.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 18.Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK, 3rd, Wu T, Li QZ, Davis LS, Mohan C, Perlman H. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity. 2008;28:206–217. doi: 10.1016/j.immuni.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 19.Hughes PD, Belz GT, Fortner KA, Budd RC, Strasser A, Bouillet P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity. 2008;28:197–205. doi: 10.1016/j.immuni.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen M, Wang YH, Wang Y, Huang L, Sandoval H, Liu YJ, Wang J. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 2006;311:1160–1164. doi: 10.1126/science.1122545. [DOI] [PubMed] [Google Scholar]
- 21.Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, Hartig H, Sundberg JP, Servick S, Kaufmann G, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hase K, Takahashi D, Ebisawa M, Kawano S, Itoh K, Ohno H. Activation-induced cytidine deaminase deficiency causes organ-specific autoimmune disease. PLoS One. 2008;3:e3033. doi: 10.1371/journal.pone.0003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rehakova Z, Capkova J, Stepankova R, Sinkora J, Louzecka A, Ivanyi P, Weinreich S. Germ-free mice do not develop ankylosing enthesopathy, a spontaneous joint disease. Hum Immunol. 2000;61:555–558. doi: 10.1016/s0198-8859(00)00122-1. [DOI] [PubMed] [Google Scholar]
- 24.Abdollahi-Roodsaz S, Joosten LA, Koenders MI, Devesa I, Roelofs MF, Radstake TR, Heuvelmans-Jacobs M, Akira S, Nicklin MJ, Ribeiro-Dias F, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008;118:205–216. doi: 10.1172/JCI32639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Croker BA, Lawson BR, Rutschmann S, Berger M, Eidenschenk C, Blasius AL, Moresco EM, Sovath S, Cengia L, Shultz LD, et al. Inflammation and autoimmunity caused by a SHP1 mutation depend on IL-1, MyD88, and a microbial trigger. Proc Natl Acad Sci U S A. 2008;105:15028–15033. doi: 10.1073/pnas.0806619105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu CC, Tsui HW, Ngan BY, Shulman MJ, Wu GE, Tsui FW. B and T cells are not required for the viable motheaten phenotype. J Exp Med. 1996;183:371–380. doi: 10.1084/jem.183.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–741. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 29 •.Palm NW. Medzhitov R: Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227:221–233. doi: 10.1111/j.1600-065X.2008.00731.x. Comprehensive review of the most important findings in the field 20 years after publication of Janeway’s theory.
- 30.Kim HS, Han MS, Chung KW, Kim S, Kim E, Kim MJ, Jang E, Lee HA, Youn J, Akira S, et al. Toll-like Receptor 2 Senses beta-Cell Death and Contributes to the Initiation of Autoimmune Diabetes. Immunity. 2007;27:321–333. doi: 10.1016/j.immuni.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 31.Wong FS, Wen L. Toll-like receptors and diabetes. Ann N Y Acad Sci. 2008;1150:123–132. doi: 10.1196/annals.1447.063. [DOI] [PubMed] [Google Scholar]
- 32.Zipris D. Innate immunity and its role in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. 2008;15:326–331. doi: 10.1097/MED.0b013e3283073a46. [DOI] [PubMed] [Google Scholar]
- 33.Lien E, Zipris D. The role of Toll-like receptor pathways in the mechanism of type 1 diabetes. Curr Mol Med. 2009;9:52–68. doi: 10.2174/156652409787314453. [DOI] [PubMed] [Google Scholar]
- 34.Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S, Odermatt B, Conrad C, Ittner LM, Bauer S, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11:138–145. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 35.Jung A, Kato H, Kumagai Y, Kumar H, Kawai T, Takeuchi O, Akira S. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J Virol. 2008;82:196–206. doi: 10.1128/JVI.01640-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richer MJ, Horwitz MS. Viral infections in the pathogenesis of autoimmune diseases: focus on type 1 diabetes. Front Biosci. 2008;13:4241–4257. doi: 10.2741/3002. [DOI] [PubMed] [Google Scholar]
- 37.Richer MJ, Lavallee DJ, Shanina I, Horwitz MS. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PLoS One. 2009;4:e4127. doi: 10.1371/journal.pone.0004127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 39.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 40.Manicassamy S, Ravindran R, Deng J, Oluoch H, Denning TL, Kasturi SP, Rosenthal KM, Evavold BD, Pulendran B. Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat Med. 2009;15:401–409. doi: 10.1038/nm.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41 •.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annu Rev Immunol. 2009;27:621–668. doi: 10.1146/annurev.immunol.25.022106.141627. Important review helping to delinate the role of PRRs in triggering autoimmune or autoinflammatory responses.
- 42.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fritz JH, Le Bourhis L, Sellge G, Magalhaes JG, Fsihi H, Kufer TA, Collins C, Viala J, Ferrero RL, Girardin SE, et al. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity. 2007;26:445–459. doi: 10.1016/j.immuni.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 44.van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, Muller FJ, Hommes DW, Zaat SA, Kapsenberg ML, de Jong EC. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. 2007;27:660–669. doi: 10.1016/j.immuni.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 45.Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, Guja C, Ionescu-Tirgoviste C, Widmer B, Dunger DB, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38:617–619. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- 46 •.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. Suggests (not proves) a role for RLRs in triggering ‘adaptive” autoimmunity.
- 47.Stetson DB. Connections between antiviral defense and autoimmunity. Curr Opin Immunol. 2009;21:244–250. doi: 10.1016/j.coi.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong FS, Hu C, Zhang L, Du W, Alexopoulou L, Flavell RA, Wen L. The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann N Y Acad Sci. 2008;1150:146–148. doi: 10.1196/annals.1447.039. [DOI] [PubMed] [Google Scholar]
- 50.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, Eberl G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–510. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 52.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, O’Shea JJ. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. 2009;206:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinkorova Z, Capkova J, Niederlova J, Stepankova R, Sinkora J. Commensal intestinal bacterial strains trigger ankylosing enthesopathy of the ankle in inbred B10.BR (H-2(k)) male mice. Hum Immunol. 2008;69:845–850. doi: 10.1016/j.humimm.2008.08.296. [DOI] [PubMed] [Google Scholar]
- 55.Moro JR, Iwata M, von Andriano UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008;8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 57 •.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. The study links diet, intestinal commensals and the host’s health.
- 58.Shamshiev AT, Ampenberger F, Ernst B, Rohrer L, Marsland BJ, Kopf M. Dyslipidemia inhibits Toll-like receptor-induced activation of CD8alpha-negative dendritic cells and protective Th1 type immunity. J Exp Med. 2007;204:441–452. doi: 10.1084/jem.20061737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59 •.Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106:3698–3703. doi: 10.1073/pnas.0812874106. See commnet on [60].
- 60 ••.Oresic M, Simell S, Sysi-Aho M, Nanto-Salonen K, Seppanen-Laakso T, Parikka V, Katajamaa M, Hekkala A, Mattila I, Keskinen P, et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J Exp Med. 2008;205:2975–2984. doi: 10.1084/jem.20081800. Together with [59] make possible longitudinal analysis of the influence of gut microbiota on metabolism and disease development.
- 61.Wilson NS, Young LJ, Kupresanin F, Naik SH, Vremec D, Heath WR, Akira S, Shortman K, Boyle J, Maraskovsky E, et al. Normal proportion and expression of maturation markers in migratory dendritic cells in the absence of germs or Toll-like receptor signaling. Immunol Cell Biol. 2008;86:200–205. doi: 10.1038/sj.icb.7100125. [DOI] [PubMed] [Google Scholar]
- 62.Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, Bourget K, Plagnol V, Field S, Atkinson M, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet. 2007;39:1074–1082. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- 63.Grant SF, Qu HQ, Bradfield JP, Marchand L, Kim CE, Glessner JT, Grabs R, Taback SP, Frackelton EC, Eckert AW, et al. Follow-up analysis of genome-wide association data identifies novel loci for type 1 diabetes. Diabetes. 2009;58:290–295. doi: 10.2337/db08-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di Genova G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 65.Chan AY, Westcott JM, Mooney JM, Wakeland EK, Schatzle JD. The role of SAP and the SLAM family in autoimmunity. Curr Opin Immunol. 2006;18:656–664. doi: 10.1016/j.coi.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 66.Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH, Howson JM, Stevens H, McManus R, Wijmenga C, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767–2777. doi: 10.1056/NEJMoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]