

Abstract

Abstraction of chloride anion from Au(I) complexes such as JohnPhosAuCl in noncoordinating solvents with 1 equiv of a silver salt, or even larger amounts, leads to the formation of chloride-bridged dinuclear gold(I) complexes, irrespective of the counteranion, which are substantially less reactive as catalysts. This incomplete removal of chloride ligand could lead to false negative results when using the in situ generation of the gold(I) active species by silver-promoted chloride abstraction.
Silver salts play an important role in the generation of the more reactive cationic species in homogeneous catalysis.1 Thus, chloride abstraction from neutral [LAuCl] complexes with a silver salt AgX is a standard procedure for the in situ formation of cationic complexes [LAuL′]X. When X is a weakly coordinating anion such as Tf2N–, neutral complexes [LAuX] are formed.2 On the other hand, silver salts with less coordinating anions such as AgBF4 and AgSbF6 lead to complexes [LAuL′]X, where L′ is a weakly coordinating ligand (i.e., MeCN, PhCN).3 Although so-called “naked gold complexes” [AuL]+ are often invoked in catalytic transformations, there is yet no structural proof for their existence as stable, isolable species.4
Silver salts are not totally innocent in gold catalysis.5,6 Intriguingly, it has recently been proposed that the reactivity of gold catalysts generated in situ by silver(I) abstraction from [LAuCl] complexes can differ dramatically if they are filtered through Celite.7 Since filtration removed AgCl, the contrasting results suggested that a different active gold species was formed. Recently, aquo gold(I) complexes were identified when AgOTf was used in analytical grade CH2Cl2.8 The interference of Ag(I) in gold(I) catalysis was also recently demonstrated by the isolation of a chloride-bridged trimetallic dication with a triangular mixed gold/silver core after mixing neutral complex [JohnPhosAuCl] (1)3 with AgSbF6, followed by filtration through Celite.9
Complex 1, and closely similar complexes,3 have often been used as precatalysts in gold(I)-catalyzed reactions assuming that their activation with a silver(I) salt results in the formation of the active catalytic species. Herein, we report that the abstraction of chloride anion from 1 with silver salts leads to the formation of chloride-bridged dinuclear gold(I) complexes, which are substantially less reactive as catalysts. This incomplete removal of chloride ligand by silver could give “false negative” results in gold-catalyzed reactions when using the in situ generation protocol.
We started by mixing in a 1:1 ratio [JohnPhosAuCl] (1) with different silver salts (AgNTf2, AgOTf, AgBF4, and AgSbF6) in CD2Cl2. The resulting 1H and 31P NMR spectra were examined before and after the filtration though Celite between −80 and +23 °C. In all cases, dinuclear [((JohnPhos)Au)2Cl]X complexes 2a–d were the major species (Scheme 1) that were isolated and characterized by X-ray diffraction (Figure 1).10 Crystalline complexes 2a–d showed weak aurophilic interactions11 (Au–Au distances = 3.48–3.54 Å, Au–Cl–Au angles = 94.7–97.5°),12 which have also been observed in related gold(I) complexes with different phosphine ligands.13
Scheme 1.

Figure 1.
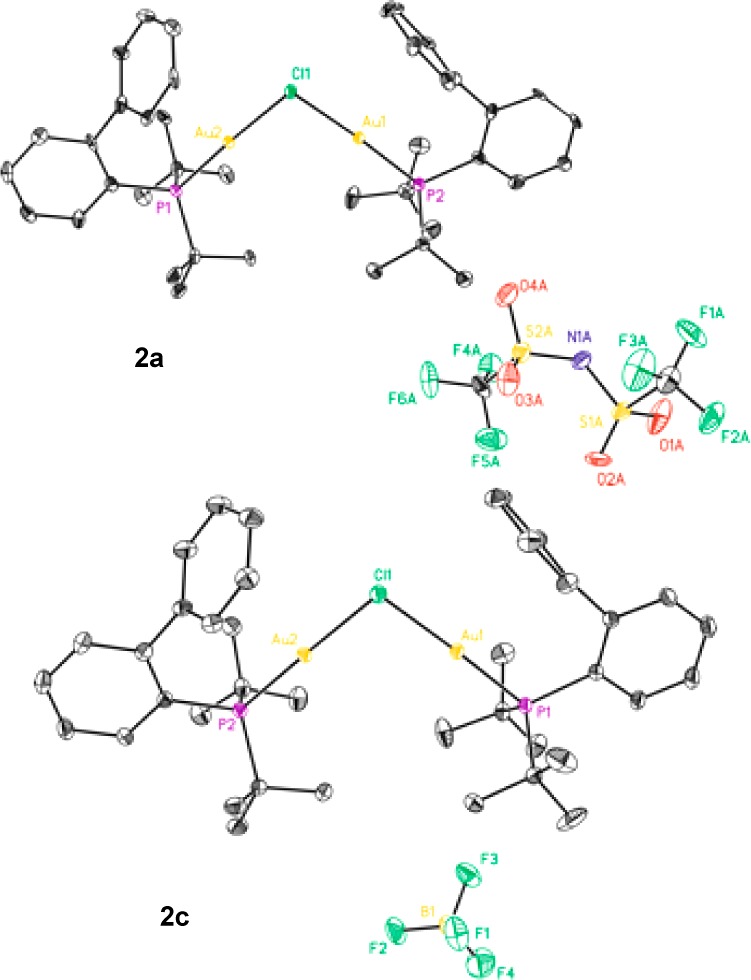
Selected X-ray structures for complexes 2. ORTEP plots (50% thermal ellipsoids). Hydrogen atoms and solvate molecules omitted for clarity.
Reaction of 1 with 1 equiv of AgNTf2 leads to a 1:1 mixture of 2a and neutral [JohnPhosAuNTf2] (3a). Dinuclear complex 2b was the only complex observed after mixing 1 with AgOTf, which evidenced the lower coordinating nature of the triflate anion. After filtration through Celite, a 5:1.5:1 mixture of 2b, [JohnPhosAuOTf] (3b),14 and [JohnPhosAu(H2O)]OTf (4) was obtained. Aquo complex 4 is related to [Ph3PAu(H2O)]OTf, recently observed by treatment of [Ph3PAuCl] with AgOTf in wet CH2Cl2.8 For the clean formation of neutral complex 3b from 1, 5 equiv of AgOTf was required.
Digold complex 2c was the only complex obtained from 1 and AgBF4. In this case, addition of up to 10 equiv of AgBF4 was not enough to cleave the chloride bridge of this dinuclear complex. Nevertheless, filtration through Celite gave small amounts of the dinuclear hydroxy species [(JohnPhosAu)2(OH)]BF4 (5) (Figure 2).15,16 Finally, when AgSbF6 was added to 1 in CD2Cl2, a broad signal corresponding to complex 2d was observed.
Figure 2.
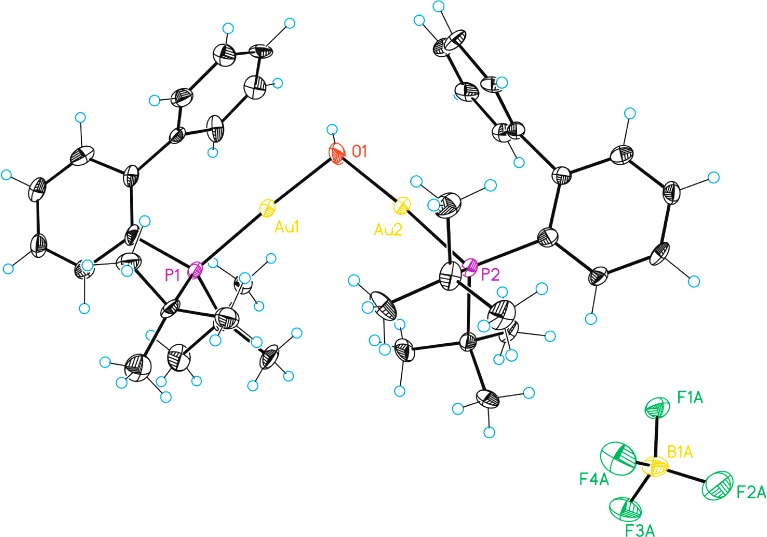
Structure of digold(I) complex 5. ORTEP plot (50% thermal ellipsoids).
To determine the relevance of the formation of complexes 2a–d in catalysis, we examined three reactions under three different conditions: (A) in situ generation of the catalyst from 1 in the presence of the substrate(s), (B) in situ generation of the catalyst followed by the addition of the substrate(s), and (C) filtration of the catalyst through Celite after its generation in situ followed by the addition of the substrate(s). All the experiments were carried out with 3 mol % of 1 and AgX in CD2Cl2. We tested an intermolecular reaction of alkynes with alkenes17 and two relatively challenging cycloisomerizations of enynes bearing a disubstituted alkyne, a class of substrates known to react more sluggishly in gold-catalyzed reactions.3a,18
(i) [2 + 2] Intermolecular Cycloaddition Reaction. The intermolecular reaction between phenylacetylene and α-methylstyrene gives cyclobutene 6 in 80% yield in the presence of [tBuXPhosAu(MeCN)]SbF6 as the catalyst.17 However, when the active gold(I) catalyst was generated in situ using AgSbF6, under conditions A and B, no cyclobutene 6 was formed. Filtration through Celite of the in situ formed catalyst (conditions C), led to 6 in 26% yield. Better results (46% yield) were obtained using digold complex 2d, although the addition of 2.5 mol % of AgSbF6 failed to give any 6. These poor results are due to fast dimerization17 and oligomerization of α-methylstyrene in the presence of AgSbF6.19 The fact that the filtration of the in situ generated complex through Celite (conditions C) gave less cyclobutene 6 (26%) than using 2d (46%) suggested that AgSbF6 is not totally removed by this filtration procedure. This reaction proceeded in low yield with the [JohnPhosAu(MeCN)]X (X = SbF6, BF4).
Different results were observed by using AgBF4. Under conditions A–C, a low yield (32 ± 2%) was consistently obtained (Table 1). Poorer results were obtained with complex 2c as catalyst, although the addition of 2.5 mol % of AgBF4 increased the yield up to 34%.
Table 1. Formation of Cyclobutene 6 by [2 + 2] Intermolecular Cycloadditiona.
| yield of 6 (%) |
||
|---|---|---|
| conditions | AgSbF6 | AgBF4 |
| A | 0 | 31 |
| B | 0 | 34 |
| C | 26 | 30 |
| 2c or 2db | 46 | 11 |
| 2c or 2d + AgX | 0 | 34 |
| [JohnPhosAu(MeCN)]Xb,c | 17 | 30 |
Yields determined by 1H NMR using diphenylmethane as internal standard.
Reactions in the absence of added AgX.
X = SbF6– (first column), X = BF4– (second column)
(ii) Cycloisomerization of 1,6-Enyne 7a. The previous experiments show that chloride-bridged complexes 2c,d are moderately active as catalysts for the [2 + 2] cycloaddition. We also examined the reaction of 1,6-enyne 7a, which undergoes cycloisomerization to form bicyclo[3.2.0]hept-6-ene derivative 8 (Table 2).3a,18,20 When AgSbF6 was added to a solution containing the enyne and 1, bicyclic product 8 was obtained in 77% yield after 2 h (conditions A). However, under conditions B, 8 was obtained in only 12% yield. No reaction was observed under conditions C. The reaction also failed using complex 2d as catalyst. However, a 67% yield was achieved after the addition of AgSbF6 to complex 2d. The reaction was found to be much less effective when using AgBF4.
Table 2. Cycloisomerization of 1,6-Enyne 7aa.
Yields determined by 1H NMR using diphenylmethane as internal standard. No reaction took place with [JohnPhosAu(MeCN)]X (X = SbF6 or BF4).
5 mol %, 18 h.
Reactions in the absence of added AgX.
(iii) Cycloisomerization of 1,6-Enyne 7b. Finally, we compared the performance of complexes 2c–d with other gold(I) catalysts in the [4 + 2] cycloaddition of enyne 7b to form tricyclic derivative 9 (Figure 3).3a,18 Cationic complex [JohnPhosAu(MeCN)]SbF6 (10) and a 1:1 mixture of [JohnPhosAuCl] (1) and AgSbF6 (in the presence of substrate 7b; conditions A) gave tricyclic product 9 in a similar yield after 2.5 h. The reaction was faster with 1 in the absence of acetonitrile, which presumably competes with the substrate 7b to form the active [LAu(substrate)]+ species.21 As expected, neutral [JohnPhosAuCl] (1) was inactive. However, when the addition of the silver salt to complex 1 was completed prior to the addition of the substrate 7b (conditions B), cycloadduct 9 was formed in only 32% yield after 2.5 h. Dinuclear complex 2d (1.5 mol %) led to 9 in even lower yield (11%) after 2.5 h. Addition of 1.5 mol % of AgSbF6 to 2d enhanced the yield up to 39%. Digold complex 2c was a much poorer catalyst than 2d even after long reaction periods.
Figure 3.
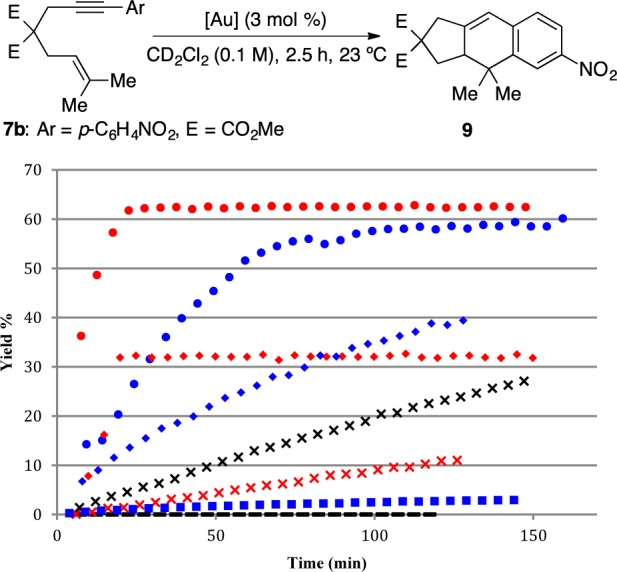
Cycloisomerization of 1,6-enyne 7b: (red dot)[7b + 1] + AgSbF6; (blue dot) 7b + 10; (red diamond) [1 + AgSbF6] + 7b; (blue diamond) [7b + 2d (1.5 mol %)] + AgSbF6 (1.5 mol %); (×) 7b + 2d; (red ×) 7b + 2d (1.5 mol %); (blue box) (7b + 2c (1.5 mol %); (−) 7b + 1. Yields determined by 1H NMR using diphenylmethane as internal standard.
All these results can be explained by the formation of the chloride-bridged complexes 2 when the catalyst is generated in situ in the absence of the coordinating substrate (conditions B). The order of addition of the silver salt is less important when the substrate is able to cleave the dinuclear complexes, as in the case of the [2 + 2] cycloaddition, but it has a significant impact if the substrate is less coordinating22 and therefore cannot enter the catalytic cycle by cleaving the chloride-bridge of complexes 2, as happens in the cycloisomerizations of 7a and 7b.
In summary, we have shown that formation of robust chloride-bridged dinuclear gold(I) complexes [(LAu)2Cl]X occurs readily when mixing [LAuCl] precatalysts with silver salts in the absence of the substrate in non coordinating solvents such as CH2Cl2. These dinuclear complexes are significantly less active than cationic complexes [LAu(NCMe)]X. As a mnemonic, it might be useful to remind that the associative law is not followed in the preparation of the active catalysts by chloride abstraction with a silver salt: (LAuCl + substrate) + AgX ≠ (LAuCl + AgX) + substrate. In other words, the order of addition of the silver salt can have a significant effect in reactivity if the substrate is not able to efficiently cleave the chloride-bridge. This could explain, at least in part, the somewhat erratic results attributed to “silver effects”. Therefore, we recommend that if the gold catalyst is generated from a neutral [LAuCl] complex, then the silver salt should be added last, after premixing [LAuCl] with the substrate to minimize the formation of chloride-bridged dinuclear gold(I) complexes. Although the ligand of study has been JohnPhos in all the experiments, probably the aforementioned structures also form when using carbenes or other phosphines as ligands. Further experiments are ongoing in our laboratories.
Acknowledgments
We thank MEC (project CTQ2010-16088/BQU and FPI predoctoral Fellowship to A.H.), the European Research Council (Advanced Grant No. 321066), and the ICIQ Foundation for financial support. We are grateful to the ICIQ X-ray diffraction unit for the X-ray crystal structures and the ICIQ NMR unit.
Supporting Information Available
Experimental details, characterization data, and additional results. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Additional address: Departament de Química Analítica i Química Organica, Universitat Rovira i Virgili, 43007 Tarragona, Spain.
The authors declare no competing financial interest.
Supplementary Material
References
- a Jiménez-Núñez E.; Echavarren A. M. Chem. Rev. 2008, 108, 3326–3350. [DOI] [PubMed] [Google Scholar]; b Gorin D. J.; Sherry B. D.; Toste F. D. Chem. Rev. 2008, 108, 3351–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Michelet V.; Toullec P. Y.; Genêt J. P. Angew. Chem., Int. Ed. 2008, 47, 4268–4315. [DOI] [PubMed] [Google Scholar]; d Fürstner A. Chem. Soc. Rev. 2009, 38, 3208–3221. [DOI] [PubMed] [Google Scholar]; e Rudolph M.; Hashmi A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. [DOI] [PubMed] [Google Scholar]
- Mézailles N.; Ricard L.; Gagosz F. Org. Lett. 2005, 7, 4133–4136. [DOI] [PubMed] [Google Scholar]
- a Nieto-Oberhuber C.; López S.; Echavarren A. M. J. Am. Chem. Soc. 2005, 127, 6178–6179. [DOI] [PubMed] [Google Scholar]; b Herrero-Gómez E.; Nieto-Oberhuber C.; López S.; Benet-Buchholz J.; Echavarren A. M. Angew. Chem., Int. Ed. 2006, 45, 5455–5459. [DOI] [PubMed] [Google Scholar]; c Pérez-Galán P.; Delpont N.; Herrero-Gómez E.; Maseras F.; Echavarren A. M. Chem.—Eur. J. 2010, 16, 5324–5332. [DOI] [PubMed] [Google Scholar]
- A recent report claimed the formation of [IPr**Au]BArF24 with the bulky ligand IPr** = 2,6-bis(di-4-tert-butylphenyl)methyl-4-methylphenyl, although no structural proof was provided:Weber S. D.; Zahner D.; Rominger F.; Straub B. F. Chem. Commun. 2012, 48, 11325–11327. [DOI] [PubMed] [Google Scholar]
- Nevado C.; Echavarren A. M. Chem.–Eur. J. 2005, 11, 3155–3164. [DOI] [PubMed] [Google Scholar]
- Weber D.; Gagné M. R. Org. Lett. 2009, 11, 4962–4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Cai R.; Sharma S.; Jirak J.; Thummanapelli S. K.; Akhmedov N. G.; Zhang H.; Liu X.; Petersen J. L.; Shi X. J. Am. Chem. Soc. 2012, 134, 9012–9019. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Yu B. RSC Adv. 2012, 2, 12686–12689. [Google Scholar]
- This work, published when our manuscript was in preparation, reported the formation of a different solvate of complex 2d from 1:Zhu Y.; Day C. S.; Zhang L.; Hauser K. J.; Jones A. C. Chem.—Eur. J. 2013, 19, 12264–12271. [DOI] [PubMed] [Google Scholar]
- X-ray crystal structures: 2a, CCDC 963480; 2b, CCDC 963484; 2c, CCDC 963479; 2d, CCDC 963481; 3a, CCDC 963485; 3b, CCDC 963486; 5, CCDC 963482. See the Supporting Information for details on the X-ray structures.
- a Schmidbaur H.; Schier A. Chem. Soc. Rev. 2012, 41, 370–412. [DOI] [PubMed] [Google Scholar]; b Schmidbaur H.; Schier A. Chem. Soc. R ev. 2008, 37, 1931–1951. [DOI] [PubMed] [Google Scholar]
- The shortest Au–Au distance (3.48 Å) and more acute Au–Cl–Au angle (94.7°) were found for 2d·(CH2Cl2)2.25, in contrast with that reported in ref (9) for a different solvate 2d·(H2O)0.5 (Au–Au = 3.54 Å, Au–Cl–Au = 97.7°), which shows that packing and/or hydrogen bonding are also significant in these structures.
- a Usón R.; Laguna A.; Castrillo M. B. Synth. React. Inorg.-Met.-Org. Chem. 1979, 9, 317–324. [Google Scholar]; b Bayler A.; Bauer A.; Schmidbaur H. Chem. Ber. 1997, 130, 115–118. [Google Scholar]
- a Zhdanko A.; Ströbele M.; Maier M. E. Chem.—Eur. J. 2012, 18, 14732–14744. [DOI] [PubMed] [Google Scholar]; b Although 3b has been occasionally depicted in the literature as [JohnPhosAu]OTf with a LAu+ cation, complex 3b is neutral with covalently bound triflate (Au–O bond distance = 2.11 Å, P–Au–O bond angle 174.2). See the Supporting Information for details.
- a The Au–Au distance and the Au–O–Au angle are 3.38 Å and 105.0°. In the analogous complex [(JohnPhosAu)2(OH)]SbF6, these parameters are 3.35 Å and 108.1°:Adriaenssens L.; Escribano-Cuesta A.; Homs A.; Echavarren A. M.; Ballester P. Eur. J. Org. Chem. 2013, 1494–1500. [Google Scholar]
- Analogous gold species with N-heterocyclic carbenes as ligands have been reported and characterized by X-ray difraction; a Gaillard S.; Bosson J.; Ramón R. S.; Nun P.; Slawin A. M. Z.; Nolan S. P. Chem.—Eur. J. 2010, 16, 13729–13740. [DOI] [PubMed] [Google Scholar]; b Gómez-Suárez A.; Oonishi Y.; Meiries S.; Nolan S. P. Organometallics 2013, 32, 1106–1111. [Google Scholar]
- López-Carrillo V.; Echavarren A. M. J. Am. Chem. Soc. 2010, 132, 9292–9294. [DOI] [PubMed] [Google Scholar]
- Nieto-Oberhuber C.; Pérez-Galán P.; Herrero-Gómez E.; Lauterbach T.; Rodríguez C.; López S.; Bour C.; Rosellón A.; Cárdenas D. J.; Echavarren A. M. J. Am. Chem. Soc. 2008, 130, 269–279. [DOI] [PubMed] [Google Scholar]
- Grirrane A.; Garcia H.; Corma A.; Álvarez E. ACS Catal. 2011, 1, 1647–1653. [Google Scholar]
- Recent mechanistic work on this type of cycloisomerization:; Brooner R. E. M.; Brown T. J.; Widenhoefer R. A. Angew. Chem., Int. Ed. 2013, 52, 6259–6261. [DOI] [PubMed] [Google Scholar]
- The reaction was slower using complex [t-BuXPhosAu(MeCN)]SbF6 bearing a bulkier phosphine ligand (only 15% yield after 2.5 h), although the yield after 24 h reached 60%.
- Phenylacetylene coordinates more efficiently to [(JohnPhos)Au]+ than 7a or 7b as observed by 31P NMR.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.