

Abstract
Background
Enteric pathogens utilize a distinct set of proteins to modulate host cell signaling events that promote host cell invasion, induction of the inflammatory response, and intracellular survival. Human infection with Campylobacter jejuni, the causative agent of campylobacteriosis, is characterized by diarrhea containing blood and leukocytes. The clinical presentation of acute disease, which is consistent with cellular invasion, requires the delivery of the Campylobacter invasion antigens (Cia) to the cytosol of host cells via a flagellar Type III Secretion System (T3SS). We identified a novel T3SS effector protein, which we termed CiaD that is exported from the C. jejuni flagellum and delivered to the cytosol of host cells.
Results
We show that the host cell kinases p38 and Erk 1/2 are activated by CiaD, resulting in the secretion of interleukin-8 (IL-8) from host cells. Additional experiments revealed that CiaD-mediated activation of p38 and Erk 1/2 are required for maximal invasion of host cells by C. jejuni. CiaD contributes to disease, as evidenced by infection of IL-10 knockout mice. Noteworthy is that CiaD contains a Mitogen-activated protein (MAP) kinase-docking site that is found within effector proteins produced by other enteric pathogens. These findings indicate that C. jejuni activates the MAP kinase signaling pathways Erk 1/2 and p38 to promote cellular invasion and the release of the IL-8 pro-inflammatory chemokine.
Conclusions
The identification of a novel T3SS effector protein from C. jejuni significantly expands the knowledge of virulence proteins associated with C. jejuni pathogenesis and provides greater insight into the mechanism utilized by C. jejuni to invade host cells.
Keywords: Invasion, Erk 1/2, p38, IL-8 secretion, Type III secretion system
Lay abstract
In contrast to the conventional view that the flagellum is only used for bacterial motility, Campylobacter jejuni also uses the flagellum as a Type III Secretion System (T3SS). A subset of proteins exported from the flagellum are delivered to the cytosol of host cell, where they modify host cell signaling events to the benefit of the bacterial pathogen. Here we report that C. jejuni, which is the leading bacterial cause of food-borne disease worldwide, possesses a novel T3SS virulence protein that we termed CiaD. We show that CiaD is required for the invasion of host cells and for the secretion of the inflammatory chemokine interleukin-8 from host cells. We also show that the newly identified virulence protein CiaD is required for the development of disease. The fact that CiaD is required of the development of disease provides a significant advancement in the understanding of C. jejuni pathogenesis.
Background
Campylobacter jejuni is a leading cause of gastroenteritis worldwide, causing ~1.4 to 2.3 million cases each year in the United States [1,2]. A serious complication of C. jejuni infection is the development of Guillain–Barré syndrome (GBS), an autoimmune disease affecting the peripheral nervous system [3]. The ability of C. jejuni to cause acute disease is a complex multifactorial process, requiring cell adherence, invasion, and intracellular survival [4-10]. Key to host cell invasion and survival is the synthesis of the Campylobacter invasion antigens (Cia) [11]. Cia proteins are exported from the bacterium’s flagellar Type III Secretion System (T3SS) and are delivered to the host cell to promote maximal cell invasion [12].
Only three (CiaB, CiaC, and CiaI) of ~18 Cia proteins have been identified to date [4,13,14]. While the precise functions of these proteins are not known, in vivo studies suggest that they contribute to disease. More specifically, inoculation of piglets with a C. jejuni wild-type strain and ciaB mutant results in different clinical signs. Piglets infected with C. jejuni develop diarrhea within 24 hours, and exhibit severe histological lesions, including shortening of the villi and production of an exudate in the lumen. In contrast, piglets inoculated with a C. jejuni ciaB-knockout mutant do not develop diarrhea until 3 days post-infection and exhibit only minor histological lesions [14]. Introducing a wild-type copy of the ciaB gene into the C. jejuni ciaB mutant restored the isolate’s virulence [15]. Additional work is needed to identify and characterize the Cia proteins to gain a better understanding of C. jejuni pathogenesis.
C. jejuni invades the cells lining the gastrointestinal tract and induces a potent inflammatory response characterized by the secretion of interleukin-8 (IL-8). However, little is known about the mechanism underlying the induction of IL-8 secretion by C. jejuni. Bacterial factors, such as the flagellum and CpG dinucleotide, are typical immune stimulators [16]. However, Toll-like receptor 5 (TLR5) is not stimulated by the C. jejuni flagellum [17,18]. Similarly, TLR9, which recognizes CpG dinucleotides, is not efficiently stimulated by C. jejuni[16]. While C. jejuni is clearly stimulating a proinflammatory response, the mechanism of immune activation is not completely understood.
Researchers have found that the NF-κB pathway is activated by C. jejuni. The NF-κB pathway is activated by JlpA, CdtABC, and peptidoglycan (intracellular NOD-1 receptors) [19-21]. While both the NF-κB and AP-1 transcriptional activators are required for expression of the gene encoding IL-8, also known as huCXCL8 [22], the mechanism of activation of AP-1 by C. jejuni is not known. In addition to their role in cell growth and differentiation, Erk 1/2 and p38 serve as important activators of the immune response in non-phagocytic cells through the activation of AP-1. Several labs have reported that Erk 1/2 and p38 signaling pathways are activated by C. jejuni, and that the activation of these pathways is dependent on bacterial de novo protein synthesis and a functional flagellum [18,23,24]. The C. jejuni factors necessary for Erk 1/2 and p38 mediated IL-8 secretion are not known.
We hypothesized that C. jejuni delivers one or more of the Cia proteins to host cells where they trigger the induction of IL-8 secretion from host cells. Here we identify a novel protein (Cj0788), which we termed Campylobacter invasion antigen D (CiaD), that is secreted via the flagellar T3SS. CiaD is required for maximal C. jejuni invasion and IL-8 secretion from human INT 407 epithelial cells. We also show that CiaD is required for the development of acute disease in vivo. Specifically, the C. jejuni wild-type strain resulted in disease characterized by a thickening of the gastrointestinal tract wall, enlarged ileocecocolic lymph nodes, and bloody lumen contents in cecum and colon, which was absent in mice infected with the C. jejuni ciaD mutant. These data are significant, as this is the first time that a C. jejuni effector protein has been shown to contribute to the development of disease in a mouse model.
Results
The flagellum is required for CiaD delivery to host epithelial cells
Previous work in our lab led to the identification of 42 proteins that contain a putative C. jejuni flagellar T3SS export signal [4]. We sought to determine if one of these proteins, Cj0788, designated Campylobacter invasion antigen D (CiaD), is secreted by C. jejuni. We tested if CiaD is secreted from a C. jejuni wild-type strain and ciaD mutant harboring a plasmid encoding CiaD fused to the adenylate cyclase domain (ACD) of the CyaA protein from Bordetella pertussis. The CiaD-ACD fusion protein was secreted from the C. jejuni wild-type strain and a ciaD mutant but not the flgBC flagellar mutant, as judged by immunoblot analysis using an ACD specific antibody (Figure 1A and Additional file 1: Figure S1A). To determine if CiaD is required for Cia secretion, we tested if a second Cia protein (CiaC) could be exported from the ciaD mutant transformed with a construct harboring CiaC-ACD. In addition, the ciaD mutant was transformed with a construct harboring MetK-ACD (a known cytosolic protein), as a negative control. In contrast to MetK, the CiaC effector protein was secreted from the ciaD mutant (Additional file 1: Figure S1A). These assays show that CiaD is secreted from a C. jejuni wild-type strain and is not required for the secretion of other Cia proteins.
Figure 1.
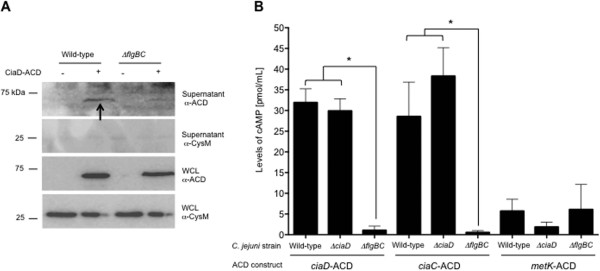
CiaD is delivered to the cytosol of human epithelial cells. (A) CiaD is secreted from C. jejuni. A C. jejuni wild-type strain transformed with the pRY111 vector harboring CiaD fused to the adenylate cyclase domain (CiaD-ACD, + designation) was analyzed by immunoblot analysis. CysM is a cytoplasmic protein (O-acetylserine sulfhydrylase B) involved in cysteine biosynthesis (Mr = 32.4 kDa). Also included was a C. jejuni wild-type strain without the pRY111 shuttle vector as a negative control (- designation) and a C. jejuni flgBC mutant expressing CiaD-ACD and a pRY111 shuttle vector. (B) CiaD is delivered to host cells. A C. jejuni wild-type strain, ciaD mutant, and flgBC mutant were transformed with pRY111 vector harboring the ciaD-ACD, ciaC-ACD, and metK-ACD inserts. C. jejuni harboring the metK-ACD vector was used as a negative control. The CiaC-ACD, a known delivered protein, was included as a positive control. The delivery of the CiaD-ACD, CiaC-ACD, and MetK-ACD fusion proteins to the cytosol of human INT 407 cells was determined using the ACD delivery assay described in Methods. The asterisk indicates that the amount of cAMP produced in the wild-type strain was significantly greater than the value obtained from the flgBC mutant, as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Each error bar represents ± the standard error of the mean (SEM) of N = 9 samples from 3 independent experiments.
To determine if CiaD is delivered to the cytosol of human epithelial cells, INT 407 human intestinal cells were infected with C. jejuni transformed with the CiaD-ACD construct and host cell cAMP levels were measured via ELISA as described previously [12]. The C. jejuni wild-type strain transformed with the CiaC-ACD and MetK-ACD constructs were included as positive and negative controls, respectively. All of the fusion proteins were synthesized in the transformed C. jejuni isolates (Additional file 1: Figure S1B). However, in comparison to the negative control, a significant increase (P < 0.05) in the level of cAMP was observed in cells inoculated with the C. jejuni CiaD-ACD strain (Figure 1B). The delivery of CiaD was found to be dependent on a functional flagellum, as infection of INT 407 cells with a C. jejuni flgBC mutant (secretion negative control) transformed with the CiaD-ACD construct resulted in a significant decrease in cAMP detected as compared to the C. jejuni wild-type strain (Figure 1B). We utilized the Cia delivery assay to determine if the ciaD mutant could deliver CiaC to a host cell. Again, in contrast to MetK, the CiaC effector protein was delivered from the ciaD mutant to host cells, as judged by a significant increase in cAMP versus the controls.
CiaD induces the secretion of IL-8 from epithelial cells
An effector protein is defined as a protein delivered from a pathogen to a host cell that ultimately functions to alter host cell behavior. Based on the finding that CiaD is delivered to host cells, we postulated that CiaD acts as an effector protein. In-silico analysis was used to look for eukaryotic domains in CiaD. The web-based program Eukaryotic Linear Motif (ELM) revealed that CiaD contained a Mitogen-activated protein kinase docking (MKD) motif (probability score = 0.0043) and a proline-directed phosphorylation ((S/T)P) motif (probability score = 0.0154) (Figure 2A) [25,26]. In addition, the Phyre2 protein folding prediction software [27] revealed that CiaD contains a putative nucleotidyltransferase folding domain (52.6% confidence) (not shown). Nucleotidyltransferase domains are commonly found in bacterial effector proteins involved in adenylylation of RhoGTPase leading to actin remodeling [28]. The presence of these eukaryotic domains raised the possibility that CiaD might alter host cell behavior.
Figure 2.

CiaD is required for the maximal secretion of interleukin-8 from host cells. A) Schematic representation of the CiaD Mitogen-activated protein kinase docking (MKD) motif residues 137–147 and the proline-directed phosphorylation site ((S/T)P) motif residue 54. (B) CiaD is required for the maximal secretion of IL-8 by INT 407 cells. The flgBC flagellar mutant was included as a negative control. The values obtained from uninfected cells were subtracted from all other values. The asterisks indicate a significant reduction in the amount of IL-8 detected in the supernatants compared to the value obtained using the C. jejuni wild-type strain, using a one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM of N = 12 samples from 3 independent experiments. (C) CiaD is secreted from a C. jejuni ciaD mutant transformed with pRY111 vector harboring the CiaD-FLAG-tagged protein. Blots were probed with FLAG and CysM antibodies. Also included was a C. jejuni ciaD mutant without the pRY111 shuttle vector (-) as a negative control. (D) The C. jejuni ciaD mutant is motile. Motility assays were performed as outlined in Methods. (E) Deletion of ciaD does not have an effect on C. jejuni binding to host cells at 30 min post-infection.
Investigators have reported that C. jejuni de novo protein synthesis is required for maximal secretion of IL-8 from host cells [18,29]. Consistent with these reports, we found that incubation of C. jejuni with chloramphenicol (a protein synthesis inhibitor) for 30 min prior to inoculation of human INT 407 cells reduced the amount of IL-8 secreted from the host cells as well as C. jejuni invasion (Additional file 2: Figure S2A and S2B). Noteworthy is that INT 407 cells are responsive to innate immune signaling molecules that engage TLR4 and TLR2 [30]. Previous work has also indicated that the genes encoding the Cia proteins are induced when C. jejuni are cultured with epithelial cells [11]. Taken together, these findings raised the possibility that a Cia protein was required for IL-8 induction. Based on the presence of the MKD site in CiaD and the link between IL-8 induction and MAP kinase signaling, we measured the IL-8 pro-inflammatory chemokine in the supernatants collected from INT 407 cells inoculated with the C. jejuni wild-type strain and ciaD mutant. The C. jejuni flgBC mutant was included as a negative control; this mutant binds to the cells at levels similar to that of a C. jejuni wild-type strain but is deficient in Cia protein secretion and delivery (not shown). A significant decrease (P < 0.01) in secreted IL-8 was observed in cells inoculated with the C. jejuni ciaD and flgBC mutants when compared to a C. jejuni wild-type strain (Figure 2B). Additionally, IL-8 secretion from host cells was not altered by removal of non-adherent bacteria by washing or in the presence of non-adherent bacteria (not shown).
Several additional findings indicate that CiaD is, in part, responsible for inducing IL-8 secretion from host cells. First, insertion of a wild-type copy of the ciaD gene driven by the constitutive promoter hupB into the ciaD mutant in trans (complemented strain) resulted in an isolate that displayed an IL-8 secretion phenotype indistinguishable from that of the wild-type strain (Figure 2B). Second, ectopic expression of a gene that encodes CiaD-EGFP in host INT 407 cells resulted in a moderate increase in IL-8 secretion as compared to ectopic expression of EGFP only (Additional file 3: Figure S3). We further demonstrated that: a) CiaD protein is secreted from the ciaD complemented isolate, as judged by immunoblot analysis (Figure 2C); b) the C. jejuni ciaD mutant and ciaD complemented isolate are both motile, as judged by motility assays, indicating a functional flagellum (Figure 2D); and c) the C. jejuni wild-type strain, ciaD mutant, and ciaD complemented isolate bound to INT 407 cells with equal efficiency, as judged by a cell binding assay (Figure 2E). Moreover, no difference was observed in the binding of all of the isolates to the host cells over an eight-hour period (not shown). In separate assays, we found that the inoculation of INT 407 cells with an isolate that contained a knockout in Cj0789, which is the gene immediately downstream of ciaD, resulted in a similar level of secreted IL-8 as that of the wild-type strain (Additional file 4: Figure S4). Consistent with the proposal that the delivery of CiaD to host cells requires bacteria-host cell contact, the addition of supernatants containing the Cia proteins to INT 407 cells did not induce IL-8 secretion (not shown). Based on these data, we concluded that CiaD is an effector protein that is involved in the induction of IL-8 secretion.
CiaD is required for cell invasion
We performed experiments to determine if there is a possible link between IL-8 induction and C. jejuni invasion of host cells. The ability of the C. jejuni ciaD mutant to invade INT 407 cells was determined using the gentamicin-protection assay. The C. jejuni ciaC mutant was included as a control, as this mutant displays a significant reduction in cell invasion compared to a wild-type strain of C. jejuni. Both the C. jejuni ciaD and ciaC mutants exhibited a reduction in cell invasion when compared to a wild-type strain (Figure 3A). To determine if cell invasion is required to induce IL-8 secretion from a host cell, INT 407 cells were inoculated with the C. jejuni ciaD and ciaC mutants and the amount of IL-8 secreted into the supernatants was determined. Consistent with our previous findings, CiaD was required to induce maximal IL-8 secretion (Figure 2B). We also found that the ciaC mutant induced levels of IL-8 secretion indistinguishable from the C. jejuni wild-type strain (Figure 3B). This finding suggested that invasion and IL-8 secretion are not directly linked.
Figure 3.

IL-8 secretion from INT 407 cells is not invasion dependent, but MAPK activation is required for host cells invasion by C.jejuni. (A) A C. jejuni ciaD mutant is deficient in maximal invasion of INT 407 cells. The C. jejuni ciaC mutant was included as an invasion deficient control. (B) IL-8 secretion does not require bacterial invasion. INT 407 cells were inoculated with various C. jejuni isolates, incubated for 24 hr, and IL-8 quantified using an IL-8 ELISA as described in Methods. The C. jejuni flgBC flagellar mutant (Cia secretion negative) and C. jejuni ciaC mutant (CiaC secretion negative and invasion deficient) were included as negative controls. (C) MAP kinase activation is required for C. jejuni invasion. Inhibitors to Erk 1/2 (PD98059) and p38 (SB 202190) were added to INT 407 cells for 30 min prior to the addition of a C. jejuni wild-type strain. (D) MAP kinase activation is required for IL-8 secretion. Inhibitors of Erk 1/2 and p38 were added to INT 407 cells for 30 min prior to inoculation with a C. jejuni wild-type strain. The values obtained from uninfected cells were subtracted from all other values. The asterisks indicate a significant reduction compared to the value obtained for the C. jejuni wild-type strain, according to one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM of N = 12 samples from 3 independent experiments.
To address the role of MAP kinase signaling in C. jejuni induction of IL-8 secretion and host cell invasion, assays were performed in the presence of cellular inhibitors to Erk 1/2 (PD98059) and p38 (SB202190). Inhibition of Erk 1/2 and p38 resulted in a significant reduction (P < 0.01) in the number of C. jejuni internalized (Figure 3C) and the amount of secreted IL-8 (Figure 3D). Consistent with these findings, we found that the amount of IL-8 secreted by the host cells inoculated with the CiaD mutant was reduced significantly when the activation of Erk 1/2 and p38 were inhibited (Additional file 5: Figure S5). Specifically, inhibition of Erk 1/2 results in a 70% reduction in the amount of IL-8 secreted from host cells infected with a C. jejuni wild-type strain, similarly inhibition of Erk 1/2 resulted also in a reduction in IL-8 secreted from host cells that were infected with the C. jejuni ciaD mutant. These results are consistent with the fact that the C. jejuni ciaD mutant activates Erk 1/2 to a level that is slightly above that of cells only. Moreover, the addition of exogenous IL-8 to Caco-2 cells, an intestinal cell line that is responsive to IL-8 because of the presence of the CXCR1 and CXCR2 receptors, did not restore the invasiveness of the C. jejuni ciaD mutant to that of a C. jejuni wild-type strain (Additional file 6: Figure S6). This finding suggests that the invasion phenotype of the ciaD mutant is due to a lack in the initiation of cellular signaling events specific to invasion, and not from the failure to induce the secretion of IL-8 from host cells. We also confirmed that Caco-2 cells are responsive to IL-8, using immunoblot analysis to quantify phospho-Akt; Akt is a downstream target of the CXCR1/2 receptors and is activated by IL-8 [31] (Additional file 6: Figure S6B). Together, these experiments revealed that C. jejuni must activate components of the MAP kinase signaling pathway for both cellular invasion and the secretion of IL-8, and that CiaD contributes to this activation.
CiaD activates of the MAP kinase signaling pathway
Based on the presence of the Mitogen-activated protein kinase docking (MKD) motif in CiaD, experiments were performed to determine if C. jejuni activated the MAP kinase signaling pathway in a CiaD dependent manner. Prior to performing these experiments, we determined that the C. jejuni wild-type strain induces significantly more IL-8 than the ciaD mutant at 4 hr post-inoculation of INT 407 cells (Figure 4A). Based on the kinetics of IL-8 secretion, we performed experiments to identify the cellular signaling pathways that are activated by C. jejuni at 3 hr post inoculation. The use of a MAP kinase phosphor-array screen revealed that the C. jejuni ciaD mutant did not activate Erk 1/2 and p38 to the same extent as inoculation of the INT 407 cells with the wild-type strain (Additional file 7: Figure S7). These results were confirmed by immunoblot analysis (Figure 4B and Figure 4C). The partial activation (above cells only) of Erk 1/2 by the ciaD mutant is consistent with the fact that Erk 1/2 is also partially activated in response to host cell binding mediated by FlpA [32]. In contrast to the C. jejuni ciaD and flgBC (Cia protein secretion-negative) mutants, the C. jejuni wild-type strain and ciaD mutant harboring a wild-type copy of the ciaD gene (complemented ciaD isolate) resulted in the activation of Erk 1/2 and p38 as judged by an increase in band intensity of the phosphorylated form of the protein. Even though the C. jejuni wild-type strain, ciaD mutant, and complemented ciaD isolate resulted in greater activation of NF-κB (p65 subunit phospho-S536) compared to the flgBC mutant, significant differences were not observed in the amount of NF-κB activation between the isolates (Figure 4B and 4C). This finding suggested that a protein or other bacterial component other than CiaD is responsible for the activation of NF-κB. Taken together, our results indicate that CiaD participates in the activation of the MAP kinase signaling molecules Erk 1/2 and p38.
Figure 4.

CiaD is required for activation of the MAP kinase signaling cascade. (A) The amount of IL-8 in supernatants is significantly increased 4 hr post infection in the C. jejuni wild-type strain when compared to the ciaD mutant. (B) Maximal activation of p38 and Erk 1/2 requires CiaD, as judged by immunoblots. Immunoblots were probed with phospho-specific antibodies to Erk 1/2 (Mr = 42 and 44 kDa), p38 (Mr = 38 kDa), and NF-κB p65 subunit phospho-S536 (Mr = 65 kDa). All blots were stripped and re-probed with an anti-tubulin (Mr = 52 kDa) antibody. (C) Maximal activation of p38 and Erk 1/2 requires CiaD. Densitometry was performed on the immunoblots shown in Panel B. The value obtained for cells only was subtracted from all other values. The asterisks indicate a significant difference in values compared to the controls, as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM of N = 6 samples from 6 independent experiments.
The MAP Kinase docking motif of CiaD is required for IL-8 secretion and host cell invasion
Mutational analysis was used to determine whether the putative eukaryotic domains of CiaD are functional. Two C. jejuni ciaD mutants were generated; the MAP kinase docking motif was deleted (ΔMKD) in one mutant and the proline-directed phosphorylation motif was changed to an alanine (ΔS/T)P) in the other mutant (Figure 5A). Immunoblot analysis revealed that both CiaD recombinant proteins were synthesized in the ciaD mutant (Additional file 8: Figure S8A). Importantly, all of the isolates were motile (Additional file 8: Figure S8B). Experiments were then performed to evaluate the ability of the CiaD ΔMKD mutant and CiaD (ΔS/T)P mutant to induce IL-8 secretion and to invade human INT 407 epithelial cells. The CiaD ΔMKD mutant was unable to induce secretion of IL-8 from host cells, and was equally impaired in its ability to invade host cells (Figure 5B and C). In contrast, the CiaD (ΔS/T)P mutant induced the secretion of IL-8 and invaded host cells to a level that was indistinguishable from the C. jejuni wild-type strain (Figure 5B and C). These results indicate that the MKD motif of CiaD is required to modify a host cell signaling pathway(s), and that the interaction of CiaD with a host protein leads to IL-8 secretion and cell invasion.
Figure 5.
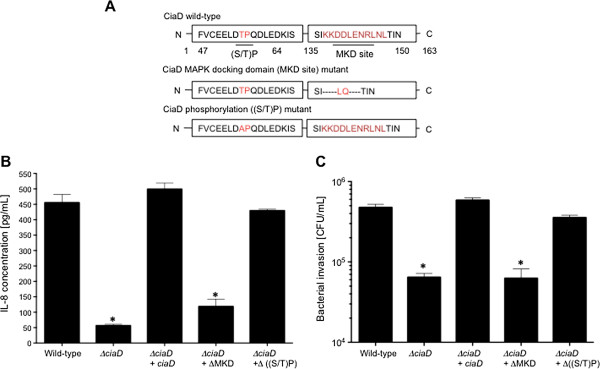
The MKD site of CiaD is necessary for cellular invasion and IL-8 secretion. (A) Schematic representation of the CiaD wild-type protein, the CiaD MAP kinase docking motif (ΔMKD site) mutant protein, and CiaD proline directed-phosphorylation site (Δ(S/T)P) mutant protein. (B) The MKD site of CiaD is required for C. jejuni to induce maximal IL-8 secretion from INT 407 cells. The value obtained from cells only was subtracted from all other values. (C) The MKD site of CiaD is required for maximal C. jejuni invasion of INT 407 cells. The asterisks indicate a significant difference compared to the C. jejuni wild-type strain, as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM of N = 12 samples from 3 independent experiments.
The phenotype of the ciaD mutant is the same in C. jejuni F38011 and 11168 strains
Prior to assessing whether ciaD contributes to the development of acute disease in vivo, it was first necessary to regenerate the ciaD mutant in the C. jejuni 11168 mouse adapted strain and confirm if the mutants behaved in a similar fashion as the C. jejuni F38011 ciaD mutant. More specifically, four isolates were used in these experiments: a) the C. jejuni 11168 wild-type strain; b) the C. jejuni 11168 ciaD mutant; c) the ciaD mutant that synthesizes a CiaD wild-type protein; and d) the ciaD mutant that synthesizes the CiaD ΔMKD recombinant protein. We did not regenerate a ciaD mutant that synthesizes the CiaD Δ(S/T)P mutant protein, as the C. jejuni F38011 isolate that synthesizes this recombinant protein did not yield a unique phenotype. The CiaD protein was readily detected in the C. jejuni 11168 ciaD mutant transformed with the vectors that encode for the CiaD wild-type protein and CiaD ΔMKD-site protein (Additional file 8: Figure S8A). All of the isolates tested were motile (Additional file 8: Figure S8B). We then measured the amount of IL-8 and MIP-2 (a homologue of IL-8 in mice) secreted from human INT 407 cells and mouse CT-26 cells inoculated with the various C. jejuni strains, respectively. The results obtained with the C. jejuni 11168 isolates in INT 407 cells mirrored those obtained with the C. jejuni F38011 strain in INT 407 cells for IL-8 secretion and cell invasion (Figure 6A and B). Similarly, we observed that the ciaD mutant and ciaD mutant that synthesize the CiaD ΔMKD-site recombinant protein were deficient in the ability to induce MIP-2 secretion and invade CT-26 cells (Figure 6C and D). These data indicate that the phenotypes of the 11168 mouse adapted isolates are indistinguishable to that of the C. jejuni F38011 isolates, and that CiaD is required for MIP-2 secretion and cell invasion. Given these findings, we then performed in vivo experiments to determine the contribution of CiaD to campylobacteriosis.
Figure 6.

CiaD function is conserved between strains of C. jejuni and different cell types. (A) The MAP kinase docking motif of CiaD is required for IL-8 secretion from human INT 407 cells. Cells only were subtracted from all shown values. (B) The MAP kinase docking motif of CiaD is required for maximal invasion of human INT 407 cells. (C) The MAP kinase docking motif of CiaD is required for MIP-2 secretion from murine CT-26 cells. MIP-2 concentrations were quantified using an MIP-2 ELISA as described in Methods. Cells only were subtracted from all shown values. (D) The MAP kinase docking motif of CiaD is required for maximal invasion of murine CT-26 cells. The asterisks indicate a significant difference in the value versus that of the C. jejuni wild-type strain, as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM of N = 12 samples from 3 independent experiments.
CiaD is required for the development of disease
C57BL/6 IL-10-/- mice were infected with a C. jejuni 11168 wild-type strain, a ciaD mutant, and a ciaD complemented isolate to assess the contribution of CiaD to the development of disease. Mice sham-inoculated with tryptic soya broth (TSB) were included as a negative control. We found that mice infected with the C. jejuni ciaD mutant exhibited less severe disease when compared to the C. jejuni wild-type strain, as judged by mouse survival (Additional file 9: Figure S9A), gross pathology (Figure 7A), histopathology (Figure 7B and C) and plasma IgG2b anti-C. jejuni antibody levels (Additional file 10: Figure S10). C. jejuni was only recovered at necropsy (either 35 or 36 days after inoculation) from mice inoculated with the wild-type strain (Additional file 9: Figure S9B); however, the fact that IL-10-/- mice are unable to down-regulate inflammatory processes once they are initiated makes it possible to detect disease after pathogen clearance. Importantly, the C. jejuni ciaD mutant expressing a wild-type copy of ciaD produced more severe clinical signs of disease in some mice compared to the ciaD mutant (Figure 7A-C). The C. jejuni ciaD mutant produced no GI lesions, comparable to the TSB controls (Figure 7C, panels 1 and 3) and in contrast to the 11168 wild-type strain that produced severe end stage typhlocolitis with neutrophilic exudates, increased crypt height, crypt abscesses, mononuclear cell infiltration and massive subcutaneous edema (Figure 7C, panel 2). Specifically, the ciaD mutant expressing a wild-type copy of ciaD produced a marked increase in gross pathology (i.e., thickening of GI tract wall and/or enlarged ileocecocolic lymph node) and histopathology (i.e., increased crypt height, goblet cell hyperplasia, and mononuclear cell infiltration) (Figure 7C, panel 4). Application of an established histopathologic scoring system to ileocecocolic sections from mice in all groups confirmed these observations (Figure 7B). Specifically, the ciaD mutant expressing a wild-type copy of CiaD exhibited a significant increase in gross pathology (i.e., thickening of GI tract wall and/or enlarged ileocecocolic lymph node). Theoretically, the ciaD complemented isolated would have achieved the same level of histopathology as the wild-type if it had been allowed to go longer. These results suggest that CiaD is contributing to the development of disease in vivo, but that colonization of IL-10-/- mice is a multi-factorial process. Together, these data indicate that CiaD is necessary for the development of disease in the IL-10-/- mouse model of campylobacteriosis.
Figure 7.

CiaD is required for the development of severe disease. (A) Gross pathology of IL-10-/- mice infected with C. jejuni. Significant differences in gross pathology were observed between the C. jejuni wild-type strain and the ciaD mutant. The C. jejuni ciaD complemented isolate partially restored the gross pathology to levels intermediate between the C. jejuni wild-type strain and ciaD mutant. (B) Ileocecocolic histopathologic lesion scores of all mice in the in vivo oral challenge study. Lesion scores for the ciaD mutant group and the C. jejuni wild-type group were significantly different, based on nonparametric Kruskal Wallis one-way ANOVA followed by the Mann–Whitney test for pairwise comparisons and the Holm-Šidák procedure for correction for multiple comparisons. Lesion scores in the ciaD complemented group showed that 3/13 (23%) of mice had restoration of the inflammatory phenotype. One mouse in the trypticase soy broth (TSB) treated group had spontaneous colitis. (C) Representative histopathology of ileocecocolic junctions. Bars represent 100 μm. Treatment groups were as follows: (1) mouse sham inoculated with trypticase soy broth, (2) mouse infected with the C. jejuni wild-type strain 11168, (3) mouse infected with the ciaD mutant, and (4) mouse infected with the ciaD complemented isolate. Note that the knockout of ciaD prevented tissue inflammation (panel 3) with tissues appearing similar to the TSB controls (panel 1); brackets in panels 1 and 3 show the normal crypt height. Mice receiving the C. jejuni wild-type strain (panel 2) had severe end stage typhlocolitis with significant increases in the crypt height of the lamina propria (upper bracket) and submucosa due to edema (lower bracket). Mononuclear cell infiltrates (dark purple cells) can be seen throughout the tissue. Complementation with ciaD restored the phenotype to that of severe typhlocolitis in the mouse shown in panel 4.
Discussion
In this study we identified and characterized a novel C. jejuni effector protein that is secreted and delivered to host cells via the flagellar T3SS. We found that CiaD is delivered to the cytosol of human INT 407 epithelial cells via a flagellar T3SS, where it is involved in maximal activation of the MAP kinase signaling pathways Erk 1/2 and p38. Interestingly, CiaD contains a MKD that is necessary for subversion of the host cell signaling pathways, leading to the secretion of IL-8 and invasion of host cells. MAP kinases are activated by the phosphorylation of their tyrosine and threonine residues by their specific MAP kinase kinase (MAPKK). The phosphorylation of MAP kinases are facilitated by a specific MKD motif, which allows the proper interaction to occur without crosstalk to inappropriate signaling molecules. We also report that CiaD is required for the development of acute disease and colon lesions.
MKD’s have been identified in several bacterial effector proteins, including SpvC, OspF, and VP1680 from Salmonella enterica, Shigella spp. and V. parahaemolyticus, respectively [33-36]. The SpvC and OspF MKD canonical (K/R)1–2-(X)2–6-OA-X-OB sequence [37] are located near the N-terminus in a disordered region. Similarly, both proteins have been shown to interact with Erk 1/2 to facilitate the irreversible dephosphorylation of threonine residues crucial for Erk 1/2 activation [38]. However, the MKD is located centrally within a region of low-complexity in the V. parahaemolyticus effector VP1680. VP1680 is necessary for the activation of the p38 and Erk 1/2 signaling pathways [36]. Bioinformatics analysis of CiaD shows that the MKD is near the C-terminus in an area of low-complexity and is required for maximal activation of Erk 1/2 and p38. These data also suggest that there are two distinct classes of MKD containing effector proteins. One class inhibits the activation of these pathways (i.e., SpvC and OspF) and the other class stimulates the activation of the MAP kinases (i.e., VP1680 and CiaD). The CiaD (ΔS/T)P mutant induced the secretion of IL-8 and invaded host cells to a level that was indistinguishable from a C. jejuni wild-type strain. This finding suggests that CiaD does not need to be phosphorylated at this motif, but does not exclude the possibility CiaD is phosphorylated at another site.
Inhibition of either Erk 1/2 or p38 has been reported to lead to a significant decrease in the amount of C. jejuni internalized into host cells [23]. However, others have shown that these pathways do not affect bacterial invasion [18]. Our results support the findings that Erk 1/2 and p38 are necessary for maximal invasion by C. jejuni. While Erk 1/2 and p38 have also been shown to be necessary for invasion of Shigella, Chlamydia, and Salmonella spp. [39-41], the molecular mechanism by which Erk 1/2 and p38 promote bacterial invasion of intestinal cells is not known. It may be that the activation of Erk 1/2 and p38 alter gene transcription and the synthesis of proteins necessary for bacterial uptake or that Erk 1/2 and p38 activate other cellular proteins involved in bacterial invasion.
Acute inflammation caused by C. jejuni or other invasive enteric pathogens, such as Shigella spp. and Salmonella enterica serotype Typhimurium (S. Typhimurium), is associated with extensive epithelial injury and necrosis of the colon and the terminal ileum, with neutrophils being responsible for the tissue damage accompanying exudative inflammation. These processes all contribute to intestinal fluid accumulation and exudative diarrhea often with hemorrhage. Similarly, the inflammatory response is associated with changes in the gut microbiota, including an increased abundance of bacteria belonging to the family Enterobacteriaceae [42]. We show that the development of disease requires CiaD. The introduction of the wild-type copy of ciaD into the ciaD mutant resulted in greater gross- and histopathologic lesions than infection with the ciaD mutant, but somewhat less changes than the C. jejuni wild-type strain. One possible reason for this intermediate phenotype is that the expression of the wild-type copy of ciaD in the mutant may be altered. The partial restoration in virulence observed with the C. jejuni ciaD complemented isolate is consistent with the results of complementation experiments performed by others using in vitro challenge studies [14,43]. Noteworthy is that this is the first time that a C. jejuni effector protein has been found to be required for the development of disease in a mouse model. This is also in agreement with our previous results indicating that the Cia proteins contribute to the development of disease in piglets [14]. More specifically, infection of piglets with a ciaB mutant exhibited less severe disease as compared to a C. jejuni wild-type strain, as judged by clinical presentation and histopathology [14].
We discovered CiaD is required for the maximal invasion of human epithelial cells, induction of the inflammatory response, and the activation of the cellular signaling components Erk 1/2 and p38. We demonstrate that CiaD is necessary for the development of disease and colon inflammatory lesions in vivo using the IL-10 KO mouse model for campylobacteriosis. In addition, we observed a plasma IgG2b anti-C. jejuni antibody response in mice infected with the C. jejuni wild-type strain and complemented ciaD isolate, suggesting that C. jejuni stimulates a general proinflammatory response in IL-10-/- mice. We believe that CiaD is, in part, responsible for the generation of plasma IgG2b anti-C. jejuni antibody response, as this response was not observed in mice inoculated with the ciaD mutant (Additional file 10: Figure S10). This finding is consistent with the fact that: 1) T-cell derived IL-10 is an inhibitor of the Th1 immune response, so an absence of IL-10 promotes a robust Th1 directed antibody response (predominantly IgG2b) [44]; and 2) The C. jejuni wild-type strain and complemented ciaD isolate are more invasive than the ciaD mutant. Noteworthy is that the Th1 “cell mediated” immune response is observed with invasive pathogens, including C. jejuni[44-47]. In conclusion, this is the first time that a C. jejuni effector protein has been shown to contribute to the development of acute disease. Future work will be directed towards the identification of a host cell-binding partner of CiaD, and to understand the role of Erk 1/2 and p38 in the larger scope of bacterial invasion.
Methods
Bacterial strains, tissues culture types, and plasmids
Campylobacter jejuni strains for in vitro experiments were cultured on Muller-Hinton agar plates supplemented with bovine citrated 5% (v/v) blood (MHB). Cultures for mouse inoculation studies were grown on trypticase soy agar containing 5% sheep’s blood. When appropriate, chloramphenicol (8 μg/ml) and tetracycline (2 μg/ml) were added to the media. Cultures were grown at 37°C in microaerobic conditions (5% O2, 10% CO2, and 85% N2). Cell lines used in this study were obtained from the American Type Tissue Culture Collection. INT 407 (ATCC CCL 6) and Caco-2 (ATCC HTB-37) cells were cultured in Minimal Essential Medium (MEM) supplemented with 10 mM sodium pyruvate, 20 mM glutamine, and 10% (v/v) fetal bovine serum (FBS). CT-26 (ATCC CRL-2638) cells were cultured in RPMI supplemented with 10% (v/v) fetal bovine serum (FBS). Plasmids and bacterial isolates used in this study are described in the Supplemental Methods (Additional file 1) and Additional file 11: Table S1. Primers used in this study are described in Supplemental Methods (Additional file 1) and Additional file 12: Table S2.
Secretion assay
Secretion assays were performed as previously described [12]. Briefly, overnight cultures of C. jejuni grown in 0.05% DOC MH broth were harvested and a bacterial suspension was added to 3 mL MEM +1% FBS for a final OD540 of 0.3/ml. Cultures were then incubated at 37°C for 3 hr under microaerobic conditions. Following incubation, supernatants were concentrated 100-fold by precipitation with 4 volumes of ice-cold 1 mM HCl-acetone. Samples were separated by SDS-PAGE, transferred to polyvinylidene fluoride (PVDF) membranes, and blots were probed with antibodies against FLAG, CysM, and the ACD of cyaA.
Cia protein delivery assays
Secretion assays were performed as previously described [12]. C. jejuni overnight cultures grown in 0.05% DOC MH broth were harvested, and the bacterial suspension was added to MEM + 1% FBS for a final OD540 of 0.015/ml. INT 407 cells were washed once with PBS and 1 mL of a 0.015 OD540 mixture was added to each well of the 24-well tissue culture tray. The trays were centrifuged for 5 min at 800 × g and incubated for 30 min at 37°C. The media in the wells were removed and the wells were washed 3 times with PBS followed by the addition of 95°C 0.1 M HCl. Each tray was boiled for 15 min by placing it on a platform directly above boiling water. Lysates were collected and transferred to a clean centrifuge tube. cAMP levels were assessed using the Direct Cyclic AMP Enzyme Immunoassay Kit (Assay Designs) according to manufacturer’s specifications.
Immunoblot analysis, cellular inhibitors, antibodies, and densitometry analysis
Bacterial whole cell lysates, serum supernatant samples, and INT 407 cellular lysates were collected and analyzed by SDS-polyacrylamide gel electrophoresis. The proteins were transferred to a PVDF membrane and probed with the indicated antibodies. The antibodies used in this work are described in the Supplemental Methods (Additional file 1). Band intensity was quantified using a LAS 4000 mini (GE healthcare) and the Multi Gauge V3.0 (Fujifilm, Valhalla, NY) software package. Inhibitors were added to the cells 30 min prior to infection and maintained thorough the experiment. The Erk 1/2 (PD98059) and the p38 (SB202190) inhibitors were used at a concentration of 50 M. Cell death was quantified by trypan blue straining. No significant death was observed with any treatment conditions. C. jejuni was pretreated with chloramphenicol (1024 μg/ml) for 30 min prior to infection of host cells and maintained throughout the experiment. Chloramphenicol treatment did not effect bacterial viability. This concentration of chloramphenicol completely inhibited bacterial protein synthesis, as judged by 35[S]-methionine incorporation assays (not shown and [7]).
Ectopic expression
EGFP plasmids were purified using the Qiagen Plasmid Purification Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocols and normalized to 200 ng/μl. Purified plasmids where transfected into INT 407 cells seeded on glass coverslips at a confluencey of 3 × 105. Transfections where performed using the Qiagen Effectene Transfection reagent (Qiagen, Valencia, CA), according to the manufacturer’s specifications.
Invasion and motility assays
Binding and internalization assays [48] and motility assays [12] were performed as previously described.
IL-8 quantification
Interleukin-8 levels in cellular supernatants were quantified with a commercial ELISA kit (OptEIA Set, Becton Dickinson, Cowley, Oxford, UK) using the manufacturer’s protocol. Briefly, cells were inoculated with 3 × 107 bacteria and centrifuged for 5 min at 800 × g to promote cell contact. After 30 min of incubation, the cells were washed one time and fresh media was added to each well (containing inhibitors where indicated). The cells were incubated at 37°C for 24 hr and the media collected. Supernatants were frozen at -20°C or used immediately.
Bioinformatics
Operon prediction was performed using MicrobesOnline [49]. Euckaryotic linear motif analysis was performed by query of the ELM website (http://elm.eu.org, [25]).
Animals
All animal experiments were conducted according to NIH guidelines under Michigan State University Animal Use Form approval 06/012-107-00. Two replicate experiments were conducted. A breeding colony of C57BL/6 IL-10-/- mice (B6.129P2-IL10 tm1Cgn /J) was maintained in a specific-pathogen-free colony at MSU with monitoring for genotype and colitogenic bacteria as previously described [44]. For experiments, mice were transferred to the University Research Containment Facility at MSU where they were individually housed in filter top cages, on sterile food and water ad libitum.
C. jejuni inoculation of IL-10-/- mice
Mice 8–12 weeks old were infected with ~1 × 1010 cfu C. jejuni by oral gavage and observed daily for clinical signs using standardized scoring criteria as previously described [44]. Mice were humanely euthanized and necropsied promptly when clinical signs of severe disease developed or at thirty-five days post-infection. Blood samples were obtained by cardiac puncture immediately following death of the mouse. The gastrointestinal (GI) tract was removed in its entirety and placed on clean absorbent bench paper. Observations on gross pathological changes were recorded during necropsy.
Gross pathology and histopathology
Gross pathology examination in all portions of the GI tract was performed by trained personnel as previously described [44]. The following criteria were used for scoring: no gross pathological changes (None, 0); either thickening of the GI tract wall or enlarged ileocecocolic lymph node (ICC or TW, 1); both thickening of GI tract wall and enlarged ileocecocolic lymph node (ICC and TW, 2); and bloody lumen contents in cecum or colon or both (BLC, 3). For histopathology, the ileocecocolic junction was removed and injected with 10% phosphate buffered formalin (Fisher Scientific, Pittsburgh, PA, USA), placed in a histological cassette and submerged in 10% phosphate buffered formalin. After 24 hr, the cassette was drained and transferred to 60% ethanol. Tissue processing, paraffin embedment, sectioning, and hematoxylin and eosin staining were carried out at the Investigative Histopathology Laboratory, Division of Human Pathology, Department of Physiology, Michigan State University. After randomization and coding to conceal identity, slides were read in a blinded fashion by a single investigator (LSM) according to a previously described scoring system [44].
Plasma IgG2b anti-C. jejuni antibody
Plasma IgG2b anti-C. jejuni levels were determined via ELISA as previously described [44].
Statistical analysis
All data was evaluated using a one-way ANOVA followed by post-hoc Tukey’s or Dunnet’s analysis of the means, using Prism 6 (GraphPad Software, La Jolla, CA). Statistical significance was defined by a maximum value of *P < 0.05 for all in vitro experiments and *P < 0.10 for all in vivo experiments. All experiments were performed a minimum of three times to ensure reproducibility. Kaplan Meier log rank analyses were performed using SigmaStat 3.1 (Systat Software, Port Richmond, CA) to assess the survival data. Gross pathology was analyzed using SigmaStat 3.1. The nonparametric Kruskal Wallis one-way ANOVA was used for gross pathology scoring. Scores for analysis of gross pathology data were assigned as follows: no gross pathological changes (None, 0); either thickening of GI tract wall or enlarged ileocecocolic lymph node (ICC or TW, 1); thickening of GI tract wall and enlarged ileocecocolic lymph node (ICC and TW, 2); and thickening of GI tract wall and enlarged ileocecocolic lymph node plus bloody lumen contents in cecum or colon or both (BLC, 3). Kruskal Wallis nonparametric one-way ANOVA was performed. Mann Whitney pairwise comparisons with Holm-Sidak correction for multiple comparisons was performed to evaluate the difference between the means of the samples, as described previously [44]. Plasma IgG2b anti-C. jejuni levels were evaluated with Kruskal Wallis one-way non-parametric ANOVA, followed by Mann Whitney pairwise comparisons with Holm-Sidak correction for multiple comparisons of the means.
Supplemental information
The data sets supporting the results of this article are included within the article and its additional files.
Abbreviations
Cia: Campylobacter invasion antigens; Erk 1/2: Extracellular regulated kinase.
Competing interests
The authors declare that they have no competing interests
Authors’ contributions
DRS planned, performed, and wrote the manuscript. MEK and LSM planned experiments, analyzed data and contributed to manuscript preparation. TPE, LD, JAB planned and performed experiments. All authors read and approved the final manuscript.
Supplementary Material
The C. jejuni ciaD mutant secretes a known effector protein. (A) The ciaD mutant is Cia secretion competent. A C. jejuni ciaD mutant was transformed with the pRY111 vector harboring CiaD, CiaC and MetK fused to the ACD and the isolates were analyzed by immunoblot analysis. Supernatant and whole cell lysates were separated by SDS-PAGE, proteins transferred to PVDF membranes, and blots probed with an ACD antibody and CysM antibody. A C. jejuni wild-type strain without a plasmid and the MetK-ACD (S-adenosylmethionine synthetase) protein, which is localized in the bacterial cytoplasm, were included as negative controls. Molecular mass standards, in kilodaltons (kDa), are indicated on the left. Arrows indicate the CiaD-ACD and CiaC-ACD secreted proteins. (B) The C. jejuni CiaC-ACD, CiaD-ACD, and CiaC-ACD fusion proteins are synthesized in similar levels. The C. jejuni wild-type strain, ciaD, and a flgBC mutant transformed with the pRY111 vector harboring CiaD, CiaC and MetK fused to the ACD were analyzed by immunoblot analysis. Protein levels were quantified by BCA, normalized to ensure equal loading, separated by SDS-PAGE, transferred to PVDF membranes, and blots probed with an ACD antibody.
C. jejuni requires de novo protein synthesis for bacterial invasion and induction of secretion. (A) Pre-treatment of C. jejuni with chloramphenicol inhibits INT 407 cell invasion. C. jejuni were pretreated with chloramphenicol (1024 μg/mL) for 30 min prior to infection of INT 407 cells. Cell invasion was assessed using a gentamicin-protection assay as outlined in Supplemental Methods (Additional file 1). (B) C. jejuni requires de novo protein synthesis for maximal IL-8 secretion. An IL-8 secretion time course assay was performed by infecting INT 407 cells with a C. jejuni wild-type strain that had been pretreated for 30 min with chloramphenicol (1024 μg/mL) and harvesting the supernatants at various times post-infection. IL-8 in the supernatant samples was quantified by ELISA as described in Methods. Gray bars indicate IL-8 quantities from INT 407 cells infected with an untreated C. jejuni wild-type strain. The black bars indicate IL-8 quantities from INT 407 cells infected with a C. jejuni wild-type strain that was pre-treated with chloramphenicol. The asterisks indicate the time points (4 and 6 hr) at which there are significant differences in the amount of IL-8 produced compared to the untreated samples, as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM.
Ectopic expression of CiaD in host INT 407 cells induces IL-8 secretions. INT 407 cells were transfected with CiaD-EGFP and EGFP-only eukaryotic expression vectors. Cells treated with the transfection reagent Effectene were also included as a vehicle control. IL-8 levels were assessed by ELISA 24 hr following transfection. The asterisks indicate that the amount of IL-8 produced was significantly increased compared to the EGFP-only control, as judged by student’s t-test (P < 0.05). Error bars represent ± SEM.
Mutation of Cj0789, which is the gene downstream of ciaD, does not have an effect on IL-8 secretion, indicating the Cj0788 mutation is not polar. INT 407 cells were infected with C. jejuni for 24 hr. Following infection, supernatants were collected and IL-8 levels quantified using an IL-8 ELISA. The asterisks indicate that the amount of IL-8 produced was significantly decreased compared to the C. jejuni wild-type strains (F38011 and NCTC 11168), as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM.
MAP kinase inhibition leads to an additive effect in IL-8 secretion by the C. jejuni ciaD mutant. Inhibitors to Erk 1/2 and p38 were added to INT 407 cells for 30 min prior to the addition of the C. jejuni ciaD mutant. The C. jejuni wild-type strain was included as a positive control. The mean value calculated for ‘cells only’ was subtracted from all other values. The asterisk indicates a significant reduction in the amount of IL-8 secreted form INT 407 cells infected with the ciaD mutant in the presence of the Erk 1/2 and p38 inhibitors as compared to the value obtained for the untreated INT 407 cells infected with the C. jejuni ciaD mutant, as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM.
The pro-inflammatory cytokine IL-8 is not required for bacterial invasion. (A) Caco-2 cells were infected with C. jejuni for 30 min followed by the addition of 300 pg/ml of IL-8 to the C. jejuni ciaD mutant and wild-type strain. The bars represent the mean of bacterial invasion of the wild-type and the ciaD mutant with the addition of IL-8 or no treatment. (B) The activation status of Akt was determined via immunoblot to confirm IL-8 induced signaling. 300 pg/ml of IL-8 was added to INT 407 cells for 15 min and cellular lysates were prepared. Blots were probed with phospho-specific antibodies to Akt (Mr = 62 kDa). All blots were stripped and re-probed with an anti-Akt (Mr = 60 kDa) antibody. The asterisk indicates a significant decrease compared to the C. jejuni wild-type strain, as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM.
The C. jejuni ciaD mutant has reduced MAP kinase signaling. (A) Maximal activation of MAP kinase signaling requires CiaD. The activation status of the MAP kinase signaling components was determined using a phospho-spot array assay as outlined in Supplemental Methods (Additional file 1). INT 407 cells were infected with the C. jejuni wild-type strain and C. jejuni ciaD mutant for 3 hr. Cellular lysates were assayed using the spot array. Pictured are the spot array profiles of the C. jejuni wild-type and C. jejuni ciaD mutant. (B) Maximal activation of MAP kinase signaling requires CiaD. Densitometry was performed on the phospho-spot arrays performed in Panel B. Significance was not assessed, as this experiment was used as a screen for activation.
The CiaD ΔMKD site and Δ(S/T)P proteins are synthesized and the isolates that produce these variant proteins are motile. (A) Deletion of the MKD site and the (S/T)P site does not significantly effect protein synthesis. The C. jejuni ciaD mutant transformed with a pRY111 vector encoding either a wild-type copy of the CiaD protein, the MAP kinase docking motif (ΔMKD site) mutant protein, or the (Δ(S/T)P) mutant protein fused to a FLAG-tag were analyzed by immunoblot analysis. The blots were probed with a FLAG antibody. Blots were also stripped and re-probed with an anti-CysM antibody to ensure equal loading of each sample. (B) C. jejuni strains synthesizing the CiaD MAP kinase docking motif (ΔMKD site) mutant protein and CiaD proline directed-phosphorylation site (Δ(S/T)P) mutant protein are motile.
C. jejuni colonization of IL10-/- mice. (A) Percent survival of C57BL/6 IL-10-/- mice infected with the C. jejuni 11168 wild-type strain, ciaD mutant, and the ciaD complemented isolate. No significant differences were observed in the number of surviving mice as judged by Kaplan Meier log rank analysis. The mouse inoculated with the C. jejuni ciaD complemented isolate that died 8 days post-infection was excluded from the gross pathology evaluation (Figure 7A), as the cause of death was not known. (B) Colonization of mice infected with the C. jejuni wild-type strain, ciaD mutant, and the ciaD complement isolate was assessed 35 days post-infection by bacterial CFU determination of colon content and Campylobacter PCR on DNA extracted from frozen cecal tips of all mice that were negative by culture.
C. jejuni stimulates the production of plasma IgG2b. Plasma IgG2b anti-C. jejuni antibody levels in mice infected with a C. jejuni wild-type strain, ciaD mutant, and the ciaD complemented isolate. Levels of IgG2B were evaluated via ELISA. We found that the C. jejuni wild-type strain had a significant increase in the amount of detectable IgG2b as judged by nonparametric Kruskal Wallis one-way ANOVA, followed by post hoc comparisons using Mann Whitney pairwise comparisons. Corrections were made for multiple comparisons using the Holm-Šidák test.
Bacterial isolates and plasmids used in this study.
Primers used in this study.
Contributor Information
Derrick R Samuelson, Email: dsamuelson@vetmed.wsu.edu.
Tyson P Eucker, Email: teucker@vetmed.wsu.edu.
Julia A Bell, Email: bellj@msu.edu.
Leslie Dybas, Email: dybasl@msu.edu.
Linda S Mansfield, Email: mansfie4@cvm.msu.edu.
Michael E Konkel, Email: konkel@vetmed.wsu.edu.
Acknowledgments
We thank the members of the Konkel lab, including Levi O'Loughlin, Jason Neal-McKinnney, Chris Gourley, Jennifer Stone, and Mark Nissen, for reviewing the manuscript. This work was supported by funds awarded to MEK from the National Institutes of Health grant (NIH) #R56 AIO88518-01A1 and from the Agriculture and Food Research Initiative Competitive (Grant NO. 2011-67015-30772) from the USDA National Institute of Food and Agriculture. DRS was supported, in part, by the National Institutes of General Medical Sciences Training Grant #T32GM083864. TPE was supported, in part, by the National Institutes of General Medical Sciences Training Grant #T32GM008336. Funding for animal work: DHHS, NIH, NIAID Cooperative Agreement U19-AI-09087. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
References
- Samuel MC, Vugia DJ, Shallow S, Marcus R, Segler S, McGivern T, Kassenborg H, Reilly K, Kennedy M, Angulo F, Tauxe RV. Epidemiology of sporadic Campylobacter infection in the United States and declining trend in incidence, FoodNet 1996–1999. Clin Infect Dis. 2004;38(Suppl 3):S165–174. doi: 10.1086/381583. [DOI] [PubMed] [Google Scholar]
- Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. Foodborne illness acquired in the United States–major pathogens. Emerg Infect Dis. 2011;17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwerer B. Antibodies against gangliosides: a link between preceding infection and immunopathogenesis of Guillain-Barre syndrome. Microbes Infect. 2002;4:373–384. doi: 10.1016/S1286-4579(02)01550-2. [DOI] [PubMed] [Google Scholar]
- Christensen JE, Pacheco SA, Konkel ME. Identification of a Campylobacter jejuni-secreted protein required for maximal invasion of host cells. Mol Microbiol. 2009;73:650–662. doi: 10.1111/j.1365-2958.2009.06797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel ME, Joens LA. Adhesion to and invasion of HEp-2 cells by Campylobacter spp. Infect Immun. 1989;57:2984–2990. doi: 10.1128/iai.57.10.2984-2990.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel ME, Corwin MD, Joens LA, Cieplak W. Factors that influence the interaction of Campylobacter jejuni with cultured mammalian cells. J Med Microbiol. 1992;37:30–37. doi: 10.1099/00222615-37-1-30. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Kim BJ, Rivera-Amill V, Garvis SG. Bacterial secreted proteins are required for the internaliztion of Campylobacter jejuni into cultured mammalian cells. Mol Microbiol. 1999;32:691–701. doi: 10.1046/j.1365-2958.1999.01376.x. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Kim BJ, Rivera-Amill V, Garvis SG. Identification of proteins required for the internalization of Campylobacter jejuni into cultured mammalian cells. Adv Exp Med Biol. 1999;473:215–224. doi: 10.1007/978-1-4615-4143-1_22. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Klena JD, Rivera-Amill V, Monteville MR, Biswas D, Raphael B, Mickelson J. Secretion of virulence proteins from Campylobacter jejuni is dependent on a functional flagellar export apparatus. J Bacteriol. 2004;186:3296–3303. doi: 10.1128/JB.186.11.3296-3303.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel ME, Mead DJ, Cieplak W Jr. Kinetic and antigenic characterization of altered protein synthesis by Campylobacter jejuni during cultivation with human epithelial cells. J Infect Dis. 1993;168:948–954. doi: 10.1093/infdis/168.4.948. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Cieplak W Jr. Altered synthetic response of Campylobacter jejuni to cocultivation with human epithelial cells is associated with enhanced internalization. Infect Immun. 1992;60:4945–4949. doi: 10.1128/iai.60.11.4945-4949.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal-McKinney JM, Konkel ME. The Campylobacter jejuni CiaC virulence protein is secreted from the flagellum and delivered to the cytosol of host cells. Front Cell Infect Microbiol. 2012;2:31. doi: 10.3389/fcimb.2012.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buelow DR, Christensen JE, Neal-McKinney JM, Konkel ME. Campylobacter jejuni survival within human epithelial cells is enhanced by the secreted protein CiaI. Mol Microbiol. 2011;80:1296–1312. doi: 10.1111/j.1365-2958.2011.07645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel ME, Monteville MR, Rivera-Amill V, Joens LA. The pathogenesis of Campylobacter jejuni-mediated enteritis. Curr Issues Intest Microbiol. 2001;2:55–71. [PubMed] [Google Scholar]
- Raphael RH, Monteville MR, Klena JD, Joens LA, Konkel ME. In: Campylobacter molecular and cellular biology. JM K, ME K, editor. Norfolk: Horizon Bioscience; 2005. Interactions of Campylobacter jejuni with non-professional phagocytic cells; pp. 397–413. [Google Scholar]
- Janssen R, Krogfelt KA, Cawthraw SA, Van Pelt W, Wagenaar JA, Owen RJ. Host-pathogen interactions in Campylobacter infections: the host perspective. Clin Microbiol Rev. 2008;21:505–518. doi: 10.1128/CMR.00055-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zoete MR, Keestra AM, Wagenaar JA, Van Putten JP. Reconstitution of a functional Toll-like receptor 5 binding site in Campylobacter jejuni flagellin. J Biol Chem. 2010;285:12149–12158. doi: 10.1074/jbc.M109.070227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RO, Galan JE. Signal transduction in Campylobacter jejuni-induced cytokine production. Cell Microbiol. 2005;7:655–665. doi: 10.1111/j.1462-5822.2004.00498.x. [DOI] [PubMed] [Google Scholar]
- Zilbauer M, Dorrell N, Elmi A, Lindley KJ, Schuller S, Jones HE, Klein NJ, Nunez G, Wren BW, Bajaj-Elliott M. A major role for intestinal epithelial nucleotide oligomerization domain 1 (NOD1) in eliciting host bactericidal immune responses to Campylobacter jejuni. Cell Microbiol. 2007;9:2404–2416. doi: 10.1111/j.1462-5822.2007.00969.x. [DOI] [PubMed] [Google Scholar]
- Jin S, Song YC, Emili A, Sherman PM, Chan VL. JlpA of Campylobacter jejuni interacts with surface-exposed heat shock protein 90alpha and triggers signalling pathways leading to the activation of NF-kappaB and p38 MAP kinase in epithelial cells. Cell Microbiol. 2003;5:165–174. doi: 10.1046/j.1462-5822.2003.00265.x. [DOI] [PubMed] [Google Scholar]
- Hickey TE, McVeigh AL, Scott DA, Michielutti RE, Bixby A, Carroll SA, Bourgeois AL, Guerry P. Campylobacter jejuni cytolethal distending toxin mediates release of interleukin-8 from intestinal epithelial cells. Infect Immun. 2000;68:6535–6541. doi: 10.1128/IAI.68.12.6535-6541.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos HC, Rumbo M, Sirard JC. Bacterial flagellins: mediators of pathogenicity and host immune responses in mucosa. Trends Microbiol. 2004;12:509–517. doi: 10.1016/j.tim.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Hu L, McDaniel JP, Kopecko DJ. Signal transduction events involved in human epithelial cell invasion by Campylobacter jejuni 81–176. Microb Pathog. 2006;40:91–100. doi: 10.1016/j.micpath.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Li YP, Vegge CS, Brondsted L, Madsen M, Ingmer H, Bang DD. Campylobacter jejuni induces an anti-inflammatory response in human intestinal epithelial cells through activation of phosphatidylinositol 3-kinase/Akt pathway. Vet Microbiol. 2011;148:75–83. doi: 10.1016/j.vetmic.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Gould CM, Diella F, Via A, Puntervoll P, Gemund C, Chabanis-Davidson S, Michael S, Sayadi A, Bryne JC, Chica C. ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 2010;38:D167–180. doi: 10.1093/nar/gkp1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puntervoll P, Linding R, Gemünd C, Chabanis-Davidson S, Mattingsdal M, Cameron S, Martin DMA, Ausiello G, Brannetti B, Costantini A. et al. ELM server: a new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Research. 2003;31:3625–3630. doi: 10.1093/nar/gkg545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Itzen A, Blankenfeldt W, Goody RS. Adenylylation: renaissance of a forgotten post-translational modification. Trends Biochem Sci. 2011;36:221–228. doi: 10.1016/j.tibs.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Hickey TE, Baqar S, Bourgeois AL, Ewing CP, Guerry P. Campylobacter jejuni-stimulated secretion of interleukin-8 by INT407 cells. Infect Immun. 1999;67:88–93. doi: 10.1128/iai.67.1.88-93.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Kim MS, Choi SH, Kim TS. Involvement of capsular polysaccharide via a TLR2/NF-kappaB pathway in Vibrio vulnificus-induced IL-8 secretion of human intestinal epithelial cells. Int J Mol Med. 2010;25:581–591. doi: 10.3892/ijmm_00000380. [DOI] [PubMed] [Google Scholar]
- Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- Larson CL, Samuelson DR, Eucker TP, O’Loughlin JL, Konkel ME. The fibronectin-binding motif within FlpA facilitates Campylobacter jejuni adherence to host cell and activation of host cell signaling. Emerging Microbes & Infections. 2013. In Press. [DOI] [PMC free article] [PubMed]
- Li H, Xu H, Zhou Y, Zhang J, Long C, Li S, Chen S, Zhou JM, Shao F. The phosphothreonine lyase activity of a bacterial type III effector family. Science. 2007;315:1000–1003. doi: 10.1126/science.1138960. [DOI] [PubMed] [Google Scholar]
- Zurawski DV, Mumy KL, Faherty CS, McCormick BA, Maurelli AT. Shigella flexneri type III secretion system effectors OspB and OspF target the nucleus to downregulate the host inflammatory response via interactions with retinoblastoma protein. Mol Microbiol. 2009;71:350–368. doi: 10.1111/j.1365-2958.2008.06524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurkiewicz P, Thomas J, Thompson JA, Liu M, Arbibe L, Sansonetti P, Holden DW. SpvC is a Salmonella effector with phosphothreonine lyase activity on host mitogen-activated protein kinases. Mol Microbiol. 2008;67:1371–1383. doi: 10.1111/j.1365-2958.2008.06134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlawska-Wasowska K, Finn R, Mustel A, O'Byrne CP, Baird AW, Coffey ET, Boyd A. The Vibrio parahaemolyticus Type III Secretion Systems manipulate host cell MAPK for critical steps in pathogenesis. BMC Microbiol. 2010;10:329. doi: 10.1186/1471-2180-10-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Li H, Long C, Hu L, Xu H, Liu L, Chen S, Wang DC, Shao F. Structural insights into the enzymatic mechanism of the pathogenic MAPK phosphothreonine lyase. Mol Cell. 2007;28:899–913. doi: 10.1016/j.molcel.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Dean P. Functional domains and motifs of bacterial type III effector proteins and their roles in infection. FEMS Microbiol Rev. 2011;35:1100–1125. doi: 10.1111/j.1574-6976.2011.00271.x. [DOI] [PubMed] [Google Scholar]
- Coombes BK, Mahony JB. Identification of MEK- and phosphoinositide 3-kinase-dependent signalling as essential events during Chlamydia pneumoniae invasion of HEp2 cells. Cell Microbiol. 2002;4:447–460. doi: 10.1046/j.1462-5822.2002.00203.x. [DOI] [PubMed] [Google Scholar]
- Evans DJ, Maltseva IA, Wu J, Fleiszig SM. Pseudomonas aeruginosa internalization by corneal epithelial cells involves MEK and ERK signal transduction proteins. FEMS Microbiol Lett. 2002;213:73–79. doi: 10.1111/j.1574-6968.2002.tb11288.x. [DOI] [PubMed] [Google Scholar]
- Romero S, Grompone G, Carayol N, Mounier J, Guadagnini S, Prevost MC, Sansonetti PJ, Van Nhieu GT. ATP-mediated Erk1/2 activation stimulates bacterial capture by filopodia, which precedes Shigella invasion of epithelial cells. Cell Host Microbe. 2011;9:508–519. doi: 10.1016/j.chom.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Keo T, Collins J, Kunwar P, Blaser MJ, Iovine NM. Campylobacter capsule and lipooligosaccharide confer resistance to serum and cationic antimicrobials. Virulence. 2011;2:30–40. doi: 10.4161/viru.2.1.14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield LS, Bell JA, Wilson DL, Murphy AJ, Elsheikha HM, Rathinam VA, Fierro BR, Linz JE, Young VB. C57BL/6 and congenic interleukin-10-deficient mice can serve as models of Campylobacter jejuni colonization and enteritis. Infect Immun. 2007;75:1099–1115. doi: 10.1128/IAI.00833-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger A. Th1 and Th2 responses: what are they? BMJ. 2000;321:424.1. doi: 10.1136/bmj.321.7258.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar JC, Lobaina Y, Muzio V, Garcia D, Penton E, Iglesias E, Pichardo D, Urquiza D, Rodriguez D, Silva D. et al. Development of a nasal vaccine for chronic hepatitis B infection that uses the ability of hepatitis B core antigen to stimulate a strong Th1 response against hepatitis B surface antigen. Immunol Cell Biol. 2004;82:539–546. doi: 10.1111/j.0818-9641.2004.01278.x. [DOI] [PubMed] [Google Scholar]
- Rathinam VA, Hoag KA, Mansfield LS. Dendritic cells from C57BL/6 mice undergo activation and induce Th1-effector cell responses against Campylobacter jejuni. Microbes Infect. 2008;10:1316–1324. doi: 10.1016/j.micinf.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eucker TP, Konkel ME. The cooperative action of bacterial fibronectin-binding proteins and secreted proteins promote maximal Campylobacter jejuni invasion of host cells by stimulating membrane ruffling. Cell Microbiol. 2012;14:226–238. doi: 10.1111/j.1462-5822.2011.01714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehal PS, Joachimiak MP, Price MN, Bates JT, Baumohl JK, Chivian D, Friedland GD, Huang KH, Keller K, Novichkov PS. et al. MicrobesOnline: an integrated portal for comparative and functional genomics. Nucleic Acids Res. 2010;38:D396–400. doi: 10.1093/nar/gkp919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The C. jejuni ciaD mutant secretes a known effector protein. (A) The ciaD mutant is Cia secretion competent. A C. jejuni ciaD mutant was transformed with the pRY111 vector harboring CiaD, CiaC and MetK fused to the ACD and the isolates were analyzed by immunoblot analysis. Supernatant and whole cell lysates were separated by SDS-PAGE, proteins transferred to PVDF membranes, and blots probed with an ACD antibody and CysM antibody. A C. jejuni wild-type strain without a plasmid and the MetK-ACD (S-adenosylmethionine synthetase) protein, which is localized in the bacterial cytoplasm, were included as negative controls. Molecular mass standards, in kilodaltons (kDa), are indicated on the left. Arrows indicate the CiaD-ACD and CiaC-ACD secreted proteins. (B) The C. jejuni CiaC-ACD, CiaD-ACD, and CiaC-ACD fusion proteins are synthesized in similar levels. The C. jejuni wild-type strain, ciaD, and a flgBC mutant transformed with the pRY111 vector harboring CiaD, CiaC and MetK fused to the ACD were analyzed by immunoblot analysis. Protein levels were quantified by BCA, normalized to ensure equal loading, separated by SDS-PAGE, transferred to PVDF membranes, and blots probed with an ACD antibody.
C. jejuni requires de novo protein synthesis for bacterial invasion and induction of secretion. (A) Pre-treatment of C. jejuni with chloramphenicol inhibits INT 407 cell invasion. C. jejuni were pretreated with chloramphenicol (1024 μg/mL) for 30 min prior to infection of INT 407 cells. Cell invasion was assessed using a gentamicin-protection assay as outlined in Supplemental Methods (Additional file 1). (B) C. jejuni requires de novo protein synthesis for maximal IL-8 secretion. An IL-8 secretion time course assay was performed by infecting INT 407 cells with a C. jejuni wild-type strain that had been pretreated for 30 min with chloramphenicol (1024 μg/mL) and harvesting the supernatants at various times post-infection. IL-8 in the supernatant samples was quantified by ELISA as described in Methods. Gray bars indicate IL-8 quantities from INT 407 cells infected with an untreated C. jejuni wild-type strain. The black bars indicate IL-8 quantities from INT 407 cells infected with a C. jejuni wild-type strain that was pre-treated with chloramphenicol. The asterisks indicate the time points (4 and 6 hr) at which there are significant differences in the amount of IL-8 produced compared to the untreated samples, as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM.
Ectopic expression of CiaD in host INT 407 cells induces IL-8 secretions. INT 407 cells were transfected with CiaD-EGFP and EGFP-only eukaryotic expression vectors. Cells treated with the transfection reagent Effectene were also included as a vehicle control. IL-8 levels were assessed by ELISA 24 hr following transfection. The asterisks indicate that the amount of IL-8 produced was significantly increased compared to the EGFP-only control, as judged by student’s t-test (P < 0.05). Error bars represent ± SEM.
Mutation of Cj0789, which is the gene downstream of ciaD, does not have an effect on IL-8 secretion, indicating the Cj0788 mutation is not polar. INT 407 cells were infected with C. jejuni for 24 hr. Following infection, supernatants were collected and IL-8 levels quantified using an IL-8 ELISA. The asterisks indicate that the amount of IL-8 produced was significantly decreased compared to the C. jejuni wild-type strains (F38011 and NCTC 11168), as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM.
MAP kinase inhibition leads to an additive effect in IL-8 secretion by the C. jejuni ciaD mutant. Inhibitors to Erk 1/2 and p38 were added to INT 407 cells for 30 min prior to the addition of the C. jejuni ciaD mutant. The C. jejuni wild-type strain was included as a positive control. The mean value calculated for ‘cells only’ was subtracted from all other values. The asterisk indicates a significant reduction in the amount of IL-8 secreted form INT 407 cells infected with the ciaD mutant in the presence of the Erk 1/2 and p38 inhibitors as compared to the value obtained for the untreated INT 407 cells infected with the C. jejuni ciaD mutant, as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM.
The pro-inflammatory cytokine IL-8 is not required for bacterial invasion. (A) Caco-2 cells were infected with C. jejuni for 30 min followed by the addition of 300 pg/ml of IL-8 to the C. jejuni ciaD mutant and wild-type strain. The bars represent the mean of bacterial invasion of the wild-type and the ciaD mutant with the addition of IL-8 or no treatment. (B) The activation status of Akt was determined via immunoblot to confirm IL-8 induced signaling. 300 pg/ml of IL-8 was added to INT 407 cells for 15 min and cellular lysates were prepared. Blots were probed with phospho-specific antibodies to Akt (Mr = 62 kDa). All blots were stripped and re-probed with an anti-Akt (Mr = 60 kDa) antibody. The asterisk indicates a significant decrease compared to the C. jejuni wild-type strain, as judged by one-way ANOVA followed by post-hoc Tukey’s analysis (P < 0.05). Error bars represent ± SEM.
The C. jejuni ciaD mutant has reduced MAP kinase signaling. (A) Maximal activation of MAP kinase signaling requires CiaD. The activation status of the MAP kinase signaling components was determined using a phospho-spot array assay as outlined in Supplemental Methods (Additional file 1). INT 407 cells were infected with the C. jejuni wild-type strain and C. jejuni ciaD mutant for 3 hr. Cellular lysates were assayed using the spot array. Pictured are the spot array profiles of the C. jejuni wild-type and C. jejuni ciaD mutant. (B) Maximal activation of MAP kinase signaling requires CiaD. Densitometry was performed on the phospho-spot arrays performed in Panel B. Significance was not assessed, as this experiment was used as a screen for activation.
The CiaD ΔMKD site and Δ(S/T)P proteins are synthesized and the isolates that produce these variant proteins are motile. (A) Deletion of the MKD site and the (S/T)P site does not significantly effect protein synthesis. The C. jejuni ciaD mutant transformed with a pRY111 vector encoding either a wild-type copy of the CiaD protein, the MAP kinase docking motif (ΔMKD site) mutant protein, or the (Δ(S/T)P) mutant protein fused to a FLAG-tag were analyzed by immunoblot analysis. The blots were probed with a FLAG antibody. Blots were also stripped and re-probed with an anti-CysM antibody to ensure equal loading of each sample. (B) C. jejuni strains synthesizing the CiaD MAP kinase docking motif (ΔMKD site) mutant protein and CiaD proline directed-phosphorylation site (Δ(S/T)P) mutant protein are motile.
C. jejuni colonization of IL10-/- mice. (A) Percent survival of C57BL/6 IL-10-/- mice infected with the C. jejuni 11168 wild-type strain, ciaD mutant, and the ciaD complemented isolate. No significant differences were observed in the number of surviving mice as judged by Kaplan Meier log rank analysis. The mouse inoculated with the C. jejuni ciaD complemented isolate that died 8 days post-infection was excluded from the gross pathology evaluation (Figure 7A), as the cause of death was not known. (B) Colonization of mice infected with the C. jejuni wild-type strain, ciaD mutant, and the ciaD complement isolate was assessed 35 days post-infection by bacterial CFU determination of colon content and Campylobacter PCR on DNA extracted from frozen cecal tips of all mice that were negative by culture.
C. jejuni stimulates the production of plasma IgG2b. Plasma IgG2b anti-C. jejuni antibody levels in mice infected with a C. jejuni wild-type strain, ciaD mutant, and the ciaD complemented isolate. Levels of IgG2B were evaluated via ELISA. We found that the C. jejuni wild-type strain had a significant increase in the amount of detectable IgG2b as judged by nonparametric Kruskal Wallis one-way ANOVA, followed by post hoc comparisons using Mann Whitney pairwise comparisons. Corrections were made for multiple comparisons using the Holm-Šidák test.
Bacterial isolates and plasmids used in this study.
Primers used in this study.