

Abstract

Protein kinases constitute an attractive family of enzyme targets with high relevance to cell and disease biology. Small molecule inhibitors are powerful tools to dissect and elucidate the function of kinases in chemical biology research and to serve as potential starting points for drug discovery. However, the discovery and development of novel inhibitors remains challenging. Here, we describe a structure-based de novo design approach that generates novel, hinge-binding fragments that are synthetically feasible and can be elaborated to small molecule libraries. Starting from commercially available compounds, core fragments were extracted, filtered for pharmacophoric properties compatible with hinge-region binding, and docked into a panel of protein kinases. Fragments with a high consensus score were subsequently short-listed for synthesis. Application of this strategy led to a number of core fragments with no previously reported activity against kinases. Small libraries around the core fragments were synthesized, and representative compounds were tested against a large panel of protein kinases and subjected to co-crystallization experiments. Each of the tested compounds was active against at least one kinase, but not all kinases in the panel were inhibited. A number of compounds showed high ligand efficiencies for therapeutically relevant kinases; among them were MAPKAP-K3, SRPK1, SGK1, TAK1, and GCK for which only few inhibitors are reported in the literature.
Phosphorylation is the most important and widespread covalent modification of proteins. It is used to control enzyme activity in cellular processes and thereby plays a major role in cell signaling and is fundamental to all aspects of cell behavior and organization.1 Protein kinases catalyze the transfer of the γ-phosphate group from ATP to recognized amino acids of proteins. Kinases have implications for many diseases including cancer, diabetes, and Alzheimer’s disease and constitute the second most exploited group of drug targets with many ongoing drug discovery efforts.2 Despite the extensive research over the past two decades, selective chemical tools are still needed to dissect the complex nature of kinase regulation.2,3
A wealth of structural information has revealed the general architecture of protein kinases, their binding sites, and complex regulation.4,5 The ATP-binding sites of most protein kinases share similar features (Figure 1a).6,7 A key recognition motive is the hinge region that forms hydrogen bonds to the adenine moiety of ATP and is targeted by many kinase inhibitors. Often, inhibitors also address one or both of the adjacent hydrophobic pockets I and II. These are more variable between different kinases than the hinge region, and the differences can be exploited to achieve selectivity.8
Figure 1.
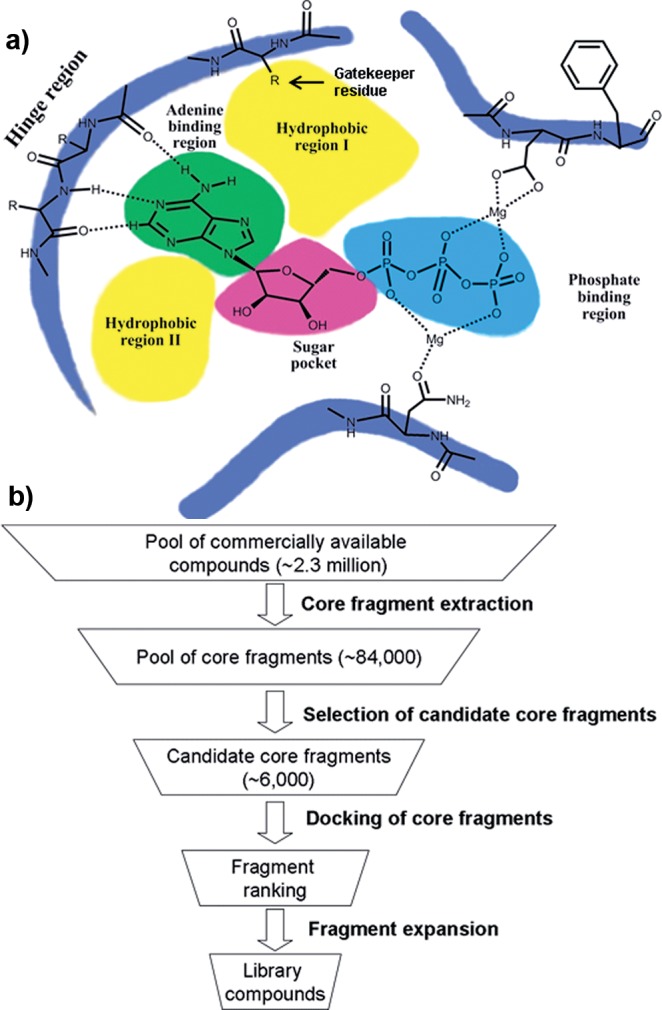
(a) ATP binding site of a typical protein kinase (adapted from ref (7)). (b) In silico screening cascade used to design novel kinase inhibitor libraries.
Kinase inhibitors are commonly discovered by high-throughput, virtual or fragment-based screening, often using compound libraries sourced from commercial suppliers.9−13 While successful in delivering hit compounds, they have only limited template diversity. In order to tackle this issue, various research groups have developed approaches to expand their libraries with proprietary compounds.14−20 Libraries that contained compounds with heterocycles, which have the potential to interact with the hinge region of the kinase binding site but no previous reported activity against kinases, were of particular high value.14−18 A difficulty in expanding the kinase libraries was to assess synthetic feasibility of the suggested compounds, especially if they contained novel cores.14,17
Here, we report on the structure-based de novo design of protein kinase inhibitors. The approach is centered on fragments that have precedence for synthesis but are not commercially available with the required substitution pattern. Libraries around six core fragments without previous reported activity against kinases were synthesized, and selected compounds were screened against a panel of 117 kinases. In addition, the crystal structure of one novel inhibitor in complex with cSrc was determined. Every tested compound was active against at least one kinase. While predicting general activity against kinases on a scaffold level was highly successful, predicting selectivity on a compound level failed. Ligand efficient inhibitors were identified for a number of kinases, which have implications in a range of diseases but for which only a few inhibitors have been reported to date.
Results and Discussion
Structure-Based Design of Novel Protein Kinase Inhibitor Libraries
An in silico screening cascade was established for the design of novel kinase inhibitor libraries (Figure 1b). This approach consisted of the following four principle steps: core fragment extraction out of commercially available compounds, selection of candidate core fragments, docking of core fragments, and fragment expansion.
A core fragment was defined as a ring system plus the directly attached heteroatom containing functional groups.11 Starting from over two million compounds, about 84,000 unique core fragments were extracted. In the next step these core fragments were filtered for fragment-like properties, the absence of unwanted functionalities, and limited complexity. The resulting 11,000 core fragments were subsequently filtered using a 3D pharmacophore to remove scaffolds that did not contain a hinge-binding motif (Supplementary Figure S1). About 6,000 core fragments that passed this filter step were docked into the binding sites of a panel of 46 different protein kinases (Supplementary Table S1). These proteins were chosen based on the availability in the MRC Protein Phosphorylation Unit in Dundee for compound testing and access to a crystal structure in the public domain.21 In order to make the docking scores comparable for the different kinases, they were normalized relative to the best score for any fragment in the active site of each kinase in a similar way as carried out previously.22 Following this approach the top 100 nonselective core fragments were visually inspected. The vast majority of these were predicted to form two or three hydrogen bonds with the kinase hinge region. Six out of the top scoring core fragments or fragments closely resembling these scaffolds had been co-crystallized with a kinase as part of a larger compound (Figure 2). Comparing the docked poses with the binding modes of these compounds confirmed that they were placed correctly in the binding site in at least one kinase of the docking panel (RMSD of maximum common substructure <2 Å). The novelty of the high-ranking hinge binders was assessed by using the core fragments as substructure queries for ChEMBL23 and SciFinder (Chemical Abstracts Service, Columbus, OH). It turned out that 73 of the top 100 nonselective core fragments were already reported in the literature as part of known kinase inhibitors. Six novel fragments out of the remaining 27 fragments were short-listed for library enumeration based on synthetic considerations (Figure 3). None of the selected core fragments was available as part of commercial compounds with either the required substitution pattern or the desired diversity at the time of the study.
Figure 2.

Docking poses of six high ranking core fragments (green carbon atoms) superimposed on crystallographically determined binding modes of ligands containing the same or a closely related core fragment (cyan carbon atoms). Putative hydrogen bonds to the hinge region are shown as dashed lines. RMSD values are given for the maximum common substructure between core fragment and ligand. (The binding sites are oriented as depicted in Figure 1a.)
Figure 3.

Predicted binding modes with respect to the hinge region for six high-ranking core fragments (A–F) for which binding to protein kinases was not reported in the literature. Only the most frequent binding mode for each core fragment is shown. The substitution points that target the hydrophobic pockets I and II and that have been selected for diversifying the cores are indicated as R1 and R2, respectively. (The binding sites are oriented as depicted in Figure 1a.)
In the final step, suitable substitution points to attach additional moieties to interact with the hydrophobic pockets I and II (Figure 3) were assigned. For all short-listed core fragments, more than one binding mode was predicted for the kinases in the docking panel. Typically, in the different orientations several hydrogen bonds with the hinge region were formed, but the interacting atoms of the cores differed. It was therefore ensured that after adopting these alternative binding modes the R-groups would still be placed into desired regions of the binding site. The libraries were enumerated using commercially available building blocks. The reaction products were filtered to remove molecules with unwanted functionalities and non-drug-like molecules according to Lipinski’s rule of five.24 A final selection was made based on diversity by visual inspection and in-house availability of the building blocks. Due to synthetic considerations, libraries containing the core fragments A and F were generated by varying either R1 or R2 (Supplementary Table S2, Figure 3), whereas for core fragments B and D R1 R2 were varied simultaneously. Core fragments C and E contained only one R-group for enumeration. In total, 265 compounds were short-listed for synthesis. All libraries were synthesized using parallel synthesis in a maximum of six steps. To speed up synthesis the synthetic routes were designed to introduce diversity as late as possible (Scheme 1), and 186 compounds representing all six core fragments were successfully prepared (yield 7–98%, purity >90%).
Scheme 1.

Inhibition Profiles of 15 Compounds against a Panel of 117 Protein Kinases
A subset of library compounds was tested against a panel of 117 protein kinases as proof of concept study (Figure 4). The compounds were chosen to cover all six core fragments and to be fragment-sized (MW < 300 Da), therefore having a higher probability of inhibiting a kinase compared to more elaborated library compounds with the potential draw-back of having only moderate binding affinity.25 Any compounds showing ≥75% inhibition at 100 μM were defined as active, between 40 and 75% as moderately active, and below 40% as inactive (Supplementary Table S3). According to these definitions, all assayed compounds were at least moderately active against at least one kinase (Figure 5a). The most selective compounds were E1 and F2, which both inhibited only one kinase with >40% inhibition (MSK1 and MKK1, respectively). The least selective compounds were B1 and A2, which inhibited 63 and 55 different kinases with >40% inhibition, respectively.
Figure 4.

Structures of 15 compounds that were screened against a panel of 117 protein kinases. Core fragments are marked in blue.
Figure 5.
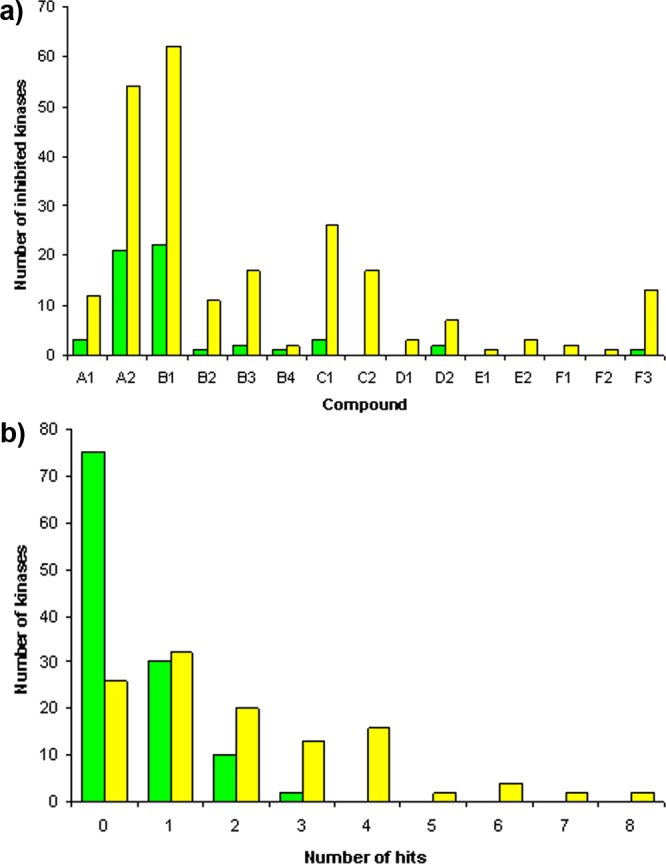
(a) Bar chart showing the number of kinases inhibited to at least 40% (yellow bars) or 75% (green bars) based on 15 compounds tested against a panel of 117 kinases. (b) Bar chart showing how many kinases were inhibited by how many compounds to at least 40% (yellow bars) or 75% (green bars) based on 15 compounds tested against a panel of 117 kinases.
The kinases presented in the kinase panel were not equally inhibited by the 15 compounds selected to present the different core fragments (Figure 5b, Supplementary Tables S3 and S4). Using a ≥40% inhibition cutoff value, 26 kinases (22%) in the panel were not inhibited by any of the tested compounds. When a cutoff of ≥75% inhibition was used, this number increased to 75 (64%).
Potency Determinations for Selected Compounds
Six compounds (A1, A2, B1, C2, D1, and F3) with inhibition of >50% for certain kinases were selected for IC50 determinations (Table 1). Activity for all compounds was confirmed with IC50 values ranging from 4 to 470 μM. Additionally, all compounds were characterized by high ligand efficiencies (>0.30 kcal/mol heavy atom)26 for at least one protein kinase, with B1 showing the best ligand efficiencies across a range of kinases.
Table 1. IC50 Values and Ligand Efficiencies for Compounds A1, A2, B1, C2, D1, and F3 for Selected Protein Kinases.
| compound | kinase | IC50 [μM]a | LE [kcal/mol heavy atom] |
|---|---|---|---|
| A1 | CK2 | 18 | 0.33 |
| MAPKAP-K3 | 21 | 0.33 | |
| RSK2 | 196 | 0.26 | |
| Src | 97 | 0.28 | |
| SRPK1 | 114 | 0.28 | |
| A2 | Aurora A | 188 | 0.26 |
| CDK2 | 26 | 0.33 | |
| CK2 | 131 | 0.27 | |
| CLK2 | 25 | 0.32 | |
| FGF-R1 | 252 | 0.25 | |
| GCK | 4 | 0.38 | |
| IGF-1R | 47 | 0.30 | |
| TAK1 | 24 | 0.32 | |
| B1 | CHK2 | 17 | 0.42 |
| GCK | 6 | 0.46 | |
| HER4 | 20 | 0.41 | |
| IGF-1R | 25 | 0.40 | |
| Src | 20 | 0.41 | |
| TAK1 | 11 | 0.43 | |
| VEG-FR | 39 | 0.39 | |
| YES1 | 21 | 0.41 | |
| C2 | GSK-3β | 25 | 0.32 |
| SGK1 | 110 | 0.28 | |
| D1 | EPH-B3 | 158 | 0.33 |
| FGF-R1 | 469 | 0.29 | |
| F3 | PIM1 | 22 | 0.38 |
| PIM3 | 14 | 0.40 |
Hill slopes range from 0.7 to 1.3.
Confirmation of Binding Mode
The binding mode of B1 in complex with cSrc was determined using X-ray crystallography. In the most frequent binding mode that was generated for core fragment B in the kinases of the docking panel the exocyclic amino group pointed toward the gatekeeper and the nitrogen atom bridging the heterocycles was placed next to the hinge region (Figure 3). This orientation closely matches the one found in the cSrc-B1 complex structure (Figure 6, Supplementary Table S5, rmsd = 0.77 Å for non-hydrogen atoms).
Figure 6.
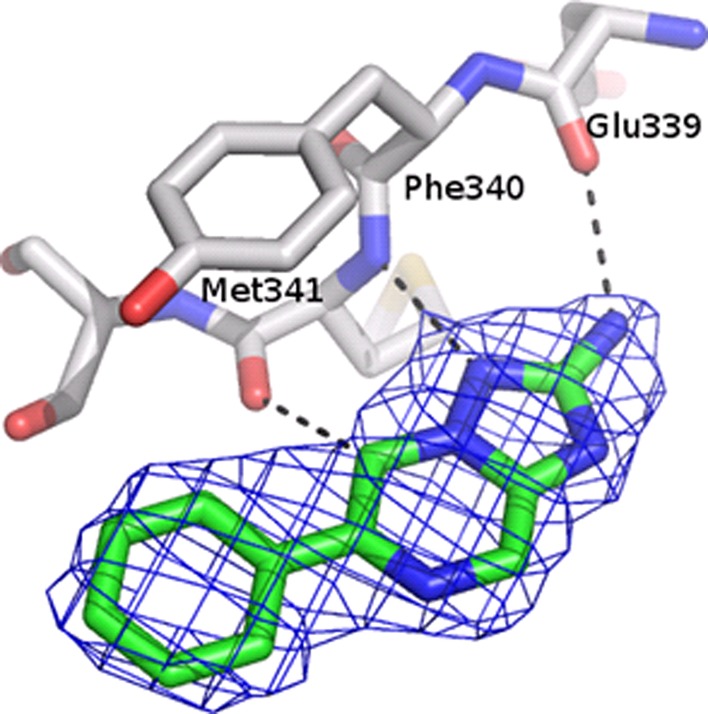
Crystallographically determined binding mode of B1 in complex with cSrc together with electron density map (2Fo – Fc contoured at 1σ). The fragment binds to the hinge region of the kinase domain. The binding site is oriented as depicted in Figure 1a. Putative hydrogen bonds are indicated as dashed lines. PDB code: 4fic.
Comparison of Predicted and Observed Inhibition Profiles
To compare the modeling results with the experimental inhibition data, we docked the 15 compounds that were profiled in our kinase panel (Figure 4) into the binding site of the kinases in the docking panel (Supplementary Table S1). The rank obtained for the least favorable scoring inhibitor among the 15 tested compounds for each kinase was compared to the number of identified hits for that target (Supplementary Table S6). If docking performance was perfect, both numbers would be identical (e.g., if two hits were identified for a kinase the ligand with the lowest score should have rank 2 with the other ligand being on rank 1). While for some targets the worst rank corresponded closely to the number of identified hits, for most targets separation between active and inactive compounds was not possible, both when a cutoff value of ≥40% (data not shown) or ≥75% inhibition was used. Similarly, docking failed to predict the selectivity profile of the inhibitors (Supplementary Table S7). For this comparison, the scores for all 15 compounds docked into all 46 protein structures were normalized relative to the best score for any fragment in the active site of each kinase obtained when docking the library of candidate core fragments. Compounds were considered to be predicted as active if their normalized score was ≥1.0 (equal or better than the best score for a core fragment in a particular kinase). At the chosen cutoff level, there was only little agreement between predicted and confirmed activity. For instance, compound B1, the least selective ligand in the panel, was predicted to be active for only nine out of the 31 kinases for which activity was found. In contrast, E1 and F2, the most selective compounds in the panel were predicted to bind to 10 and 21 kinases, respectively. However, neither compound demonstrated activity against these kinases in a biochemical assay. The only compound for which the profile was predicted correctly was B4, which did not inhibit any kinase in the docking panel.
Overall, the chosen de novo design strategy (Figure 1b) was highly successful. Starting with a database of core fragments, six scaffolds for which activity against kinases was not reported previously were identified, and synthetic routes were developed to prepare focused libraries around each core. All tested compounds were active against at least one kinase (Supplementary Table S3). We attribute this success to the following reasons: (1) Focusing on core fragments that occur in commercially available compounds but are not available with the required substitution pattern ensured that the libraries were synthetically accessible and at the same time novel. (2) Exploiting the wealth of kinase crystal structures when short-listing the cores canceled differences in the kinase binding sites and increased the chances that highly ranked cores would actually bind to a kinase when part of a larger compound.
Evaluating a subset of the library compounds in a large kinase panel confirmed some previous findings but also highlighted differences (Supplementary Table S4). For instance, we deliberately concentrated our synthetic efforts on predicted unselective fragments. It turned out that derivatives of each core displayed distinct selectivity profiles (Figure 5). This observation is in tune with the notion that kinase selectivity often does not stem from the core itself but from its substituents.13,27 Further, we observed inhibition of PIM1 and PIM3 (Table 1). These kinases possess an altered hinge binding region incompatible with forming the hydrogen-bonding interactions typically observed in kinase-ligand complexes (Figure 1a).28 It was speculated that interactions with different amino acids in the binding site require a similar pharmacophore as the interactions with the hinge region that presumably lead to high hit rates of fragment-like kinase inhibitors for these atypical kinases.13 Our data confirms this hypothesis. However, Posy et al.(27) and Bamborough et al.(29) reported low hit rates for ASK1 (0 and 0.2%, respectively). In contrast, by testing just 15 compounds against this kinase we found two hits containing two different scaffolds. Anastassiadis et al.(30) found no hits for NEK6 and MAPKAP-K3, whereas in our exercise two inhibitors containing the same scaffold and seven inhibitors containing three different scaffolds were discovered, respectively. One of the MAPKAP-K3 inhibitors had an IC50 value of 21 μM and a ligand efficiency of 0.33 kcal/mol heavy atom (Table 1). The previous studies had concluded that these kinases were less tractable. Our data contradict these findings, which is another warning that profiling data can be interpreted only in relation to the chemical space covered by the screening library.
Molecular docking was very successful when predicting binding modes of fragments and general activity against kinases on a core fragment-level but less so when individual compounds for individual kinases were considered. Six high ranking core fragments had been co-crystallized with kinases as part of more elaborated compounds (Figure 2). All of the observed binding modes for the core fragments were among the ones generated in the kinase docking panel. Also, the binding mode of B1 in cSrc was predicted correctly (Figure 6). Remarkably, 73 out of the top 100 ranked core fragments had activity against kinases reported in the literature. This unusually high enrichment was presumably caused by exclusively docking compounds that fulfilled a pharmacophore required for hinge-binding fragments (Supplementary Figure S1a) and by using consensus scoring across a panel of kinase structures (Supplementary Table S1). This success provided confidence that also the remaining high ranking fragments for which no activity against kinases was reported would be suitable hinge-binding scaffolds. At least for the selected core fragments that were chosen for library synthesis and for which examples were tested in a kinase panel this turned out to be the case. When trying to predict the selectivity profiles of the tested library compounds, docking failed to give useful guidance (Supplementary Table S7). This failure is driven by the inability to correctly rank the tested compounds according to their affinity for given kinases (Supplementary Table S6). Small changes, such as adding a nitrogen atom to the heterocycles (E1 vs F1, Figure 4), which change the selectivity of the compounds are not accurately captured by the used scoring function, and furthermore discrimination between more drastic changes, such as ranking structurally unrelated compounds, failed in most cases. In addition, predicting the effect of the subtle differences in the kinase binding sites on compound affinity remains a challenge.14 In the past, more promising results were obtained by using structural and inhibition data to train predictors.31−34 However, due to their nature, these approaches do not allow to make predictions for new compound classes or kinases.
A number of compounds were chosen for IC50 determinations against selected kinases (Table 1). All tested compounds were characterized by high ligand efficiencies (≥0.30 kcal/mol heavy atom) for at least one kinase, rendering them in general promising starting points for drug discovery for a range of targets with high medical interest.26 Among others, compounds with high ligand efficiencies were identified for MAPKAP-K3, SRPK1, SGK1, TAK1, and GCK (Table 1). For all of these kinases less than 30 compounds with affinities ≤100 μM are reported in ChEMBL Kinase SARfari (as of April 2012).23 They are thought to be targets for the treatment of cancer, systemic inflammation response syndrome, rheumatoid arthritis, other inflammatory diseases, and hypertension.35−39 The hits identified by our in silico study can serve as starting points to further explore these targets.
Summary and Conclusion
A de novo design approach for novel kinase inhibitors was established that was centered on core fragments having precedence for synthesis but no reported activity against kinases. Using a wide range of methods at the interface of chemistry and biology, e.g., structure-based design, chemical synthesis, X-ray crystallography and biological profiling, the approach was validated. Six small molecule libraries were synthesized. Selected compounds were profiled in a large kinase panel, and dose–response curves were determined. In addition, the binding mode of one compound was confirmed using X-ray crystallography. Whereas predicting binding modes and general activity against kinases was very reliable, correctly predicting selectivity solely based on docking to crystal structures failed. Nevertheless, this study demonstrated the overall success of the chosen design strategy. In addition, a number of inhibitors with good binding efficiency were identified for a range of kinases with therapeutic relevance and can now serve as starting points for chemical tools to further explore these targets.
Methods
Core Fragment Extraction
Core fragments were derived from an in-house database containing more than two million commercially available compounds using a method described previously.11 In brief, core fragments were defined as ring atoms plus the atoms of directly attached polar functional groups. Polar functional groups were specified as polar hetero atoms (S, N, P, or O) and polar hetero atoms double or triple bonded to carbon atoms or linked to other polar hetero atoms or carbonyl groups. Functional groups linking ring atoms were added to both resulting scaffolds. Fragments containing only carbon atoms or carbon atoms and aromatic sulfur or oxygen atoms were disregarded.
Fragment Selection
Three filters were used to eliminate inappropriate or undesirable scaffolds from the generated database of core fragments. First, fragments that contained unwanted functionalities as described previously were removed.11 Second, fragments that did not comply with a modified “Rule of Three” were removed from the database.40 Here, only ring fragments with MW < 300 Da, number of hydrogen bond donors ≤3, number of hydrogen bond acceptors ≤6, and clogP ≤ 3 were retained. Additionally, core fragments with the total charge ≤ −1 or ≥1 were removed. Finally, compounds containing more than two fused rings or more than six rotatable bonds were rejected.
Pharmacophore Search
The 3D pharmacophore search was carried out using the UNITY tool available with the SYBYL-X 1.0 package (Tripos Inc.). The default file sln3d_macros.def that contained the definition of predefined feature types (macros) such as hydrogen-bond acceptor or hydrogen-bond donor was modified to also account for C–H···O hydrogen bonds as often observed in protein kinases (Supplementary Figure S1b). Otherwise, default settings were used when converting the filtered core fragments to a 3D UNITY database.
The pharmacophore was generated based on the crystal structure of CDK2 in complex with an imidazole piperazine inhibitor (PDB code 2w05). The resulting pharmacophore model was made up of four features (Supplementary Figure S1a). One hydrogen-bond acceptor and two hydrogen-bond donor features (radius 0.6 Å) were defined to interact with the hinge region. To consider the directionality of the hydrogen bonds, the features were linked with their hydrogen-bond binding partners in the protein. The aromatic feature (radius 1.5 Å) was added to take into account that the vast majority of protein kinase inhibitors contain an aromatic ring system that is placed within the adenine binding region.14 To fulfill the pharmacophore, all hits were required to contain the aromatic feature and two out of the possible three hydrogen-bonding features.
Receptor Preparation
Crystal structures of protein kinases that were available in the MRC Protein Phosphorylation Unit, University of Dundee, for profiling at the beginning of the study were downloaded from the PDB (Supplementary Table S1). All crystal structures representing these kinases exhibit the DFG-in conformation.6 The structures were aligned with CDK2 (PDB code 2w05) using the hinge region as matching atoms. Hydrogen atoms of polar groups were added using MOLOC (Gerber Molecular Design), and their positions were minimized with the MAB force field as implemented in MOLOC.41 Subsequently, all nonprotein atoms were removed from the crystal structures. Compounds were placed into the binding site and scored using DOCK 3.5.54.42,43 The required sphere set to define the region with a low dielectric constant was composed of the bound ligands’ atoms. If necessary, the set was manually modified to cover the entire buried part of binding site. The sphere set used as matching points for docking was manually generated by placing matching atoms into the adenine binding region close to the hinge region. The same set was used for all protein kinases. To favor interactions between ligand and hinge region, partial charges of the carbonyl oxygen atoms in the amide backbone of the hinge region were decreased by 0.4, and the partial charge of the amide hydrogen atom was increased by 0.4 (Figure S1a). The total charge was then balanced by distributing the charge difference among the remaining hinge region atoms. Grids to store information about excluded volumes, van der Waals potential, and ligand desolvation were calculated as described previously.44,45
Ligand Preparation
Tautomers, stereoisomers, and protonation states were enumerated and converted to a suitable database format as described previously.44
Docking Protocol
Multiple conformations and orientations of each ligand were docked into the kinase binding sites using DOCK 3.5.54.42,43 Ligand and receptor overlap bins were set to 0.4 Å. Distance tolerance for matching ligand atoms to receptor was set to 1.2 Å. All fragments were docked into the ATP binding pocket, and each docking orientation was filtered for steric fit. Only fragments that were able to pass the steric fit filter were scored for electrostatic, van der Waals, and desolvation energies. The best scoring conformation and representative (tautomer, protonation state) of each compound was used for final ranking.
To compare fragments between different kinases, the originally calculated energy scores were normalized by the formula Sijnorm = Sij/Sjbest where Sijnorm is the normalized score for fragment i in the active site of kinase j, Sij is the original score, and Sjbest is the best score for any ligand in the active site of the kinase j. All negative Sijnorm scores were set to zero. Thus, Sijnorm = 1 if the fragment scores best of all docked fragments for a particular kinase, and Sijnorm = 0 if it scores worst.
Library Enumeration
The reagents were selected through a web interface based on Pipeline Pilot (Accelrys Software Inc.). Only in-house reagents that were commercially available were used for the enumeration process. Enumeration of libraries was performed using the “core plus R-groups” method from Pipeline Pilot which was again presented through a customized web interface. All reaction products were filtered to remove molecules with unwanted functionalities, >5 hydrogen-bond donors, >10 hydrogen bond acceptors, molecular weight >500 Da, clogP > 5, and polar surface area >140 Å. Finally, all remaining reaction products were docked into the binding site of three kinases (CSK, RSK1, and GSK3β) to exclude those compounds which did not sterically fit into a typical ATP binding site.
Chemistry
All synthesized compounds had a purity of greater than 90% (measured on analytical HPLC-MS system). The synthetic details for the library compounds together with M+ and 1H NMR data to confirm compound identity and purity are listed in the Supporting Information.
Kinase Assay
Selected compounds were screened against a panel of mammalian kinases routinely run in the MRC Protein Phosphorylation Unit at the University of Dundee (www.kinase-screen.mrc.ac.uk).21 Compounds were supplied in DMSO and screened in duplicates at 100 μM concentration using a radioactive (33P-ATP) filter-binding-assay. For hit validation and all subsequent IC50 determinations, selected compounds were solubilized in DMSO at a top concentration of 51 mM and serially diluted to achieve 10-point titration of final assay concentrations from 1 mM to 30 nM. All IC50 determination assays were done in duplicate and the IC50 values were calculated using Prism (GraphPad Software, Inc.). All biochemical assays were run below the Kmapp for the ATP for each enzyme allowing comparison of inhibition across the panel.
Crystallization and Structure Determination of cSrc-B1
Fragment B1 was co-crystallized with cSrc using conditions similar to those previously reported by Michalczyk et al.(46) Diffraction data of the cSrc-B1 complex crystals were collected at the PX10SA beamline of the Swiss Light Source (PSI, Villingen, Switzerland) to a resolution of 2.5 Å, using wavelengths close to 1 Å. The data set was processed and refined as described previously.46 Detailed data, refinement, and Ramachandran statistics are provided in Supplementary Table S5.
Acknowledgments
This work was supported by the Welcome Trust (WT083481) and through a CASE studentship for R.U., jointly funded by MSD&Schering-Plough and the Biotechnology and Biological Sciences Research Council. N.T.-L. was supported by a fellowship from the German Academic Exchange Service (DAAD). We thank OpenEye for free software licenses, J. Downward (University of Dundee) for administration of the workstations, A. Hinton (MSD&Schering-Plough, Newhouse, U.K.) for help with Pipeline Pilot, and the staff in the MRC Protein Phosphorylation Unit at the University of Dundee for carrying out the kinase assays. D.R. is grateful for funds by the German Federal Ministry for Education and Research, which supported this work through the German National Genome Research Network-Plus (NGFNPlus) (Grant No. BMBF 01GS08104).
Supporting Information Available
Additional tables with percent inhibition data for the discussed compounds, hit rates in various kinase screens, and data collection and refinement details for the crystal structure of cSrc-B1; a figure of the pharmacophore used for virtual screening; the synthetic routes to the described compounds; NMR spectra of key compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The crystal structure of cSrc in complex with B1 has been deposited into the PDB with the code 4fic.
The authors declare no competing financial interest.
Supplementary Material
References
- Cohen P. (2002) Protein kinases—the major drug targets of the twenty-first century?. Nat. Rev. Drug. Discovery 1, 309–315. [DOI] [PubMed] [Google Scholar]
- Cohen P.; Alessi D. R. (2013) Kinase Drug Discovery - What’s Next in the Field?. ACS Chem. Biol. 8, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp S.; Arruda P.; Blagg J.; Burley S.; Drewry D. H.; Edwards A.; Fabbro D.; Gillespie P.; Gray N. S.; Kuster B.; Lackey K. E.; Mazzafera P.; Tomkinson N. C.; Willson T. M.; Workman P.; Zuercher W. J. (2013) A public-private partnership to unlock the untargeted kinome. Nat. Chem. Biol. 9, 3–6. [DOI] [PubMed] [Google Scholar]
- Rabiller M.; Getlik M.; Klüter S.; Richters A.; Tückmantel S.; Simard J. R.; Rauh D. (2010) Proteus in the world of proteins: conformational changes in protein kinases. Arch. Pharm. (Weinheim) 343, 193–206. [DOI] [PubMed] [Google Scholar]
- Eswaran J.; Knapp S. (2010) Insights into protein kinase regulation and inhibition by large scale structural comparison. Biochem. Biophys. Acta, Proteins Proteomics 1804, 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J. J. (2007) Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 50, 409–424. [DOI] [PubMed] [Google Scholar]
- Traxler P. (1998) Tyrosine kinase inhibitors in cancer treatment (part II). Expert Opin. Ther. Pat. 8, 1599–1625. [Google Scholar]
- Fang Z.; Grütter C.; Rauh D. (2013) Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem. Biol. 8, 58–70. [DOI] [PubMed] [Google Scholar]
- Stahura F. L.; Xue L.; Godden J. W.; Bajorath J. (1999) Molecular scaffold-based design and comparison of combinatorial libraries focused on the ATP-binding site of protein kinases. J. Mol. Graph. Model. 17(1–9), 51–52. [DOI] [PubMed] [Google Scholar]
- Prien O. (2005) Target-family-oriented focused libraries for kinases--conceptual design aspects and commercial availability. ChemBioChem 6, 500–505. [DOI] [PubMed] [Google Scholar]
- Brenk R.; Schipani A.; James D.; Krasowski A.; Gilbert I. H.; Frearson J.; Wyatt P. G. (2008) Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 3, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decornez H.; Gulyas-Forro A.; Papp A.; Szabo M.; Sarmay G.; Hajdu I.; Cseh S.; Dorman G.; Kitchen D. B. (2009) Design, selection, and evaluation of a general kinase-focused library. ChemMedChem 4, 1273–1278. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Brown M. J.; Christopher J. A.; Chung C. W.; Mellor G. W. (2011) Selectivity of kinase inhibitor fragments. J. Med. Chem. 54, 5131–5143. [DOI] [PubMed] [Google Scholar]
- Akritopoulou-Zanze I.; Hajduk P. J. (2009) Kinase-targeted libraries: the design and synthesis of novel, potent, and selective kinase inhibitors. Drug Discovery Today 14, 291–297. [DOI] [PubMed] [Google Scholar]
- Erickson J. A.; Mader M. M.; Watson I. A.; Webster Y. W.; Higgs R. E.; Bell M. A.; Vieth M. (2010) Structure-guided expansion of kinase fragment libraries driven by support vector machine models. Biochem. Biophys. Acta, Proteins Proteomics 1804, 642–652. [DOI] [PubMed] [Google Scholar]
- Vieth M.; Erickson J.; Wang J.; Webster Y.; Mader M.; Higgs R.; Watson I. (2009) Kinase inhibitor data modeling and de novo inhibitor design with fragment approaches. J. Med. Chem. 52, 6456–6466. [DOI] [PubMed] [Google Scholar]
- Kettle J. G.; Ward R. A. (2010) Toward the comprehensive systematic enumeration and synthesis of novel kinase inhibitors based on a 4-anilinoquinazoline binding mode. J. Chem. Inf. Model. 50, 525–533. [DOI] [PubMed] [Google Scholar]
- Baldwin I.; Bamborough P.; Haslam C. G.; Hunjan S. S.; Longstaff T.; Mooney C. J.; Patel S.; Quinn J.; Somers D. O. (2008) Kinase array design, back to front: biaryl amides. Bioorg. Med. Chem. Lett. 18, 5285–5289. [DOI] [PubMed] [Google Scholar]
- Deanda F.; Stewart E. L.; Reno M. J.; Drewry D. H. (2008) Kinase-targeted library design through the application of the PharmPrint methodology. J. Chem. Inf. Model. 48, 2395–2403. [DOI] [PubMed] [Google Scholar]
- Lowrie J. F.; Delisle R. K.; Hobbs D. W.; Diller D. J. (2004) The different strategies for designing GPCR and kinase targeted libraries. Comb. Chem. High Throughput Screen. 7, 495–510. [DOI] [PubMed] [Google Scholar]
- Bain J.; Plater L.; Elliott M.; Shpiro N.; Hastie C. J.; McLauchlan H.; Klevernic I.; Arthur J. S.; Alessi D. R.; Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozalbes R.; Simon L.; Froloff N.; Sartori E.; Monteils C.; Baudelle R. (2008) Development and experimental validation of a docking strategy for the generation of kinase-targeted libraries. J. Med. Chem. 51, 3124–3132. [DOI] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 23, 3–25. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Greer J. (2007) A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discovery 6, 211–219. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. (2004) Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Posy S. L.; Hermsmeier M. A.; Vaccaro W.; Ott K. H.; Todderud G.; Lippy J. S.; Trainor G. L.; Loughney D. A.; Johnson S. R. (2011) Trends in kinase selectivity: insights for target class-focused library screening. J. Med. Chem. 54, 54–66. [DOI] [PubMed] [Google Scholar]
- Jacobs M. D.; Black J.; Futer O.; Swenson L.; Hare B.; Fleming M.; Saxena K. (2005) Pim-1 ligand-bound structures reveal the mechanism of serine/threonine kinase inhibition by LY294002. J. Biol. Chem. 280, 13728–13734. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Drewry D.; Harper G.; Smith G. K.; Schneider K. (2008) Assessment of chemical coverage of kinome space and its implications for kinase drug discovery. J. Med. Chem. 51, 7898–7914. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. (2011) Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P. C.; De Sapio V.; Turner K. B.; Elmer S. P.; Roe D. C.; Schoeniger J. S. (2012) Identification of binding specificity-determining features in protein families. J. Med. Chem. 55, 1926–1939. [DOI] [PubMed] [Google Scholar]
- Subramanian G.; Sud M. (2010) Computational Modeling of Kinase Inhibitor Selectivity. ACS Med. Chem. Lett. 1, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E.; Mukherjee P. (2012) Kinase-kernel models: accurate in silico screening of 4 million compounds across the entire human kinome. J. Chem. Inf. Model. 52, 156–170. [DOI] [PubMed] [Google Scholar]
- Ma X. H.; Wang R.; Tan C. Y.; Jiang Y. Y.; Lu T.; Rao H. B.; Li X. Y.; Go M. L.; Low B. C.; Chen Y. Z. (2010) Virtual screening of selective multitarget kinase inhibitors by combinatorial support vector machines. Mol. Pharm. 7, 1545–1560. [DOI] [PubMed] [Google Scholar]
- Cohen P. (2009) Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 21, 317–324. [DOI] [PubMed] [Google Scholar]
- Sakurai H.; Miyoshi H.; Toriumi W.; Sugita T. (1999) Functional interactions of transforming growth factor beta-activated kinase 1 with IkappaB kinases to stimulate NF-kappaB activation. J. Biol. Chem. 274, 10641–10648. [DOI] [PubMed] [Google Scholar]
- Zhong J.; Gavrilescu L. C.; Molnar A.; Murray L.; Garafalo S.; Kehrl J. H.; Simon A. R.; Van Etten R. A.; Kyriakis J. M. (2009) GCK is essential to systemic inflammation and pattern recognition receptor signaling to JNK and p38. Proc. Natl. Acad. Sci. U.S.A. 106, 4372–4377. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ackermann T. F.; Boini K. M.; Beier N.; Scholz W.; Fuchss T.; Lang F. (2011) EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell. Physiol. Biochem. 28, 137–146. [DOI] [PubMed] [Google Scholar]
- Hayes G. M.; Carrigan P. E.; Beck A. M.; Miller L. J. (2006) Targeting the RNA splicing machinery as a novel treatment strategy for pancreatic carcinoma. Cancer Res. 66, 3819–3827. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Carr R.; Murray C.; Jhoti H. (2003) A ‘rule of three’ for fragment-based lead discovery?. Drug Discovery Today 8, 876–877. [DOI] [PubMed] [Google Scholar]
- Gerber P. R.; Müller K. (1995) MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput. Aided Mol. Des. 9, 251–268. [DOI] [PubMed] [Google Scholar]
- Lorber D. M.; Shoichet B. K. (1998) Flexible ligand docking using conformational ensembles. Protein Sci. 7, 938–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B. Q.; Baase W. A.; Weaver L. H.; Matthews B. W.; Shoichet B. K. (2002) A model binding site for testing scoring functions in molecular docking. J. Mol. Biol. 322, 339–355. [DOI] [PubMed] [Google Scholar]
- Mpamhanga C. P.; Spinks D.; Tulloch L. B.; Shanks E. J.; Robinson D. A.; Collier I. T.; Fairlamb A. H.; Wyatt P. G.; Frearson J. A.; Hunter W. N.; Gilbert I. H.; Brenk R. (2009) One scaffold, three binding modes: novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J. Med. Chem. 52, 4454–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Shoichet B. K. (2010) Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 50, 1561–1573. [DOI] [PubMed] [Google Scholar]
- Michalczyk A.; Klüter S.; Rode H. B.; Simard J. R.; Grütter C.; Rabiller M.; Rauh D. (2008) Structural insights into how irreversible inhibitors can overcome drug resistance in EGFR. Bioorg. Med. Chem. 16, 3482–3488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.