

Abstract
In previous studies, it was shown that post-conditioning, a transient period of brief ischemia following prolonged severe ischemia in the retina, could provide significant improvement in post-ischemic recovery, attenuation of cell loss, and decreased apoptosis. These studies showed that post-conditioning effectively prevented damage after retinal ischemia when it was instituted early (within one hour) in the post-ischemic period. While post-ischemic conditioning holds high promise of clinical translation, patients often present late after the onset of retinal ischemia and therefore immediate application of this anti-ischemic maneuver is generally not feasible. In this study, we examined the hypothesis that application of a post-conditioning stimulus at 24 h or greater following the end of prolonged ischemia would decrease the extent of ischemic injury. Ischemia was induced in rat retina in vivo. Recovery after ischemia followed by 5 minutes of post-conditioning brief ischemia 24 or 48 h after prolonged ischemia was assessed functionally (electroretinography) and histologically at 7 days after ischemia and post-conditioning or sham post-conditioning. We found that the brief ischemic stimulus applied 24, but not 48 h after prolonged ischemia significantly improved functional recovery and decreased histological damage induced by prolonged ischemia. We conclude that within a defined time window, delayed post-ischemic conditioning ameliorated post-ischemic injury in rats. Compared to earlier studies, the present work demonstrates for the first time the novel ability of a significantly delayed ischemic stimulus to provide robust neuroprotection in the retina following ischemia.
INTRODUCTION
Retinal ischemia, associated with glaucoma, diabetic retinopathy, central retinal artery occlusion, and other vascular diseases, causes visual loss. Applying a brief ischemic stimulus after prolonged retinal ischemia (Post-conditioning, “post-C”) provided significant neuroprotection, with improved post-ischemic functional recovery, decreased histological damage, and attenuation of apoptosis in rats (Fernandez et al. 2009; Dreixler et al. 2010). Further, it was shown that new protein synthesis was required for post-C (Fernandez et al. 2009). These results with post-C are very exciting and directly clinically translatable to patients who have experienced retinal ischemia. Clinical studies of post-C are already underway, e.g., for acute myocardial infarction (Limalanathan et al. 2010). However, the neuroprotective effect of post-ischemic conditioning in the retina was effective only within 1 hour after ischemia and required multiple applications of brief ischemia (Fernandez et al. 2009). In a number of studies, post-C ameliorated neuronal injury in models of cerebral ischemia in rodents; however, in all of these reports post-C was initiated within minutes to just a few hours after ischemia (Zhao et al. 2006; Wang et al. 2008; Xing et al. 2008; Pignataro et al. 2009). To date, only one study has demonstrated that a delayed post-C stimulus could alter the damage after ischemia in the CNS; application of systemic hypoxia 5 days after transient middle cerebral artery occlusion in mice reduced thalamic atrophy (Leconte et al. 2009). Considering the retina’s greater degree of tolerance to ischemia compared to brain,(Hayreh et al. 1980) we theorized that application of brief ischemia 24 h or later after ischemia ended could still provide neuroprotection in the retina. Delayed application of a brief, non-damaging ischemic stimulus to alter the outcome after ischemia is a novel concept which has considerable potential for clinical translation.
MATERIALS AND METHODS
Retinal ischemia and preconditioning
Wistar rats (200–250 gm) purchased from Harlan (Indianapolis, IN) were maintained on a 12 h on/12 h off light cycle. Procedures (Roth et al. 2006; Dreixler et al. 2008) conformed to the ARVO Resolution on the Use of Animals in Research and were approved by our Animal Care Committee. For retinal ischemia, rats were anesthetized with chloral hydrate, 275 mg/kg i.p., and intraocular pressure (IOP) increased to 130 – 135 mm Hg for 55 min using a pressurized bag of sterile ocular irrigating solution (BSS: Alcon, Fort Worth, TX) connected to a 27-g needle positioned in the center of the anterior chamber. IOP was measured continuously using the needle and pressure tubing via a 3-way stopcock interposed into the line, with an electronic pressure transducer (Hewlett-Packard) zeroed to the level of the eye. IOP exceeded the systolic blood pressure, monitored non-invasively using a tail blood pressure cuff (IITC Life Science; Woodland Hills, CA). At 24 or 48 h after ischemia ended, post-C was produced by anesthetizing rats with ketamine (35 mg/kg), and xylazine (5 mg/kg), and placing a 2.0 silk suture behind the globe, which had been passed through a small length of plastic tubing (PE-200, Intramedic; Becton-Dickinson, Parsippany, NJ), and the suture was pulled to maximal tightness to occlude the retinal circulation for 5 min, as we previously described (Roth et al. 1998). In control groups, the suture was placed but not tightened.
Body temperature was maintained at 36–37 C by a servo–controlled heating blanket (Harvard Apparatus, Natick, MA) to prevent a protective effect on ischemia of hypothermia (Faberowski et al. 1989). To preclude ischemic tolerance due to hypoxia (Zhu et al. 2007), O2 saturation was measured with a pulse oximeter (Ohmeda; Louisville, CO) on the rat’s tail. Supplemental oxygen was administered when necessary to maintain O2 saturation > 94%.
Electroretinography Before and after Retina Ischemia
Procedures were similar to those we reported previously (Dreixler et al. 2010). Animals were dark-adapted for at least 2 h before recordings. For baseline and post–ischemic (i.e., after 7 days) follow–up ERG recordings, rats were injected i.p. with ketamine (35 mg/kg), and xylazine (5 mg/kg). Corneal analgesia was with 1–2 drops of 0.5% proparacaine (Alcon). Pupils were dilated with 0.5% tropicamide (Alcon), and cyclomydril (0.2% cyclopentolate HCl and 1% phenylephrine HCl (Alcon).
The ERG was recorded at baseline (prior to experiments) and 7 days after Post-C using procedures modified from those we described previously (Dreixler et al. 2010). Electrodes were placed on the cornea and sclera (Weymouth and Vingrys 2008). These custom Ag/AgCl electrodes were fashioned from 0.010” Teflon coated silver wire (Grass Technologies, West Warwick, RI), with 5 mm of silver wire exposed and fashioned into a ‘U’ shape to act as the corneal/positive electrode, while 10 mm of silver wire was exposed to form a loop, sclera/negative electrode. Electrodes were then attached to a 9V battery and placed in a 1N HCl bath for 12 seconds until coated with AgCl. All electrodes were referenced to a 12 mm, 30 gauge stainless steel, needle electrode (Grass) inserted 2/3 down the length of the tail.
Stimulus-intensity ERG analysis was achieved on a UTAS-E4000 using a full-field Ganzfeld stimulator (LKC Technologies, Gaithersburg, MD), with the rat’s head centered 7 inches from the stimulator. The low pass filter was 0.05 Hz and the high pass 500 Hz. Flash intensity varied electronically from −3.39 log cd.s/m2 to 1.89 log cd.s/m2. Settings were confirmed by photometry (EG & G Model 550 photometer, Electro-Optics, Boulder, CO). Responses were averaged for 3 to 10 flashes delivered 4 to 60 s apart depending upon flash intensity, with number of flashes decreasing and time between them increasing with flash intensity. Flashes were progressively delivered from the lowest intensity to the highest to prevent possible effect upon dark adaptation, and at least 1 min elapsed between the series of flashes for the three highest intensity settings.
Recorded time, intensity, and amplitude were exported and analyzed in Matlab (The MathWorks, Natick, MA) as we described previously (Dreixler et al. 2011a). Recordings were first baseline corrected for drift or low frequency noise. For intensity-response ERGs, peak a-wave amplitude was calculated as the negative minimum following the light stimulus, and the b wave amplitude as difference between the a-wave and the maximum b wave value recorded thereafter. The Hood and Birch phototransduction model, P3(i,t) = Rmax[1−e−i·S·(t−teff)2] was fit to the leading edge of the a-wave. The derived P3 was then subtracted from the baseline corrected ERG waveform. Oscillatory potentials (OPs) were measured by extracting OP wavelet components using Fast Fourier transformation. The sum of the root mean squares (Sum RMS) of the amplitudes of the OP wavelets was calculated (Bui et al. 2005). OPs were then subtracted from the remaining waveform leaving the scotopic P2 for the rod bipolar response.
Histology
Eyes enucleated on the seventh day after post conditioning were immediately placed in Davidson’s fixative (11% glacial acetic acid, 2% neutral buffered formalin and 32% ethanol in H2O) for 24 h, then transferred to 70% ethanol for 24 h and stored in PBS at 4°C. Eyes were embedded in paraffin, sectioned to 4 µm and stained with hematoxylin and eosin (H&E). Sections were examined by light microscopy and cell counts in the retinal ganglion cell (RGC) layer were quantified using 40× optics. Specifically, the number of putative retinal ganglion cells in the RGC layer was counted in a standardized region in all of the retinae (called a “region of interest”), centered 1280 µm distant (as measured by an eyepiece reticule) from the thinning of the neurofilaments arising from the optic nerve head and comprising the neuronal fiber layer. The counts were made, in both directions from the optic nerve head, in this same region spanning 128 µm. the RGC layer is reported as cells per region of interest.
Data handling and statistical analysis
ERG data were corrected for daily variation in the normal eye and for simultaneous reference to the baseline in the ischemic eye, as previously described (Roth et al. 2006; Dreixler et al. 2008; Dreixler et al. 2009a; Dreixler et al. 2009b). The a- and b-waves, SUM RMS of the OPs, and P2 from ischemic eyes 7 days after post conditioning in the groups for comparison were expressed as stimulus-intensity plots (intensity, log cd.s/m2 on the x-axis), and % recovery of baseline on the y-axis (Dreixler et al. 2008; Dreixler et al. 2011a). The data were normally distributed as confirmed using normality plots and skewness/kurtosis in Stata version 10.0 (College Station, TX). For histopathological data analysis, we used the Mann-Whitney non-parametric test.
RESULTS
Post-C administered 24 h after ischemia (n = 15) significantly increased the b-wave, OP RMS and P2 recovery at 7 days following the delayed ischemia and post-C, when compared to the sham post-C and 24 h ischemia control group (n = 11) over a range of flash intensities from −1.02 to 1.4 log cd.s/m2 (Fig. 1). Fig. 2 shows that the averaged ERG stimulus-intensity plots, for a-wave, b-wave, OP RMS and P2, were consistent with the results of the normalized results. The time latency for the b-wave significantly increased from 75.9 ± 3.6 ms at baseline to 111.8 ± 13.5 (p = 0.03) at day 7 at the 0.87 log cd.s/m2 flash intensity in the 24 h sham post-C group. At the same flash intensity the b-wave latency was unchanged in the 24 h post-C group (data not shown). Representative ERG traces are shown in Fig. 3A.
Figure 1.
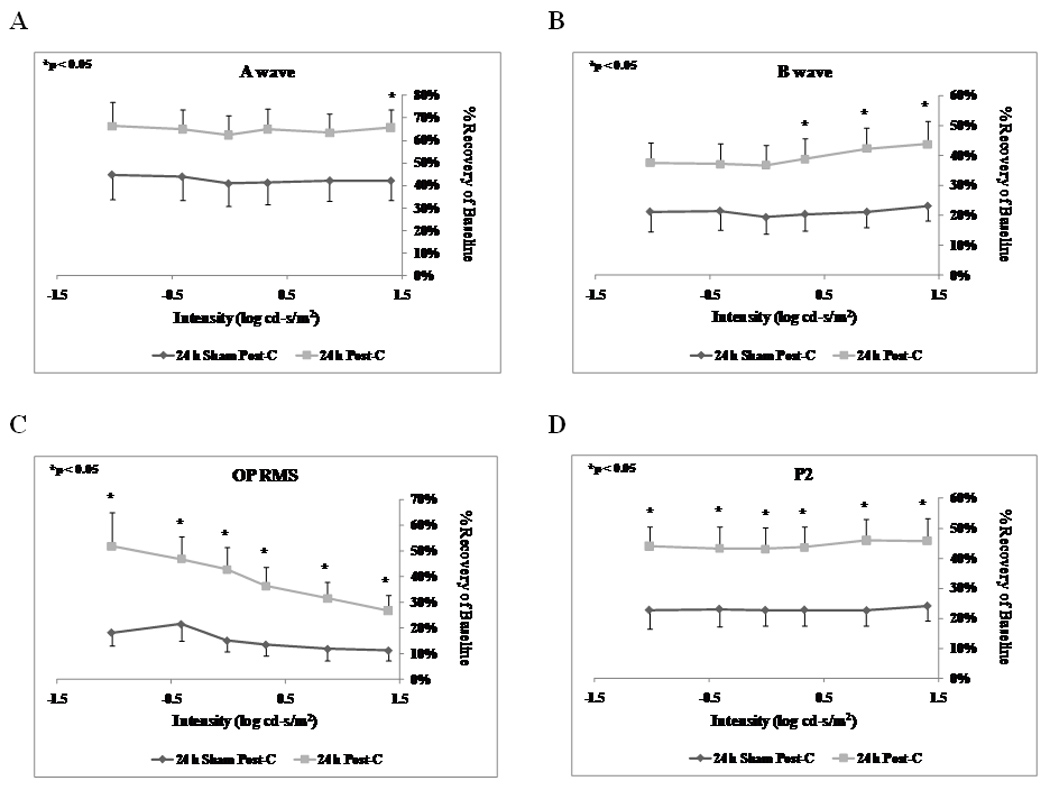
Stimulus-Intensity response in post-conditioned retinae for 24 h post-C vs 24 h sham post-C. Double normalized (corrected for the non-ischemic eye and for diurnal variation from baseline to 7 days after post-conditioning) ERG data for a-, b-wave, oscillatory potentials (sum of root mean square, OP RMS) and P2 over a range of flash intensities from −1.02 to 1.40 log cd-s/m2. The data were recorded at baseline (prior to ischemia) and at 7 days after post-conditioning; post-C was applied 24 h after ischemia. ERG data for a- (A), b-wave (B), OP RMS(C) and P2 (D) over a range of flash intensities suggests that 24 h delayed post-C was functionally protective. Solid lines with diamonds = 24 h sham post-C (n = 11). Solid lines with squares = 24 h post-C (n = 15).
Figure 2.
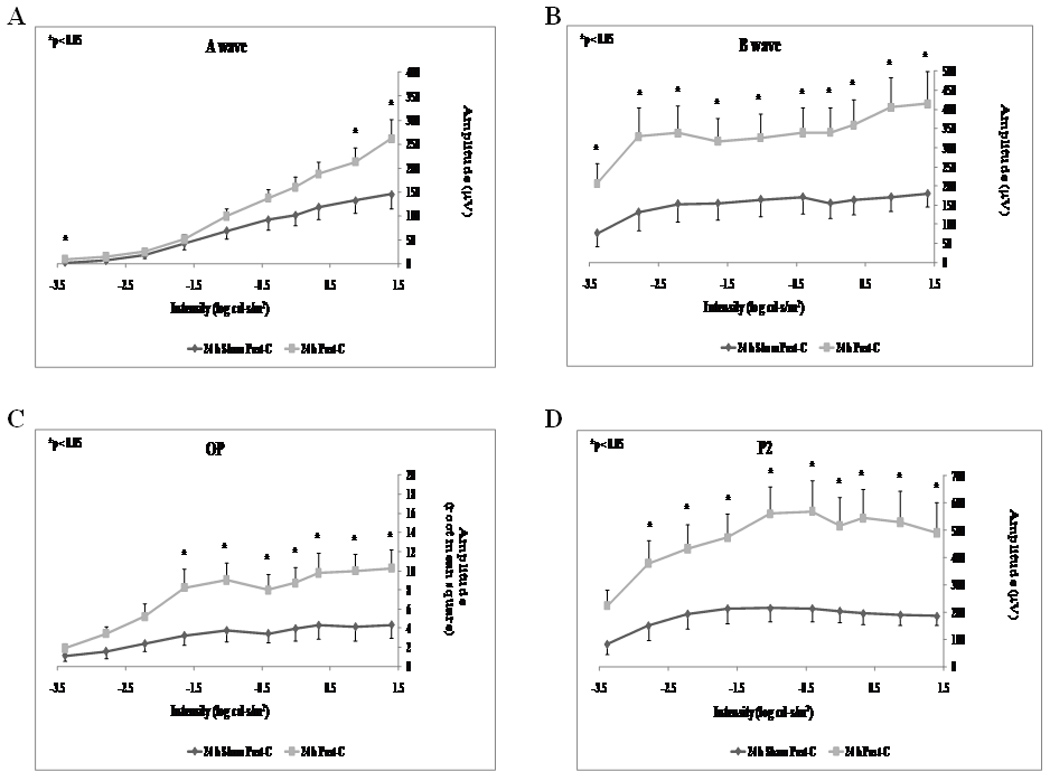
Stimulus-Intensity response in post-conditioned retinae for 24 h post-C vs 24 h sham post-C. Absolute ERG data for a-, b-wave, oscillatory potentials (sum of root mean square, OP RMS) and P2 over a range of flash intensities from −1.02 to 1.40 log cd-s/m2. The data were recorded at baseline (prior to ischemia) and at 7 days after post- conditioning; post-C was applied 24 h after ischemia. ERG data for a- (A), b-wave (B), OP RMS(C) and P2 (D) over a range of flash intensities suggests that 24 h delayed post-C was functionally protective. Solid lines with diamonds = 24 h sham post-C (n = 11). Solid lines with squares = 24 h post-C (n = 15).
Figure 3.
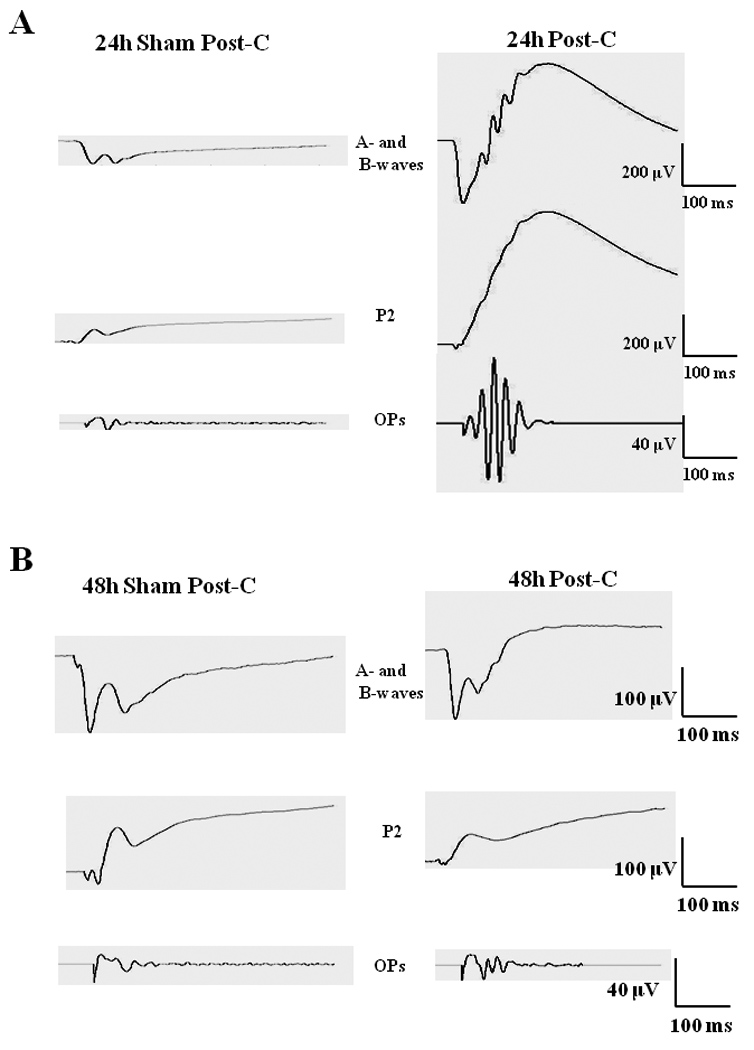
(A) Representative ERG traces at 0.87 log cd.s/m2 flash intensity for the a- and b-wave (top), P2 (middle) and the OPs (bottom) for both 24 h post-C and 24 h sham post-C groups. (B) Representative ERG traces at 0.87 log cd.s/m2 flash intensity for the a- and b-wave (top), P2 (middle) and the OPs (bottom) for both 48 h post-C and 48 h sham post-C groups.
Histological examination of the retinae 7 days after ischemia + 24 h later post-C or 24 h later sham post-C showed that the number of cells in the RGC layer in the ischemic retinae significantly increased to 8.8 ± 0.8 cells/region of interest (n = 9) in the 24 h post-C group from 6.5 ± 0.7 (n = 6) in the ischemic retinae for the 24 h sham post-C group (p = 0.02), indicating the preservation of histological structure of the retina after ischemia by post-C (Table 1; Fig. 4). There was qualitatively, decreased inflammatory cell infiltration in the 24 h post-C group compared to that of ischemia and sham post-C (Fig. 4). The normal retinae RGC layer counts were 11.2 ± 0.5 for the ischemia + 24 h later post-C group and 11.3 ± 0.5 for the ischemia + 24 h later sham post-C group.
Table 1.
Number of retinal ganglion cells per region of interest (mean +/− s.e.m.) in the ischemic retinae 7 days after 24h Sham Post-C compared to the number of retinal ganglion cells in the ischemic retinae for 24h Post-C.
| Number of RGCs |
p-value vs. Ischemia |
|
|---|---|---|
| 24h Sham Post-C | 6.5 ± 0.7 | --- |
| 24h Post-C | 8.8 ± 0.8 | 0.02 |
Figure 4.
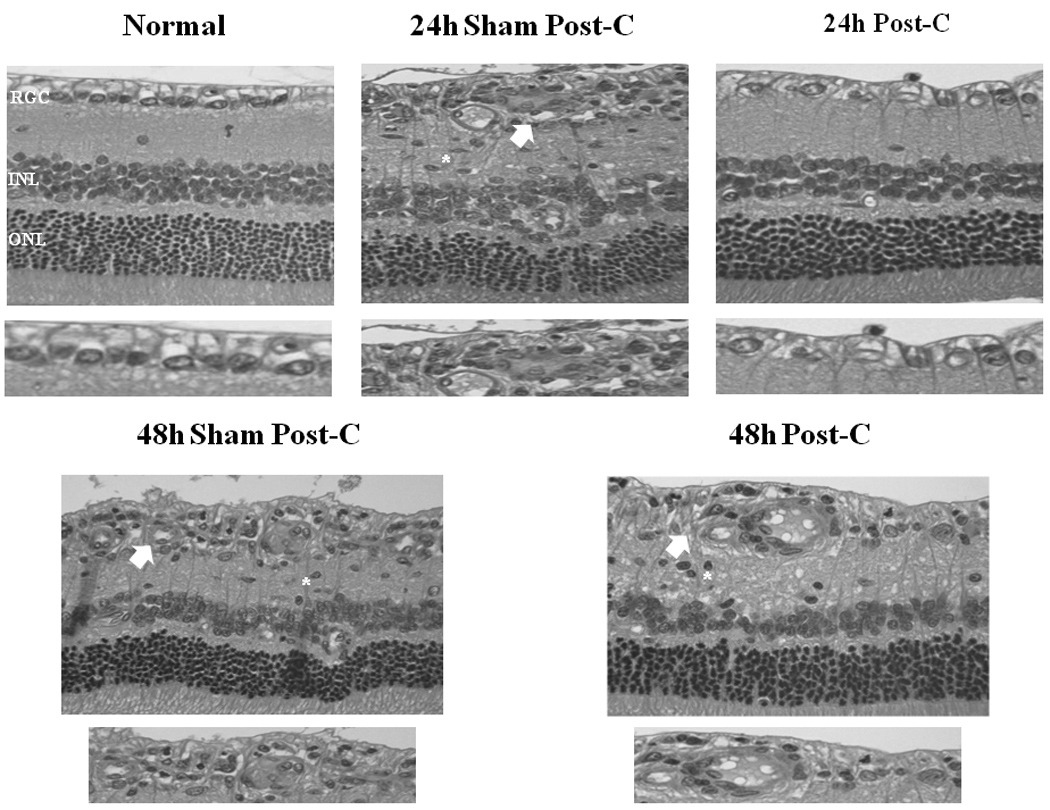
Representative histopathological images of hematoxylin and eosin-stained retinae in 4 µm thick sections for each of the experimental groups. These sections were prepared from retinae removed from the rats at 7 days following delayed post-C. Arrows indicate layers demonstrating cell loss and asterisks denote regions of inflammatory cell infiltration. Below each representative retina is a close-up of the RGC layer.
Lengthening the time between ischemia and post-C time resulted in loss of the protection afforded by delayed post-C. Post-C administered 48 h after ischemia (n = 4) did not change the a-wave, b-wave, OP RMS and P2 recovery at 7 days after the delayed post-C, when compared to the sham 48 h post-C control group (n = 4) over a range of flash intensities from −1.02 to 1.4 log cd.s/m2 (Fig. 5). Fig. 6 shows that the averaged ERG stimulus-intensity plots, for a-wave, b-wave, OP RMS and P2, were consistent with the results of the normalized results. The time latency for the b-wave was unchanged from baseline to day 7 for the 48 h post-C and 48 h sham post-C groups (data not shown). Representative ERG traces are shown in Fig. 3B.
Figure 5.
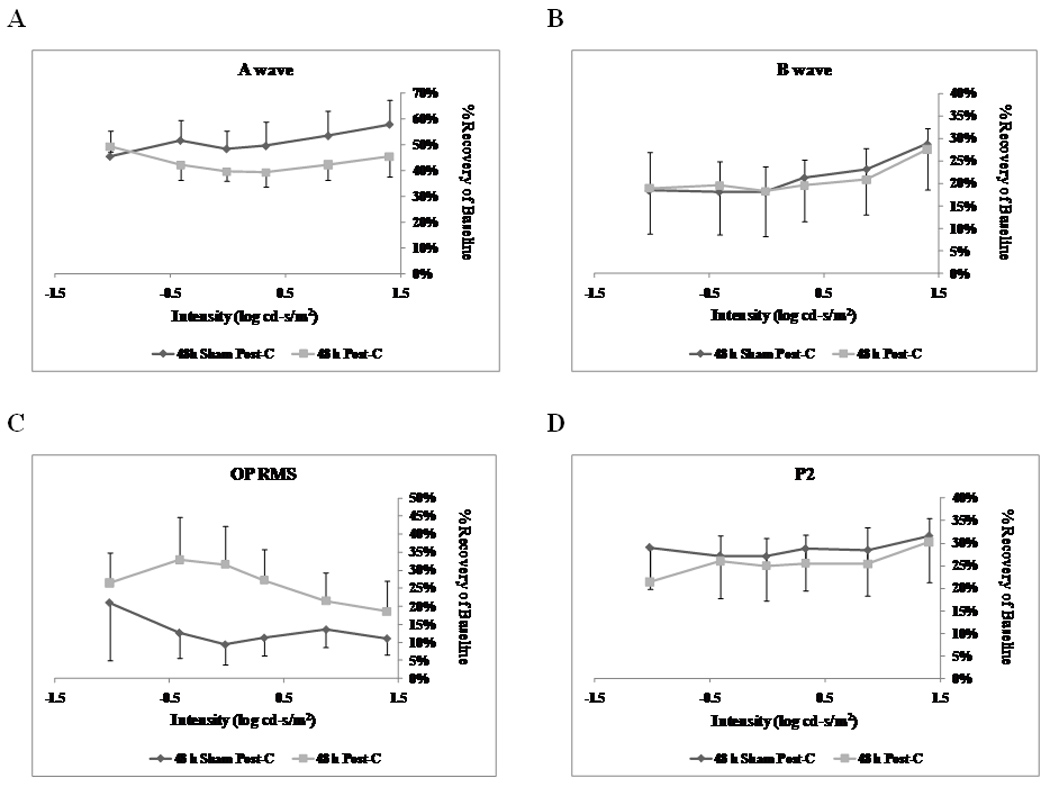
Stimulus-Intensity response in post-conditioned retinae for 48 h post-C vs 48 h sham post-C. Double normalized (corrected for the non-ischemic eye and for diurnal variation from baseline to 7 days after post-C) ERG data for a-, b-wave, oscillatory potentials (sum of root mean square, OP RMS) and P2 over a range of flash intensities from −1.02 to 1.40 log cd-s/m2. The data were recorded at baseline (prior to ischemia) and at 7 days after post-conditioning; post-C was applied 48 h after ischemia. ERG data for a- (A), b-wave (B), OP RMS(C) and P2 (D) over a range of flash intensities suggests that 48 h delayed post-C was not functionally protective. Solid lines with diamonds = 48 h sham post-C (n = 4). Solid lines with squares = 48 h post-C (n = 4).
Figure 6.
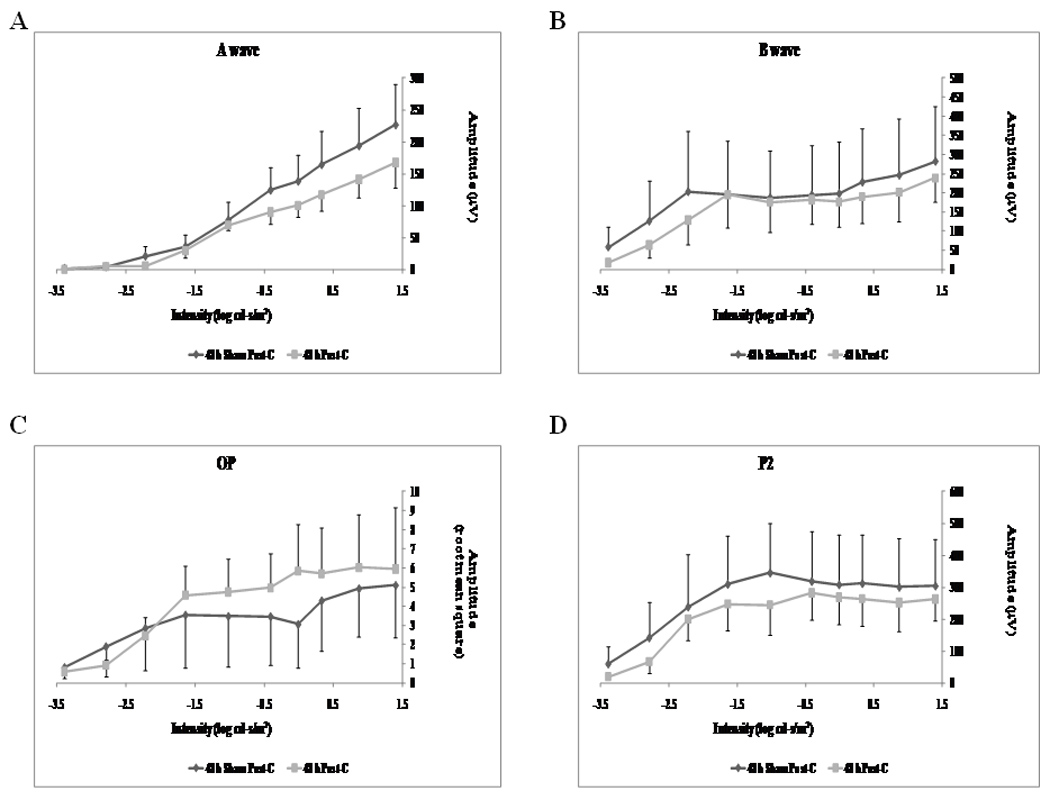
Stimulus-Intensity response in post-conditioned retinae for 48 h post-C vs 48 h sham post-C. Absolute ERG data for a-, b-wave, oscillatory potentials (sum of root mean square, OP RMS) and P2 over a range of flash intensities from −1.02 to 1.40 log cd-s/m2. The data were recorded at baseline (prior to ischemia) and at 7 days after post-conditioning; post-C was applied 48 h after ischemia. ERG data for a- (A), b-wave (B), OP RMS(C) and P2 (D) over a range of flash intensities suggests that 48 h delayed post-C was not functionally protective. Solid lines with diamonds = 48 h sham post-C (n = 4). Solid lines with squares = 48 h post-C (n = 4).
Examination of the retinae 7 days after ischemia + 48 h post-C or 48 h sham post-C showed that the number of cells in the RGC layer in the ischemic retinae did not change. The number of cells in the RGC layer in the ischemic retinae was 6.5 ± 0.8 (n = 4) in the 48 h post-C group and 5.8 ± 0.5 (n = 4) for the 48 h sham post-C group (p = 0.47; Table 1; Fig. 4). Compared to the sham post-C group, 48 h post-C did not alter inflammatory cell infiltration after ischemia (Fig. 4). The normal retinae RGC layer counts were 11.1 ± 0.5 for the ischemia + 48 h later post-C group and 10.8 ± 1.0 for the ischemia + 48 h later sham post-C group.
DISCUSSION
Our results confirmed the hypothesis that delayed post-conditioning could ameliorate the damage after retinal ischemia. As a novel strategy that could potentially protect against retinal ischemia, post-C shifts the focus of neuroprotection away from the paradigm of pre-ischemic intervention to manipulations that alter the post-ischemic state in a manner that is favorable to survival of retinal cells. In the present study we demonstrated the new finding that delayed application of a brief ischemic stimulus afforded significant neuroprotection after retinal ischemia. However, the time window of effectiveness of post-C was limited to 24 h, because no significant effect of post-conditioning was demonstrated at 48 h after ischemia.
In this study, the most apparent effect of delayed post-conditioning 24 h after ischemia was found in the inner retina, as reflected by dramatically improved recovery of the b-wave, P2, and oscillatory potentials, and decreased loss of cells in the retinal ganglion cell layer and diminished infiltration of inflammatory cells, while recovery of the a-wave showed a non-significant trend for improvement with delayed post-C. The reason for the relative enhancement of inner retinal function may be due to the nature of the ischemia produced in this model. There is relative sparing of the outer retina, perhaps due to residual blood supply from the choroidal circulation or decreased sensitivity to ischemia of photoreceptor cells (Hughes 1991). Despite the improved outcome after ischemia with 24 h delayed post-C, post-C 48 h later only produced a non-significant trend for improvement of the oscillatory potentials. This suggests there is a limited time window for effectiveness of post-C.
Our findings of the effectiveness of post-C as late as 24 h after ischemia are unique compared to earlier studies of post-conditioning in the central nervous system. With respect to cerebral ischemia, nearly all studies have examined post-C using a brief ischemic stimulus in only the first minutes after the onset of reperfusion, where it was effective in reducing infarct size and disturbed behavioral or motor function in rats (Gao et al. 2008; Pignataro et al. 2008; Jiang et al. 2009). In one study (Ren et al. 2008) delayed post-conditioning 6 h after cerebral ischemia in a mouse model reduced infarct size. The latest time window for post-conditioning shown to date was where hypoxic post-conditioning, a different type of stimulus than brief ischemia, 5 days after focal cerebral ischemia in mice reduced thalamic atrophy, but it did not alleviate ischemia-induced functional deficits (Leconte et al. 2009). Yet in our study, ischemic post-C 24h after ischemia significantly improved both retinal function and decreased cellular loss, thereby demonstrating an even more profound neuroprotective effect.
In a previous study, retinal post-C was found to reduce ischemia-induced apoptosis (Dreixler et al. 2010), and post-C was found to require new protein synthesis (Fernandez et al. 2009; Dreixler et al. 2010) Recently, we reported that post-C requires mitogen-activated protein kinase p38α and protein kinase B, or Akt (Dreixler et al. 2011b). Apart from these findings, further investigation is required to understand the mechanisms of post-C in the retina.
Our results differ from those of others. Previously, it was reported that post-C at 24 h after retinal ischemia in rats was ineffective in altering outcome (Fernandez et al. 2009). A potentially important difference in the studies is that we used a total occlusion of the central retinal artery with a suture placed behind the globe as the post-C stimulus. When intraocular pressure is elevated to produce ischemia, some residual blood flow remains in the retinal or choroidal circulation (Roth and Pietrzyk 1994; Lin and Roth 1999). It is possible that the depth of the ischemic stimulus for post-C is related to its effectiveness, similar to our demonstration that post-C is more effective following prolonged compared to shorter periods of damaging ischemia (Dreixler et al. 2010). In any event, our study demonstrates the feasibility of a brief post-conditioning ischemic stimulus to ameliorate damage as late as 24 h after ischemic insult. This stimulus could possibly be applied to patients who have sustained central retinal vascular occlusion or similar events where the outcome is often poor (Chen and Lee 2008), and may be an effective supplement or substitute to treatment attempts such as thrombolysis.
Table 2.
Number of retinal ganglion cells per region of interest (mean +/− s.e.m.) in the ischemic retinae 7 days after 48h Sham Post-C compared to the number of retinal ganglion cells in the ischemic retinae for 48h Post-C.
| Number of RGCs |
p-value vs. Ischemia |
|
|---|---|---|
| 48h Sham Post-C | 5.8 ± 0.5 | --- |
| 48h Post | 6.5 ± 0.8 | 0.47 |
Acknowledgments
Supported by National Institutes of Health (Rockville, MD) grants EY10343-15S1 and -16S1 (American Recovery and Reinvestment Act) to Dr Roth, AG029795-02 for the Medical Student Summer Research Program at the Pritzker School of Medicine, UL1RR024999 to the University of Chicago Institute for Translational Medicine; the Illinois Society for the Prevention of Blindness (Chicago, IL); France and Chicago Collaborating in the Sciences (The Chicago-France Center, Chicago, IL); and the Dean’s Research Advisory Committee of the Division of Biological Sciences of the University of Chicago. Jacqueline N. Poston was the recipient of a Medical Student Research Fellowship Award from the American Academy of Neurology, St Paul, MN.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
There is no conflict of interest or commercial interest for any of the authors.
References
- Bui BV, Edmunds B, Cioffi GA, Fortune B. The gradient of retinal functional changes during acute intraocular pressure elevation. Invest Ophthalmol Vis Sci. 2005;46:202–213. doi: 10.1167/iovs.04-0421. [DOI] [PubMed] [Google Scholar]
- Chen CS, Lee AW. Management of acute central retinal artery occlusion. Nature Clin Pract Neurol. 2008;4:376–383. doi: 10.1038/ncpneuro0811. [DOI] [PubMed] [Google Scholar]
- Dreixler J, Shaikh A, Alexander M, Savoie B, Roth S. Post-ischemic conditioning in the rat retina is dependent upon ischemia duration and is not additive with ischemic preconditioning. Exp Eye Res. 2010;91:844–852. doi: 10.1016/j.exer.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreixler JC, Bratton A, Du E, Shaikh AR, Marcet MM, Roth S. Mitogen activated protein kinase phosphatase-1 (MKP-1) in retinal ischemic preconditioning. Exp Eye Res. 2011a doi: 10.1016/j.exer.2010.10.011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreixler JC, Hagevik S, Hemmert JW, Shaikh AR, Rosenbaum DM, Roth S. Involvement of erythropoietin in retinal ischemic preconditioning. Anesthesiology. 2009a;110:774–780. doi: 10.1097/ALN.0b013e31819c4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreixler JC, Hemmert JW, Shenoy SK, Shen Y, Lee HT, Shaikh AR, Rosenbaum DM, Roth S. The role of Akt/protein kinase B subtypes in retinal ischemic preconditioning. Exp Eye Res. 2009b;88:512–521. doi: 10.1016/j.exer.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreixler JC, Sampat A, Shaikh AR, Alexander MJ, Marcet MM, Roth S. Protein kinase B (Akt) and mitogen activated protein kinase p38α in retinal ischemic post-conditioning. J Mol Neurosci. 2011b doi: 10.1007/s12031-011-9523-5. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreixler JC, Shaikh AR, Shenoy SK, Shen Y, Roth S. Protein kinase C subtypes and retinal ischemic preconditioning. Exp Eye Res. 2008;87:300–311. doi: 10.1016/j.exer.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faberowski N, Stefansson E, Davidson RC. Local hypothermia protects the retina from ischemia. A quantitative study in the rat. Invest Ophthalmol Vis Sci. 1989;30:2309–2313. [PubMed] [Google Scholar]
- Fernandez DC, Bordone MP, Chianelli MS, Rosenstein RE. Retinal neuroprotection against ischemia-reperfusion damage induced by postconditioning. Invest Ophthalmol Vis Sci. 2009;50:3922–3930. doi: 10.1167/iovs.08-3344. [DOI] [PubMed] [Google Scholar]
- Gao X, Zhang H, Takahashi T, Hsieh J, Liao J, Steinberg GK, Zhao H. The Akt signaling pathway contributes to postconditioning's protection against stroke; the protection is associated with the MAPK and PKC pathways. J Neurochem. 2008;105:943–955. doi: 10.1111/j.1471-4159.2008.05218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS, Kolder HE, Weingeist TA. Central retinal artery occlusion and retinal tolerance time. Ophthalmology. 1980;87:75–78. doi: 10.1016/s0161-6420(80)35283-4. [DOI] [PubMed] [Google Scholar]
- Hughes WF. Quantitation of ischemic damage in the rat retina. Exp Eye Res. 1991;53:573–582. doi: 10.1016/0014-4835(91)90215-z. [DOI] [PubMed] [Google Scholar]
- Jiang X, Ai C, Shi E, Nakajima Y, Ma H. Neuroprotection against spinal cord ischemia-reperfusion injury induced by different ischemic postconditioning methods: roles of phosphatidylinositol 3-kinase-Akt and extracellular signal-regulated kinase. Anesthesiology. 2009;111:1197–1205. doi: 10.1097/ALN.0b013e3181bf1d93. [DOI] [PubMed] [Google Scholar]
- Leconte C, Tixier E, Freret T, Toutain J, Saulnier R, Boulouard M, Roussel S, Schumann-Bard P, Bernaudin M. Delayed hypoxic postconditioning protects against cerebral ischemia in the mouse. Stroke. 2009;40:3349–3355. doi: 10.1161/STROKEAHA.109.557314. [DOI] [PubMed] [Google Scholar]
- Limalanathan S, Andersen GO, Hoffmann P, Klow N-E, Abdelnoor M, Eritsland J. Rationale and design of the POSTEMI (postconditioning in ST-elevation myocardial infarction) study. Cardiology. 2010;116:103–109. doi: 10.1159/000316965. [DOI] [PubMed] [Google Scholar]
- Lin J, Roth S. Retinal hypoperfusion after ischemia in rats: Attenuation by ischemic preconditioning. Invest Ophthalmol Vis Sci. 1999;40:2925–2931. [PubMed] [Google Scholar]
- Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, Gala R, Simon RP. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. [erratum appears in J Cereb Blood Flow Metab. 2008 Feb;28(2):440 Note: Gala, Rosaria [added]] J Cereb Blood Flow Metabol. 2008;28:232–241. doi: 10.1038/sj.jcbfm.9600559. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Scorziello A, Di Renzo G, Annunziato L. Post-ischemic brain damage: effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. FEBS J. 2009;276:46–57. doi: 10.1111/j.1742-4658.2008.06769.x. [DOI] [PubMed] [Google Scholar]
- Ren C, Gao X, Niu G, Yan Z, Chen X, Zhao H. Delayed postconditioning protects against focal ischemic brain injury in rats. [Erratum appears in PLoS ONE. 2009;4(2). doi: 10.1371/annotation/bbfdac40-32cc-4c3a-a049-436796875bf4.] PLoS ONE [Electronic Resource] 2008;3:e3851. doi: 10.1371/journal.pone.0003851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, Dreixler JC, Shaikh AR, Lee KH, Bindokas V. Mitochondrial potassium ATP channels and retinal ischemic preconditioning. Invest Ophthalmol Vis Sci. 2006;47:2114–2124. doi: 10.1167/iovs.05-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, Li B, Rosenbaum PS, Gupta H, Goldstein IM, Maxwell KM, Gidday JM. Preconditioning provides complete protection against retinal ischemic injury in rats. Invest Ophthalmol Vis Sci. 1998;39:775–785. [PubMed] [Google Scholar]
- Roth S, Pietrzyk Z. Blood flow after retinal ischemia in cats. Invest Ophthalmol Vis Sci. 1994;35:3209–3217. [PubMed] [Google Scholar]
- Wang J-y, Shen J, Gao Q, Ye Z-g, Yang S-y, Liang H-w, Bruce IC, Luo B-y, Xia Q. Ischemic postconditioning protects against global cerebral ischemia/reperfusion-induced injury in rats. Stroke. 2008;39:983–990. doi: 10.1161/STROKEAHA.107.499079. [DOI] [PubMed] [Google Scholar]
- Weymouth AE, Vingrys AJ. Rodent electroretinography: methods for extraction and interpretation of rod and cone responses. Prog Retinal & Eye Res. 2008;27:1–44. doi: 10.1016/j.preteyeres.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Xing B, Chen H, Zhang M, Zhao D, Jiang R, Liu X, Zhang S. Ischemic post-conditioning protects brain and reduces inflammation in a rat model of focal cerebral ischemia/reperfusion. J Neurochem. 2008;105:1737–1745. doi: 10.1111/j.1471-4159.2008.05276.x. [DOI] [PubMed] [Google Scholar]
- Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metabol. 2006;26:1114–1121. doi: 10.1038/sj.jcbfm.9600348. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Zhang Y, Ojwang BA, Brantley MA, Jr, Gidday JM. Long-term tolerance to retinal ischemia by repetitive hypoxic preconditioning: role of HIF-1alpha and heme oxygenase-1. Invest Ophthalmol Vis Sci. 2007;48:1735–1743. doi: 10.1167/iovs.06-1037. [DOI] [PubMed] [Google Scholar]