

Abstract
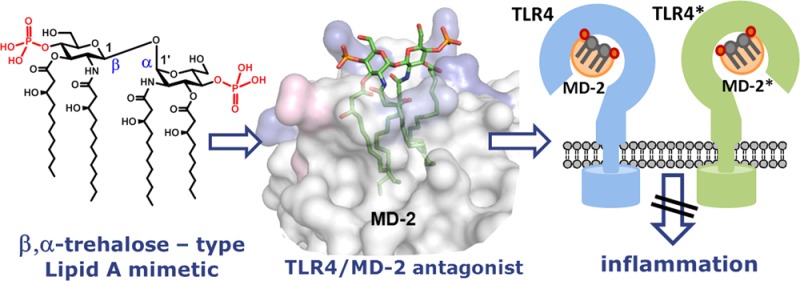
Recognition of the lipopolysaccharide (LPS), a major component of the outer membrane of Gram-negative bacteria, by the Toll-like receptor 4 (TLR4)-myeloid differentiation factor 2 (MD-2) complex is essential for the control of bacterial infection. A pro-inflammatory signaling cascade is initiated upon binding of membrane-associated portion of LPS, a glycophospholipid Lipid A, by a coreceptor protein MD-2, which results in a protective host innate immune response. However, activation of TLR4 signaling by LPS may lead to the dysregulated immune response resulting in a variety of inflammatory conditions including sepsis syndrome. Understanding of structural requirements for Lipid A endotoxicity would ensure the development of effective anti-inflammatory medications. Herein, we report on design, synthesis, and biological activities of a series of conformationally confined Lipid A mimetics based on β,α-trehalose-type scaffold. Replacement of the flexible three-bond β(1→6) linkage in diglucosamine backbone of Lipid A by a two-bond β,α(1↔1) glycosidic linkage afforded novel potent TLR4 antagonists. Synthetic tetraacylated bisphosphorylated Lipid A mimetics based on a β–GlcN(1↔1)α–GlcN scaffold selectively block the LPS binding site on both human and murine MD-2 and completely abolish lipopolysaccharide-induced pro-inflammatory signaling, thereby serving as antisepsis drug candidates. In contrast to their natural counterpart lipid IVa, conformationally constrained Lipid A mimetics do not activate mouse TLR4. The structural basis for high antagonistic activity of novel Lipid A mimetics was confirmed by molecular dynamics simulation. Our findings suggest that besides the chemical structure, also the three-dimensional arrangement of the diglucosamine backbone of MD-2-bound Lipid A determines endotoxic effects on TLR4.
The innate immune system recognizes the presence of pathogen associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs). Specific Gram-negative bacterial antigen, lipopolysaccharide (LPS), also known as endotoxin, is recognized by a complex composed of myeloid differentiation factor 2 (MD-2) and Toll-like receptor 4 (TLR4), a type I transmembrane protein characterized by an ectodomain that contains varying numbers of leucin-rich-repeat motifs and a cytoplasmic signaling domain. Intensive research in the past 15 years has shown that the activation of innate immune response by submicromolar concentrations of endotoxin necessitates a successive interaction of LPS with lipid-binding protein (LBP), CD14 (a differentiation antigen of monocytes), and TLR4/MD-2 complex.1−3 The membrane-bound glycophospholipid portion of LPS, known as Lipid A, is believed to represent the “endotoxic principle” of LPS. Upon binding of hexaacylated E. coli Lipid A by a coreceptor protein MD-2, a receptor multimer composed of two copies of the TLR4/MD-2–Lipid A complex is formed, which triggers a downstream signaling cascade and activates host innate immunity (Figure 1).3 LPS-mediated initiation of the TLR4/MD-2 signaling actively contributes to the development and maintenance of advantageous defensive host response. Though, in the conditions of uncontrolled inflammation, TLR4 activation results in the detrimental overproduction of cytokines and chemokines leading to a life-threatening sepsis syndrome (the 10th leading cause of death in developed countries, 30% mortality rate) and septic shock, the effective treatment for which is still not available.4−6 Inappropriate regulation of TLR4 signaling and ensuing overactivation of innate immune system was shown to contribute to the development and pathogenesis of chronic inflammatory, autoimmune, and infectious diseases, including asthma, arthritis, multiple sclerosis, inflammatory bowel disease, and cancer.7−10
Figure 1.
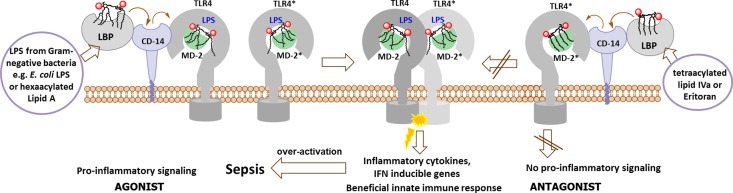
Schematic representation of E. coli Lipid A - induced activation and antagonist (Eritoran, lipid IVa)-dependent inhibition of TLR4/MD-2 signaling.
One of the most successful approaches for down-regulation of TLR4 signaling involves application of compounds which compete with endotoxic LPS in binding to the same site on MD-2 and, thereby, inhibit the induction of the signal transduction pathway by impairing LPS-initiated receptor dimerization (Figure 1). To date, several Lipid A variants that specifically block the LPS-binding site on human (h) MD-2 have been identified: lipid IVa (a biosynthetic precursor of E. coli Lipid A)11 and a nonpathogenic Lipid A from R. sphaeroides,12,13 which served as structural basis for the synthetic antisepsis drug candidate Eritoran (E5564).14,15
Ground-breaking X-ray structural analyses of variably acylated Lipid A bound to MD-2/TLR4 complexes revealed strikingly different modes of binding of agonistic and antagonistic ligands. All four acyl chains of the tetraacylated antagonists Eritoran16 and lipid IVa11 were fully inserted into the hydrophobic binding pocket of hMD-2. The binding of the ligand did not induce dimerization of hMD-2/TLR4–Lipid A complexes, therefore, the intracellular signaling cascade was not initiated (Figure 2A, 2B).
Figure 2.

Side view of PDB co-crystal structures of (A) hybrid TLR4/hMD-2 with bound antagonist Eritoran (PDB: 2Z65, TLR4 is not shown); (B) hMD-2 with bound antagonist lipid IVa (PDB: 2E59); (C) mTLR4/MD-2 with bound agonist lipid IVa (PDB: 3VQ1); (D) hTLR4/MD-2 with bound E. coli Re-LPS (PDB: 3FXI, for clarity only Lipid A portion of LPS is shown). Orientation of the ligand is inverted by 180° for parts C and D. Arg and Lys involved in ionic interactions with 1- and 4′-phosphates of Lipid A at the rim of the binding pocket of MD-2 are depicted in blue; Phe126 is in orange. Phe126 points outside in antagonist co-crystal structures (A, B) and is located inward in agonist structures (C, D) wherein Phe126 establishes hydrophobic contacts to the exposed lipid chain (in yellow). Images were generated with PyMol.
In contrast, the orientation of endotoxic hexaacylated E. coli Lipid A within the binding pocket of MD-2 was turned by 180°. Only five long-chain acyl residues were intercalated in the interior of the binding cavity, whereas the sixth acyl chain was exposed onto the surface of MD-2, constituting, together with the patch of hydrophobic amino acids (Phe126 loop), the core hydrophobic interface for the interaction with the second TLR4*/MD-2*–LPS complex (Figure 2D). Hydrophobic contacts of the exposed acyl chain of Lipid A with the second TLR4*, along with intermolecular ionic interactions of the Lipid A phosphates,17 triggered the formation of an active homodimeric signaling ligand–receptor complex (Figure 1).3,18,19
Remarkably, tetraacylated lipid IVa acts as antagonist on human but as an agonist on mouse TLR4 presenting one lipid chain on the surface of mMD-2, which reveals striking similarity to the crystal structure of hexaacylated Lipid A–hMD-2/TLR4 (Figure 2C, D).20 This species-specific activity of lipid IVa is attributed, among other factors, to the dissimilarities in the shape of the hydrophobic binding pocket of h- and mMD-2 and to the variations in the electrostatic potentials at the rim of the binding cavity of MD-2 and at the dimerization interface.20−23 However, particular structural features of the ligand, such as lipid IVa, which could be responsible for the species-specific TLR4 activation have not yet been assessed.
Despite a huge volume of accumulated data on the activity of both isolated24,25 and synthetic Lipid A derivatives,26−29 there is no universal correlation between the chemical structure of Lipid A and its function in TLR4/MD-2 complex, which would allow the prediction of biological activity of a particular Lipid A variant. Subtle differences in the length (e.g., 2 × CH2) and distribution pattern of acyl chains, changes in the volume and the overall shape of the hydrophobic portion of Lipid A, as well as the phosphorylation status of the diglucosamine backbone are known to profoundly affect the biological activity of Lipid A.27−30 In this study, we further explore the structure–function relationships in Lipid A–MD-2/TLR4 complex wherein we address the three-dimensional (3D) molecular shape of MD-2-bound Lipid A/lipid IVa as the main structural determinant of endotoxicity. Whereas all by now synthesized Lipid A analogues or Lipid A mimetics were based either on the regular β-GlcN(1→6)GlcN disaccharide backbone27,28 or on the more flexible skeletons wherein the reducing GlcN was replaced by a linear aglycon,26,29 we focused our research on restricting the internal flexibility of the diglucosamine backbone of Lipid A.
Results and Discussion
Conformational Rearrangement in Diglucosamine Backbone of Lipid A as the Key Triggering Event Required for TLR4/MD-2 Activation: Design of Trehalose-type Lipid A Mimetics
Chemically, the structure of Lipid A is based on the highly conserved bisphosphorylated β–GlcN(1→6)GlcN backbone, which exemplifies a highly flexible part of the molecule,31 whereas 2,2′-N- and 3,3′-O- long-chain acyl residues comprise a rigid hydrophobic cluster (Figure 2).32 The relative spatial orientation of GlcN rings of the diglucosamine backbone of Lipid A diverges notably for MD-2-bound agonistic and antagonistic ligands (Figure 3). The proximal GlcN moiety of the MD-2 – bound agonists (Lipid A/hMD-2 and lipid IVa/mMD-2) adopts a “twisted” orientation with respect to the distal GlcN ring (Figure 3A,B), whereas both glucosamines in antagonist ligands are positioned in nearly one plane (Figure 3C, D). These arrangements are mostly achieved by adjustment of the exocyclic oxymethyl (−CH2O−) fragment involved in the three-bond (1→6) glycosidic linkage, which represents the most “adaptable” segment of Lipid A (Figure 4A).
Figure 3.
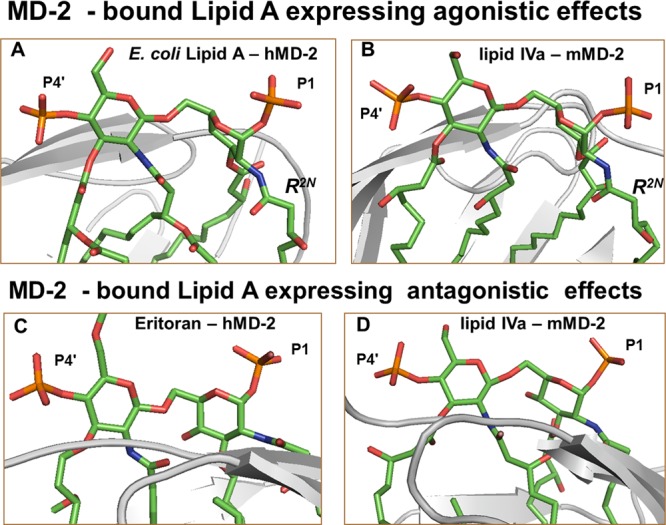
PDB co-crystal structures of (A) hMD-2 with bound E. coli Re-LPS (PDB: 3FXI, for clarity only Lipid A portion of LPS is shown). (B) mMD-2 with bound agonist lipid IVa (PDB: 3VQ1); (C) hMD-2 with bound Eritoran (PDB: 2Z65); (D) hMD-2 with bound antagonist lipid IVa (PDB: 2E59); orientation of MD-2 is inverted by 180° for parts C and D. The 3D orientation of the distal GlcN ring (the one with 4′ – phosphate P4′) of Lipid A was set similar for all four snapshots so that the difference in the relative orientation of proximal (reducing) GlcN rings between A,B (twisted) and C,D (co-planar) becomes evident. Images were generated with PyMol.
Figure 4.

(A) Conformational changes in MD-2-bound agonistic Lipid A attained through adjustments of dihedrals about glycosidic and oxymethyl (−OCH2−) linkages. Dihedral angles determining the overall conformation of the molecule are ω, ϕ, and ψ; (B) Conformationally constrained Lipid A mimetics based on (1↔1)-glycosidically connected trehalose-type scaffolds.
We assume that the exposure of the 2N-alkanoyl chain (R2N) of agonistic Lipid A out of the binding pocket of MD-2 is enabled through MD-2 driven adjustment of dihedrals ω, ϕ, and ψ about oxymethyl and glycosidic linkages (Figure 4A, Supporting Information (SI) Table 1S). Accordingly, upon binding of the agonistic ligand by MD-2, the proximal GlcN ring (the one situated at the dimerization interface) is relocated in a “tilted” position, which assists in destabilization of intramolecular hydrophobic interactions between long fatty chains (Figure 4A). This allows the exposure of a single lipid chain on the surface of MD-2 for the contact with the second TLR4*/MD-2* complex without substantial entropic loss. The torsional angles ω, ϕ, and ψ determining the linkage geometry were also shown to significantly differ between solution- and membrane-incorporated32,33 Lipid A structures.
To verify this supposition, we have designed conformationally confined Lipid A mimetics based on the two-bond linked (1↔1)-glycosidically connected disaccharides, which lack the flexible oxymethyl linkage, wherein the reducing (proximal) GlcN ring of Lipid A is replaced by a nonreducing sugar (Figure 4B). Varying anomeric configuration, phosphorylation pattern, and acyl chain distribution on the (1↔1)-trehalose-type scaffold would afford Lipid A mimetics having potential antagonistic or agonistic effects on TLR4. A compelling advantage of novel Lipid A mimetics consists in replacement of extremely labile anomeric phosphate function of natural Lipid A by a stable secondary phosphoester group.
The first series of conformationally constrained tetraacylated Lipid A mimetics was designed on the basis of β,α-pseudo-trehalose type scaffold (Figure 5). The 3D shape of the carbohydrate backbone of pseudo-trehalose found in the crystal structure34 resembles the 3D arrangement of β-GlcN(1→6)GlcN backbone of hMD-2-bound antagonists lipid IVa and Eritoran, wherein the reducing GlcN moiety is not twisted like in the agonistic Lipid A ligands (Figure 3A, B), but rather “flatly” positioned with respect to the orientation of the nonreducing (distal) GlcN ring (Figure 5).
Figure 5.
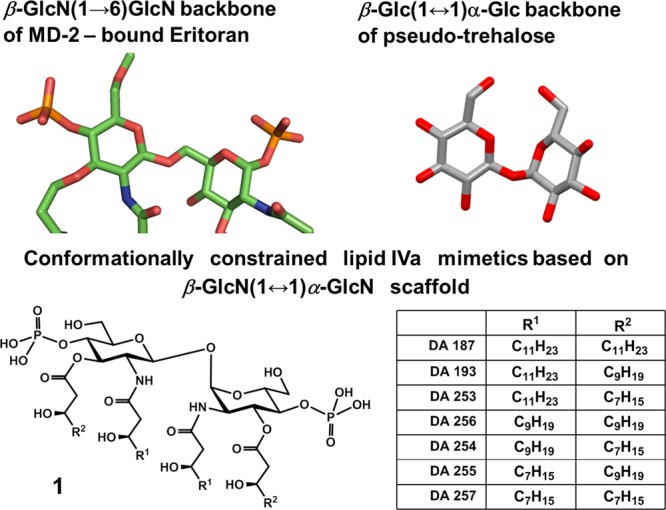
Conformationally constrained tetraacylated Lipid A mimetics 1 based on the crystal structures of Eritoran (PDB code: 2Z65) and β,α-pseudo-trehalose (doi: 10.1107/S0108270196012693).34
β,α-Pseudo-trehalose served as a model for a synthetic β-GlcN(1↔1)α–GlcN scaffold in Lipid A mimetics 1, whereas the acylation pattern (2,3; 2′,3′) and positions of the phosphates (4 and 4′) were chosen to match the corresponding distances found in the co-crystal structures of hMD-2-bound antagonists (SI Table 2S). Along with the conformationally restrained counterpart of lipid IVa having four β-hydroxymyristoyl chains (4 × C14, DA187) several short-chain (C10–C14) analogues have been similarly prepared (Figure 5).
The reduced flexibility of β,α-(1↔1) glycosidic linkage in mimetics 1, which is imposed, in part, by anomeric and exoanomeric effects, was supposed to confine conformational rearrangements in diglucosamine backbone given that only one energetically preferable conformation for β,α-trehalose was found to exist in the previous computational work.31,35,36 Therefore, it was assumed that the “proximal” GlcN moiety of mimetics 1 would not be able to adopt a “twisted” orientation upon binding by MD-2. The 3D arrangement of the carbohydrate backbone of mMD-2–bound lipid IVa (Figure 3B) would not be achieved.
The latter was expected to impede the agonistic effects on mMD-2/TLR4 by retaining the strong hydrophobic intramolecular interactions between lipid chains and abrogating the exposure of one β-hydroxyacyl chain onto the surface of mMD-2. Besides, the ligands 1 could have higher affinity to hMD-2 compared with lipid IVa, since conformationally confined β,α–(1↔1)–linked diglucosamine scaffold would resemble the overall conformation of the backbone of the potent antagonist Eritoran (Figure 5).
Chemical Synthesis of Lipid A Mimetics Based on β,α–1,1′–diglucosamine Scaffold
In contrast to conventional glycoside synthesis, the stereoselective construction of (1↔1) glycosidic bond requires control of stereochemistry at two anomeric centers. Typically, synthetic approaches to nonreducing disaccharides lead to a mixture of diastereomers, whereas the yields rarely exceed 50%. Additionally, the synthesis of Lipid A mimetics is complicated by the necessity to use a set of orthogonal protecting groups, which must be removable under very mild conditions not affecting acid-labile phosphotriester and base-labile acyloxy- functionalities, to allow for selective phosphorylation and introduction of N- and O-acyloxy fatty chains after construction of the glycosidic bond. To ensure the stereoselective formation of 1,2-trans- and 1,2-cis- anomeric linkages, a participating group at C-2 of the donor and a protecting group at C-2 of the acceptor which favors α-configuration of the anomeric hydroxyl group, respectively, were required. Among a series of 2N-protecting groups tested, the N-Troc (trichloro-ethoxycarbonyl) group was found to provide the best results in terms of both anomeric selectivity in the glycosylation step and α-anomeric preference of 1-OH group at the lactol acceptor 4 (α/β = 9:1).
For the assembly of the nonreducing β,α–disaccharide 6 an imidate donor 5 equipped with participating N-Troc protecting group and N-Troc–protected axially oriented lactol 4 as acceptor, both obtained from a single precursor 3, were applied (Scheme 1). Symmetrically protected nonreducing disaccharide 6 was obtained in excellent yield (75%) and high stereoselectivity. Reductive cleavage of N-Troc protecting groups followed by acylation with benzyl-protected β-hydroxy fatty acids of different length 8–10 furnished diacylated disaccharides 11–13. Sequential deprotection of 3-O- and 3′-O-TBDMS groups and subsequent acylation with 8–10 resulted in the tetraacylated disaccharides 17–23. Regioselective reductive opening of benzylidene acetals and successive phosphorylation of the liberated 4- and 4′-hydroxy groups afforded bis-phosphates 31–37. Finally, benzyl protecting groups were removed by hydrogenation on Pd-black to furnish, after purification by gel-permeation chromatography, conformationally constrained variably acylated lipid IVa mimetics 1. Similar to most synthetic Lipid A derivatives, the amphiphilic mimetics 1 are not water soluble and, hence, were applied for biological testing as stock solutions in DMSO, such that the final DMSO concentration on the cells did not exceed 0.1% (at a concentration 1000 ng/mL of 1).
Scheme 1. Synthesis of Conformationally Constrained Lipid A Mimetics Based on β,α-1,1′-diglucosamine Scaffold.

Reagents and conditions: (a) Me2(tBu)SiCl, Im, DMF; (b) [Ir(COD)(Ph2MeP)2]PF6, H2, THF, then I2, THF/aq.NaHCO3; (c) CF3C(NPh)Cl, K2CO3, acetone; (d) TMSOTf, MS 4 Å, CH2Cl2; (e) Zn, CH3COOH; (f) 8–10, HATU, DMF; (g) TBAF, THF; (h) 8–10, EDC, DMAP; (i) TfOH, Et3SiH, MS 4 Å, CH2Cl2; (k) (BnO)2PN(iPr)2, 1H-tetrazole, then mCPBA; (l) Pd black, H2, toluene-MeOH.
Biological Activities of Tetraacylated Lipid A Mimetics Based on β,α–1,1′–diglucosamine Scaffold
All seven variably acylated Lipid A mimetics 1 lacked pro-inflammatory activities in hTLR4/MD-2 transfected HEK293 cells and, except for 4 × C14-acylated DA187, which is a conformationally constrained counterpart of lipid IVa, were shown to potently inhibit pro-inflammatory responses in TLR4-transfected HEK293 cells stimulated with E. coli Lipid A (Figure 6A, SI Figure 1S-AB). Independent of the mode of application (preincubation with mimetics 1; simultaneous application of E. coli Lipid A and mimetics 1 or prestimulation with E. coli Lipid A) the Lipid A mimetics 1 revealed potent antiendotoxic activity at a concentration of 1000 ng/mL. Two “representative” hTLR4 antagonists DA193 and DA254 were also evaluated for their ability to inhibit E. coli Lipid A - induced expression of IL-6 in human monocytic cells. Both compounds were found to effectively suppress IL-6 production upon concurrent application with E. coli Lipid A (Figure 6B).
Figure 6.

Inhibition of hMD-2/TLR4 complex by mimetics 1 (compared to lipid IVa) in T-REx-293/hTLR4 cells transfected with hMD-2/fLuc/rLuc. (A) concurrent application of E. coli Lipid A and mimetics 1/lipid IVa; (B) DA193 and DA254 inhibit IL-6 expression in human monocytic cell line (MM6) stimulated with E. coli Lipid A.
The lack of activity of 4 × C14-acylated DA187 should not relate to the differences in solubility due to the longer fatty chain length, since its flexible counterpart, tetra-myristoylated lipid IVa, exhibits antiendotoxic properties under the same experimental conditions. We assume that the failure of DA187 to compete with E. coli Lipid A for the binding site on MD-2 could be due to minor increase of the hydrophobic volume of lipid chains, which would prevent the ligand from entering the binding groove of MD-2. The co-planar orientation of GlcN rings in (1↔1) diglucosamine scaffold of DA187 and its confined flexibility would not allow the rearrangement to a more compact conformation wherein the cluster of four C-14 acyl chains would have decreased hydrophobic volume necessary for entering rather constrained binding site of MD-2 (SI Figure 2S).
Lipid A mimetics 1 were further examined for their capacity to inhibit E. coli O111 LPS - induced activation of TLR4/MD-2 complex in TLR4-transfected HEK293 cells (Figure 7, SI Figure 3S). Upon application of DA193 and DA256 at a concentration of 500 ng/mL, the inflammatory responses to E. coli LPS were entirely abrogated; whereas the shorter chain Lipid A mimetics DA255, DA256, and DA254 allowed for 50% inhibition at the submolar concentration of 5 ng/mL. The antiendotoxic potencies of five differently acylated variants of mimetics 1 were comparable or better than those of natural lipid IVa. Surprisingly, Lipid A mimetic DA257 having four R-3̅-hydroxydecanoic acids, did not antagonize E. coli LPS, though it was shown to suppress cell activation elicited by E. coli Lipid A (Figure 6). Thus, DA257 having smaller hydrophobic volume (4 × C10-acyl residues) successfully competes with Lipid A for the binding site on hMD-2 but fails to displace the LPS, which possesses greater endotoxic potency and higher affinity for MD-2/TLR4 complex than the corresponding Lipid A.37 Similarly, synthetic short-chain (4 × C10) analogue of natural lipid IVa was found to be inactive in suppression of IL-6 induction by LPS.38
Figure 7.
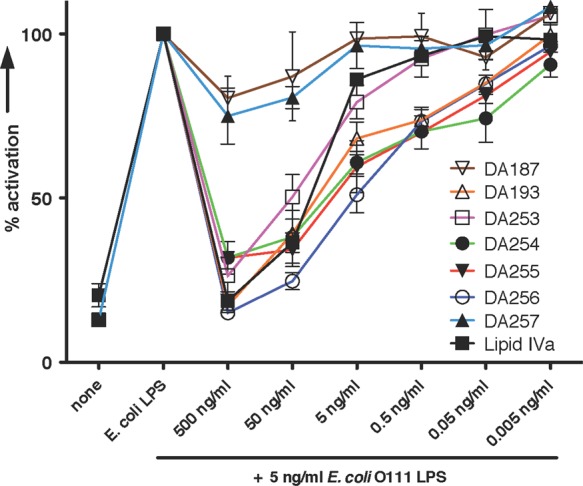
Concentration-dependent antagonistic activity of mimetics 1 (compared to lipid IVa) in hTLR4-transfected HEK293 cells (HEKBlue) stimulated with E. coli O111 LPS. Cells were preincubated with mimetics 1 for 1 h, then stimulated with E. coli LPS.
In contrast to lipid IVa, which is an agonist in mouse cells, all conformationally constrained tetraacylated mimetics 1, as predicted, did not activate mTLR4 (Figure 8A). Instead, they acted as antagonists of mTLR4 (again except for DA187), whereas the mimetics with shorter chains inhibited mTLR4/MD-2 complex most effectively (Figure 8B, SI Figure 1S-C). mTLR4 antagonist DA255 was shown to suppress the LPS - induced expression of TNF-α in mice when coadministered intraperitoneally with LPS (SI Figure 4S).
Figure 8.

(A) Activation of the mTLR4/MD-2 complex by mimetics 1 in T-REx-293/mTLR4 cells. Lipid IVa (100 ng/mL), similarly to E. coli Lipid A, activates mTLR4; mimetics 1 (100 ng/mL) do not activate mTLR4; (B) inhibition of mTLR4/MD-2 complex by mimetics 1 in T-REx-293/mTLR4 cells concurrently stimulated with E. coli Lipid A. (Lipid IVa does not inhibit (only activates) mTLR4.)
Molecular Dynamics Simulations
Molecular dynamics simulations of the Lipid A mimetic having 2 × C14, 2 × C12 acyl chains (DA193, which has been shown to display the most pronounced antagonistic effects) in nonbound state and in complex with h- and mMD-2 revealed the expected reduced flexibility of the β,α-(1↔1) diglucosamine backbone. Minor differences in the typical torsional dihedral angles of the β,α–(1↔1) glycosidic linkage of DA193 upon ligand binding by MD-2 could be observed, but no significant conformational changes.
The average values of the ϕ and ψ dihedral angles in DA193 deviated by 22° and 27°, respectively, from the X-ray conformation of β,α-trehalose34 and by 7° and 12° from the most stable conformation observed in the previous computational work (SI Table 3S, SI Figure 5S).35,36 This can be explained by replacement of Glc(1↔1)Glc disaccharide of pseudo-trehalose for GlcN(1↔1)GlcN scaffold in DA193 and by substitution of the diglucosamine with functional groups (four acyl chains and two phosphate groups). The observed conformational differences were already detected in the simulations of DA193 in the absence of the protein (SI Figure 6S), inferring that the changes in the overall conformation of the carbohydrate backbone of DA193 are mostly due to the substitutions with functional groups and not to the protein environment. Accordingly, DA193 is accommodated within the binding pocket of MD-2 in a preferred conformation, which implies that the reduced flexibility of the (1↔1) glycosidic linkage does not allow extensive structural adjustment of the carbohydrate backbone of DA193 to the geometry of the binding pocket of MD-2 (SI Figure 7S).
Structural comparison of the mode of binding of DA193 and agonistic lipid IVa within the hydrophobic cavity of mMD-2 demonstrates that DA193 is displaced by ∼3.0 Å toward the hydrophobic interior of the mMD-2 pocket, whereas DA193 modeled with hMD-2 is much more alike in its disposition to antagonists Eritoran/hMD-2 and lipid IVa/hMD-2 (Figure 9A-D). The 4- and 4′- phosphate groups of DA193 are involved in strong ionic interactions with Lys122, Lys125, and Arg90 situated at the entrance of the binding cavity of hMD-2 (Figure 9E). Unlike agonistic lipid IVa–mMD-2/TLR4 complex, wherein the exposed lipid chain is stabilized by the hydrophobic contacts with Phe126, the hydrophobic side chain of Phe126 in mMD-2 complexated with antagonistic DA193 is exposed to solvent (Figure 9F, H).
Figure 9.

Left: Superimposition of final h- and mMD-2 structures in complex with DA193 (green) with the co-crystal structures of Lipid A ligands (yellow) bound to h- or mMD-2/TLR4. (A) Comparison of Eritoran-hMD-2 (PDB: 2Z65) with DA193-hMD-2; (B) comparison of lipid IVa–hMD-2 (PDB: 2E59) with DA193-hMD-2; (C) comparison of Ra-LPS-hMD-2 (PDB: 3FXI) with DA193-hMD-2 (for clarity only Lipid A portion of LPS is shown); (D) comparison of lipid IVa-mMD-2 (PDB: 3VQ1) with DA193-mMD-2; Right: intermolecular ionic interactions of 4- and 4′- phosphates of DA193 with positively charged residues at the rim of the binding pocket of hMD-2 (E), and mMD-2 (F); ionic interactions of 4′- and 1-phosphates of lipid IVa with Lys and Arg of hMD-2 (PDB: 2E59) (G), and mMD2 (PDB: 3VQ1) (H). The “twisted” orientation of the proximal GlcN ring of mMD-2-bound lipid IVa (H) enables the exposure of a single fatty chain (stabilized by a hydrophobic interaction with Phe126) on the surface of mMD-2. Images were generated with PyMol.
Conclusions
The inability of conformationally constrained tetraacylated Lipid A mimetics 1 to activate mTLR4/MD-2 complex can be rationalized as follows. The exposure of the 2N-β–hydroxyacyl chain (R2N) of Lipid A on the surface of MD-2, secured by repositioning of the aromatic side chain of Phe126 as the result of the ligand-induced conformational changes in MD-2, is considered to be the main driving force for receptor dimerization and initiation of signaling, which has been shown for both human and murine MD-2/TLR4–LPS complexes.3,20,39,40 The dihedrals about (1↔1)-glycosidic linkage determining the relative spatial orientation of two sugar rings in mimetics 1 can only be marginally affected. Consequently, in contrast to mMD-2–bound lipid IVa, the “proximal” GlcN ring of the diglucosamine backbone of tetraacylated mimetics 1 cannot be relocated in a “tilted” orientation (seen in Figure 3A, B). As a result, none of the lipid chains can be presented on the surface of mMD-2, so that the dimerization and establishment of the active homodimeric Lipid IVa–mMD-2/TLR4 complex is not possible.
Collectively, our results define a crucial role of the inherent plasticity of the carbohydrate backbone of Lipid A, which is decisive for the relocation of a single lipid chain onto the surface of MD-2, in the ligand–receptor structure–function relationships. Replacement of the flexible three-bond (1→6)-linked backbone of Lipid A by the conformationally confined (1↔1) glycosidically connected diglucosamine scaffold resulted in abrogation of species-specific agonistic activity of lipid IVa. Manipulating conformational flexibility of the carbohydrate backbone of Lipid A represents therefore a useful tool for the rational design of potential MD-2/TLR4—specific immuno-modulating therapeutics.
Novel antiendotoxic Lipid A mimetics 1 based on the β,α–(1↔1) diglucosamine scaffold represent a lead structure for the development of pharmaceutically relevant prospective medications for sepsis as well as preventive therapeutics for other disorders resulting from an overexuberant LPS–MD-2/TLR4 mediated immune response. Profound antagonistic effects of compounds 1 not only on human but also on mouse TLR4 open wide opportunities for in vivo animal trials. One of the reasons for the rapid deactivation of the failed antisepsis drug candidate Eritoran,41 after intravenous administration, was hydrolysis of the anomeric phosphate functionality leading to inactive metabolite.42 Replacement of the labile anomeric phosphate by a hydrolytically stable secondary phosphoester group and straightforward high-yielding synthesis renders novel tetraacylated Lipid A mimetics 1 to attractive anti-inflammatory drug candidates.
Methods
Inhibition of h- and mTLR4 by Mimetics 1 in T-Rex-293 Cells Stimulated with E. coli Lipid A
T-REx cell lines stably expressing human or mouse TLR4 were made using the Flp-In system according to the manufacturer’s instructions (Invitrogen). Briefly, h- and mTLR4 nucleotide sequences were cloned from the pUNO-HA vector into pcDNA5/FRT expression vector, which was then transiently transfected into Flp-In T-REx cells along with pOG44 plasmid for expression of the Flp recombinase. After homologous recombination between the FRT sites in the T-REx cell genome and the pcDNA5/FRT vector, stable cells were selected for hygromycin resistance. T-REx-293 cells were grown in DMEM supplemented with 10% FBS. Lipid IVa mimetics 1 were dissolved in DMSO to provide 1 mg mL–1 stock solutions. Further dilutions were made prior to cell culture stimulation with the DMEM cell culture medium supplemented with 10% FBS. HEK293 and T-REx/TLR4 cells were seeded in 96-well Costar plates (Corning, NY, U.S.A.) at 3 × 104 cells/well and incubated overnight in a humidified atmosphere with 5% CO2 at 37 °C. The next day the T-Rex/TLR4 cells were cotransfected with pEF-BOS-MD-2 (10 ng/well), NF-κB-dependent luciferase (fLuc) (50 ng/well), and constitutive Renilla (rLuc) (10 ng/well) reporter plasmids using JetPEI transfection reagent. Cells were treated 6 h after transfection with the indicated concentration of ligands (E. coli Lipid A and/or mimetics 1 or lipid IVa). E. coli Lipid A was added in a total volume of 10 μL, the mimetics 1 were applied at final concentrations of 1000 ng/mL in a volume of 10 μL. In case of preincubation with antagonists, mimetics 1 were added 1 h prior to E. coli Lipid A. In case of prestimulation with agonist, E. coli Lipid A was added 1 h prior to mimetics 1. After 16 h of stimulation the cells were lysed in 1× reporter assay lysis buffer (Promega, U.S.A.) and analyzed for reporter gene activities using a dual-luciferase reporter assay system. Relative luciferase activity (RLA) was calculated by normalizing each sample’s luciferase activity (fLuc) for constitutive Renilla activity (fLuc) measured within the same sample.
Inhibition of IL-6 Expression by Antagonist Mimetics 1 in Human Monocyte Cell Line (MM6) Stimulated with E. coli Lipid A
The MonoMac6 cells (MM6) were grown in RPMI medium supplemented with 10% FBS, 2% nonessential amino acids and 1% OPI supplement. For cytokine production the MonoMac6 cells were seeded in 24-well plates at 7 × 105 cells/well and incubated overnight in a humidified atmosphere with 5% CO2 at 37 °C. The next day the cells were stimulated with the indicated concentrations of E. coli Lipid A or DA193/DA254 and E. coli Lipid A, which were added to the wells at the same time, 16 h later the cell culture supernatants were collected and human interleukin-6 concentration was determined using the “ReadySetGo” ELISA kits.
Concentration-Dependent hTLR4 Inhibition by Antagonist Mimetics 1 in HEKBlue hTLR4 Cells Stimulated with E. coli O111 LPS
HEKBlue hTLR4 cells are HEK293 cells stably expressing human TLR4, MD-2, CD14, and a secreted NFκB dependent reporter (secreted embryonic alkaline phosphatase, SEAP, InvivoGen). Mimetics 1 were dissolved in DMSO to give 1 mg mL–1 stock solutions. Further dilution was made with DMEM supplemented with 10% FCS. The compounds 1 were added in a total volume of 10 μL to 25000 HEKBlue hTLR4 cells in 180 μL, E. coli O111 LPS was applied at the concentration of 5 ng/mL in a total volume of 10 μL, and the plates were incubated at 37 °C and 5% CO2. In case of preincubation with antagonist, the solution of compounds 1 was added first, the plate was incubated for 1 h, then the cells were stimulated with E. coli O111 LPS and the plate was incubated for 20–24h. In case of simultaneous addition of E. coli LPS and 1, the antagonists were added together with E. coli O111 LPS and the plate was incubated for 20–24 h. In case of preactivation with LPS, the cells were stimulated with E. coli O111 LPS first, incubated for 1 h, then antagonists 1 were applied and the plate was incubated for 20–24 h. Detection followed the QUANTI-Blue protocol (InvivoGen). Briefly, 20 μL of challenged cells supernatant were incubated with 180 μL detection reagent. Plates were incubated at 37 °C and 5% CO2 and SEAP levels were determined at 650 nm using a spectrophotometer (SpectraMAX 190).
In Vivo Experiments
C3H/HeN (22–24 weeks old) male mice were randomly assigned into groups, weighed, and injected intraperitoneally with the appropriate amount of E. coli 055:B5 LPS, DA255, a mixture of both or the vehicle control, as indicated in SI Figure 3S. The stock solution of the LPS (5 mg mL–1) was prepared in endotoxin-free PBS, the stock solution of DA255 (1 mg mL–1) was prepared in DMSO. Dilutions of E. coli 055:B5 LPS or DA255 were done in endotoxin-free PBS. Blood was collected 1 h later from the tail vein and serum was prepared using heparin-gel tubes. All animal experiments were performed in accordance with the institutional guidelines and were approved by the Veterinary Administration of the Republic of Slovenia. Serum from the C3H/HeN mice was tested with the mouse TNF-α ELISA kit (“ReadySetGo”, eBioscience) to determine the levels of mouse TNF-α. The experiment was performed according to the manufacturer’s instructions.
Acknowledgments
Financial support from Austrian Science Foundation (FWF) (Grant No. FWF P-22116), Slovenian Research Agency, and COST action BM1003 is gratefully acknowledged.
Supporting Information Available
Supporting information includes supplementary tables and figures, methods for molecular dynamics simulation, synthetic procedures, experimental details, analytical data for all new synthetic compounds, NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Miyake K. (2006) Roles for accessory molecules in microbial recognition by Toll-like receptors. J. Endotoxin Res. 12, 195–204. [DOI] [PubMed] [Google Scholar]
- Prohinar P.; Re F.; Widstrom R.; Zhang D.; Teghanemt A.; Weiss J. P.; Gioannini T. L. (2007) Specific high affinity interactions of monomeric endotoxin–protein complexes with Toll-like receptor 4 ectodomain. J. Biol. Chem. 282, 1010–1017. [DOI] [PubMed] [Google Scholar]
- Park B. S.; Song D. H.; Kim H. M.; Choi B. S.; Lee H.; Lee J. O. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195. [DOI] [PubMed] [Google Scholar]
- Munford R. S. (2008) Sensing Gram-negative bacterial lipopolysaccharides: A human disease determinant?. Infect. Immun. 76, 454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. H.; Kim N. D.; Jung J. K.; Lee C. K.; Han S. B.; Kim Y. (2012) Myeloid differentiation 2 as a therapeutic target of inflammatory disorders. Pharmacol.Ther. 133, 291–298. [DOI] [PubMed] [Google Scholar]
- Opal S. M. (2007) The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int. J. Med. Microbiol. 297, 365–377. [DOI] [PubMed] [Google Scholar]
- Hammad H.; Chieppa M.; Perros F.; Willart M. A.; Germain R. N.; Lambrecht B. N. (2009) House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med. 15, 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdollahi-Roodsaz S.; Joosten L. A. B.; Roelofs M. F.; Radstake T. R. D. J.; Matera G.; Popa C.; Van der Meer J. W. M.; Netea M. G.; van den Berg W. B. (2007) Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 56, 2957–2967. [DOI] [PubMed] [Google Scholar]
- Abreu M. T.; Arditi M. (2004) Innate immunity and toll-like receptors: Clinical implications of basic science research. J. Pediatr. 144, 421–429. [DOI] [PubMed] [Google Scholar]
- Reisser D., and Jeannin J.-F. (2009) Lipid A in cancer therapies: Preclinical studies. In Lipid A in Cancer Therapy, pp 101–110, Springer, Landes Bioscience, New York. [Google Scholar]
- Ohto U.; Fukase K.; Miyake K.; Satow Y. (2007) Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 316, 1632–1634. [DOI] [PubMed] [Google Scholar]
- Takayama K.; Qureshi N.; Beutler B.; Kirkland T. N. (1989) Diphosphoryl lipid A from Rhodopseudomonas sphaeroides ATCC 17023 blocks induction of cachectin in macrophages by lipopolysaccharide. Infect. Immun. 57, 1336–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ W. J.; McGuinness P. D.; Asano O.; Wang Y.; Mullarkey M. A.; Perez M.; Hawkins L. D.; Blythe T. A.; Dubuc G. R.; Robidoux A. L. (1994) Total synthesis of the proposed structure of Rhodobacter sphaeroides Lipid A resulting in the synthesis of new potent lipopolysaccharide antagonists. J. Am. Chem. Soc. 116, 3637–3638. [Google Scholar]
- Hawkins L. D.; Christ W. J.; Rossignol D. P. (2004) Inhibition of endotoxin response by synthetic TLR4 antagonists. Curr. Top. Med. Chem. 4, 1147–1171. [DOI] [PubMed] [Google Scholar]
- Shirey K. A.; Lai W.; Scott A. J.; Lipsky M.; Mistry P.; Pletneva L. M.; Karp C. L.; McAlees J.; Gioannini T. L.; Weiss J.; Chen W. H.; Ernst R. K.; Rossignol D. P.; Gusovsky F.; Blanco J. C. G.; Vogel S. N. (2013) The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 497, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. M.; Park B. S.; Kim J. I.; Kim S. E.; Lee J.; Oh S. C.; Enkhbayar P.; Matsushima N.; Lee H.; Yoo O. J.; Lee J. O. (2007) Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130, 906–917. [DOI] [PubMed] [Google Scholar]
- Meng J.; Gong M.; Björkbacka H.; Golenbock D. T. (2011) Genome-wide expression profiling and mutagenesis studies reveal that lipopolysaccharide responsiveness appears to be absolutely dependent on TLR4 and MD-2 expression and is dependent upon intermolecular ionic interactions. J. Immunol. 187, 3683–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teghanemt A.; Re F.; Prohinar P.; Widstrom R.; Gioannini T. L.; Weiss J. P. (2008) Novel roles in human MD-2 of phenylalanines 121 and 126 and tyrosine 131 in activation of Toll-like receptor 4 by endotoxin. J. Biol. Chem. 283, 1257–1266. [DOI] [PubMed] [Google Scholar]
- Resman N.; Vasl J.; Oblak A.; Pristovsek P.; Gioannini T. L.; Weiss J. P.; Jerala R. (2009) Essential roles of hydrophobic residues in both MD-2 and Toll-like receptor 4 in activation by endotoxin. J. Biol. Chem. 284, 15052–15060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohto U.; Fukase K.; Miyake K.; Shimizu T. (2012) Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl. Acad. Sci. U.S.A. 109, 7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J.; Lien E.; Golenbock D. T. (2010) MD-2-mediated ionic interactions between Lipid A and TLR4 are essential for receptor activation. J. Biol. Chem. 285, 8695–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C.; Gangloff M.; Monie T.; Smyth T.; Wei B.; McKinley T. J.; Maskell D.; Gay N.; Bryant C. (2008) Elucidation of the MD-2/TLR4 interface required for signaling by lipid IVa. J. Immunol. 181, 1245–1254. [DOI] [PubMed] [Google Scholar]
- Meng J.; Drolet J. R.; Monks B. G.; Golenbock D. T. (2010) MD-2 residues Tyrosine 42, Arginine 69, Aspartic acid 122, and Leucine 125 provide species specificity for lipid IVa. J. Biol. Chem. 285, 27935–27943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zughaier S. M.; Zimmer S. M.; Datta A.; Carlson R. W.; Stephens D. S. (2005) Differential induction of the Toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect. Immun. 73, 2940–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent M. S.; Stead C. M.; Tran A. X.; Hankins J. V. (2006) Diversity of endotoxin and its impact on pathogenesis. J. Endotoxin Res. 12, 205–223. [DOI] [PubMed] [Google Scholar]
- Akamatsu M.; Fujimoto Y.; Kataoka M.; Suda Y.; Kusumoto S.; Fukase K. (2006) Synthesis of lipid A monosaccharide analogues containing acidic amino acid: Exploring the structural basis for the endotoxic and antagonistic activities. Bioorg. Med. Chem. 14, 6759–6777. [DOI] [PubMed] [Google Scholar]
- Fujimoto Y.; Adachi Y.; Akamatsu M.; Fukase Y.; Kataoka M.; Suda Y.; Fukase K.; Kusumoto S. (2005) Synthesis of lipid A and its analogues for investigation of the structural basis for their bioactivity. J. Endotoxin Res. 11, 341–347. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Gaekwad J.; Wolfert M. A.; Boons G. J. (2007) Modulation of innate immune responses with synthetic Lipid A derivatives. J. Am. Chem. Soc. 129, 5200–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazin H. G.; Murray T. J.; Bowen W. S.; Mozaffarian A.; Fling S. P.; Bess L. S.; Livesay M. T.; Arnold J. S.; Johnson C. L.; Ryter K. T.; Cluff C. W.; Evans J. T.; Johnson D. A. (2008) The “Ethereal” nature of TLR4 agonism and antagonism in the AGP class of lipid A mimetics. Bioorg. Med. Chem. Lett. 18, 5350–5354. [DOI] [PubMed] [Google Scholar]
- Seydel U.; Oikawa M.; Fukase K.; Kusumoto S.; Brandenburg K. (2000) Intrinsic conformation of lipid A is responsible for agonistic and antagonistic activity. Eur. J. Biochem. 267, 3032–3039. [DOI] [PubMed] [Google Scholar]
- Rao V. S. R., Qasba P. K., Balaji P. V., and Chandrasekaran R. (1998) Conformation of disaccharides. In Conformation of Carbohydrates, pp 91–130, OPA: Overseas Publishers Association, Harwood Academic Publishers, Amsterdam. [Google Scholar]
- Oikawa M.; Shintaku T.; Fukuda N.; Sekljic H.; Fukase Y.; Yoshizaki H.; Fukase K.; Kusumoto S. (2004) NMR conformational analysis of biosynthetic precursor-type lipid A: monomolecular state and supramolecular assembly. Org. Biomol. Chem. 2, 3557–3565. [DOI] [PubMed] [Google Scholar]
- Wang W.; Sass H.-J.; Zähringer U.; Grzesiek S. (2008) Structure and dynamics of 13C,15N-labeled lipopolysaccharides in a membrane mimetic. Angew. Chem. Int. Ed. 47, 9870–9874. [DOI] [PubMed] [Google Scholar]
- Taga T.; Miwa Y.; Min Z. (1997) α,β-Trehalose monohydrate. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 53, 234–236. [Google Scholar]
- Dowd M. K.; Reilly P. J.; French A. D. (1992) Conformational analysis of trehalose disaccharides and analogues using MM3. J. Comput. Chem. 13, 102–114. [Google Scholar]
- Peric-Hassler L.; Hansen H. S.; Baron R.; Hünenberger P. H. (2010) Conformational properties of glucose-based disaccharides investigated using molecular dynamics simulations with local elevation umbrella sampling. Carbohydr. Res. 345, 1781–1801. [DOI] [PubMed] [Google Scholar]
- Gaekwad J.; Zhang Y.; Zhang W.; Reeves J.; Wolfert M. A.; Boons G. J. (2010) Differential induction of innate immune responses by synthetic Lipid A derivatives. J. Biol. Chem. 285, 29375–29386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukase K.; Fukase Y.; Oikawa M.; Liu W. C.; Suda Y.; Kusumoto S. (1998) Divergent synthesis and biological activities of lipid A analogues of shorter acyl chains. Tetrahedron 54, 4033–4050. [Google Scholar]
- DeMarco M. L.; Woods R. J. (2011) From agonist to antagonist: Structure and dynamics of innate immune glycoprotein MD-2 upon recognition of variably acylated bacterial endotoxins. Mol. Immunol. 49, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Phillips R. L.; Zhang D.; Teghanemt A.; Weiss J. P.; Gioannini T. L. (2012) NMR studies of hexaacylated endotoxin bound to wild-type and F126A mutant MD-2 and MD-2-TLR4 ectodomain complexes. J. Biol. Chem. 287, 16346–16355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barochia A.; Solomon S.; Cui X.; Natanson C.; Eichacker P. Q. (2011) Eritoran tetrasodium (E5564) treatment for sepsis: Review of preclinical and clinical studies. Expert Opin. Drug Metab. Toxicol. 7, 479–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischer J. G.; Rossignol D.; Francis G. A.; Chan T.; Lynn M.; Wasan K. M. (2012) Deactivation of the lipopolysaccharide antagonist eritoran (E5564) by high-density lipoprotein-associated apolipoproteins. Innate Immun. 18, 171–178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.