

Abstract
The prevalence of the metabolic syndrome has increased worldwide over the past few years. Sympathetic nervous system overactivity is a key mechanism leading to hypertension in patients with the metabolic syndrome. Sympathetic activation can be triggered by reflex mechanisms as arterial baroreceptor impairment, by metabolic factors as insulin resistance, and by dysregulated adipokine production and secretion from visceral fat with a mainly permissive role of leptin and antagonist role of adiponectin. Chronic sympathetic nervous system overactivity contributes to a further decline of insulin sensitivity and creates a vicious circle that may contribute to the development of hypertension and of the metabolic syndrome and favor cardiovascular and kidney disease. Selective renal denervation is an emerging area of interest in the clinical management of obesity-related hypertension. This review focuses on current understanding of some mechanisms through which sympathetic overactivity may be interlaced to the metabolic syndrome, with particular regard to the role of insulin resistance and of some adipokines.
1. Introduction
The metabolic syndrome (MetS) is a cluster of abnormalities that include diabetes mellitus (DM), morbid obesity, dyslipidemia, and hypertension (HT) that are all risk factors for the development of cardiovascular disease (CVD) and chronic kidney disease (CKD) [1]. The National Cholesterol Education Program's Adult Treatment Panel III report (ATP III) identified six components of the MetS that relate to CVD: abdominal obesity, atherogenic dyslipidemia, raised blood pressure (BP), insulin resistance (I.R.)/glucose intolerance, and proinflammatory and prothrombotic state [2]. A major problem concerning the WHO and NCEP ATPIII definitions was their applicability to different ethnic groups, especially when obesity cutoffs were to be defined. This is particularly evident for the risk of type II DM, which may be associated with much lower levels of obesity in Asians compared to Caucasians. The International Diabetes Federation has then proposed a new set of criteria with ethnic/racial specific cutoffs [3]. The MetS central feature is obesity, and the MetS is a growing epidemic in the United States and throughout the world [4, 5]. Approximately 1 adult in 4 or 5, depending on the country, has the MetS. Incidence increases with age; it has been estimated that in people over 50 years of age, the MetS affects more than 40% of the population in the United States and nearly 30% in Europe [6, 7]. Whether the effects of the MetS are due to a sum of comorbidities or to individual features is still a matter of debate; however, there is sufficient data to support an increased risk of CVD in people affected by the MetS in the absence of other baseline risk factors [8–10]. Central obesity is an independent risk factor for CVD and is associated with MetS [11]. Central obesity predisposes to diabetic nephropathy, hypertensive nephrosclerosis, and focal segmental glomerulosclerosis and represents an independent risk factor for the development and progression of CKD [12]. Obesity and the development of I.R. are thought to be a central feature, contributing to the significant morbidity and mortality associated with the MetS and development of a particular resistant form of HT [13–15]. The development of resistant HT in individuals with MetS can be attributed to a number of factors including proinflammatory cytokines, inappropriate activation of the renin-angiotensin system (RAAS), vasoconstriction from increased sympathetic nervous system (SNS) activation, and dysregulation in adipokines production and secretion [16]. Several components of the MetS are associated with indirect or direct markers of adrenergic overdrive [17]. This review will focus on current understanding of the mechanisms through which sympathetic overactivity may be interlaced to the metabolic syndrome, with particular regard to the role of insulin resistance and of some adipokines.
2. Pathophysiology of the Mets
In 1988, Reaven first postulated “the syndrome X,” which is now named “Metabolic Syndrome” (MetS) [14]. Reaven noticed the frequent association of factors leading to the development of CVD: glucose intolerance, hyperinsulinemia, high serum triglycerides, low serum high-density lipoprotein cholesterol, and HT. I.R. was proposed as the “driving force” of the syndrome [14, 18]. Subsequently, other abnormalities, in particular prothrombotic and chronic proinflammatory states, were added to the definition of the MetS. Later on, abdominal obesity became the “core” of the syndrome [19–21]. Since metabolic abnormalities linked to I.R. are usually found in patients with abdominal obesity [22, 23] I.R. is considered to be the “core” of the MetS and central obesity its most important clinical clue [24].
3. Metabolic Syndrome and Sympathetic Overactivity
As BP and thermogenesis are both under adrenergic control, an alteration in the SNS could be part of the pathophysiology of the MetS. Also, alterations in the sympathetic control of heart rate (HR), cardiac output, peripheral vascular resistance, and renal sodium handling may promote, alone or in combination, the development and progression of HT [25, 26]. Actually, sympathetic overdrive occurs in MetS. Many components of the MetS are characterized by an increased adrenergic activity. Interestingly, sympathetic overdrive is detectable in obese patients prone to MetS before HT occurs. Also, when obesity and HT are both present in the same patient the degree of sympathetic activation is much greater than in those with either condition separately [27]. Individuals with central obesity show increased sympathetic nervous activity (SNA) when compared to individuals with subcutaneous form of obesity [28]. Increased sympathetic outflow has been reported in obese nonhypertensive individuals with the determination of circulating catecholamines, urinary norepinephrine (NE), muscle sympathetic nerve activity (MSNA) recordings of postganglionic sympathetic nerve fibers, and renal NE spillover [29], suggesting that vasoconstriction and renal mechanisms are both involved [30]. There is also evidence that SNS overactivity is not generalized in obesity [31]. Obesity causes differential activation of tissue SNS activity. Increase in HR results from a decrease in parasympathetic activity rather than an increase in sympathetic activity. On the contrary, SNA increases both in the kidney and skeletal muscles of obese hypertensive subjects. Increased SNA does not directly determine vasoconstriction but instead stimulates renin secretion and increases renal sodium reabsorption [15]. Interestingly, Kalil and Haynes showed that the presence of misleading inferences from multifiber recordings of MSNA involved vascular tone in obesity and also that increased SNA does not necessarily translate into increased vascular tone [30]. It is worth noting that MSNA was mainly measured rather than renal SNS activity, the most important pathway for SNS to cause chronic HT [15]. So MSNA activity may not reflect renal sympathetic nervous activity. Renal sympathetic nerves mediate most, if not all, of the chronic effects of SNA on BP in obesity. In obese dogs fed with a high-fat diet, bilateral renal denervation greatly attenuates sodium retention and HT. Thus, obesity may increase renal sodium reabsorption and cause HT mainly by increasing renal sympathetic activity [15].
4. Mechanisms of Sympathetic Overactivity in the Metabolic Syndrome
Among the different hypotheses which have been proposed to explain the cause of obesity and obesity-related metabolic disturbances, SNS activation plays thus a pivotal role. A large body of evidence clearly shows that sympathetic activity is increased in human obesity [32, 33]. In 1986, Landsberg suggested that sympathetic activation could represent an insulin-mediated adaptative response to overeating promoting thermogenesis and acting as a buffer against weight gain [34]. Reaven first proposed I.R. as the key abnormality leading to hyperinsulinemia, sympathetic activation, and HT [14]. Later on, other investigators stressed the important role of I.R. and hyperinsulinemia and their relationship to SNS activation [35, 36].
Interestingly, Julius et al. proposed increased sympathetic activity as the primary defect leading to I.R. and weight gain [35]. Whether SNS activation is the cause or the consequence of obesity is still matter of debate. SNS activation results in the release of norepinephrine which stimulates adrenergic receptors. Physiological responses depend upon the receptors present in the target organs, the fasting state, and the rate of neuronal firing [37]. Cardiovascular, renal, and metabolic effects of chronic and sustained SNS activation may contribute to HT and the development of I.R. over a prolonged period of time [37]. The pathophysiological mechanisms linking SNS overactivity and obesity-related MetS are complex and still need to be fully elucidated.
Multiple neurohumoral mechanisms can activate the SNS in patients with the MetS. Neural mechanisms include direct activation of the SNS in response to the activation of higher cerebral nuclei by hunger or feeding and renal afferent nerve activation mediated by perirenal fat accumulation and kidney compression [15]. Sympathetic activation can also be triggered by reflex mechanisms (arterial baroreceptor impairment), psychological stress, oxidative stress, obstructive sleep apnea, inflammation, and metabolic factors as I.R. and dysregulated production and secretion of adipokines from visceral fat with a particular important role of leptin.
Table 1 summarizes the possible causes and clinical effects of the MetS. We will first focus on the role of I.R./hyperinsulinemia, the key metabolic alteration in MetS, and then discuss the role of adipokines in the activation of SNS and their interplay with insulin. Figure 1 summarizes these interactions.
Table 1.
Effects of insulin resistance, ghrelin, and some adipokines on endocrine and metabolic functions in the pathogenesis of the MetS.
| General effects | Effects on sympathetic nervous system | In vitro/animal studies (references) | Human studies (references) | |
|---|---|---|---|---|
| Insulin resistance | Direct antinatriuretic action | (i) Its intracerebral administration increases sympathetic outflow (ii) Induces sympathetic overactivity (iii) Stimulates SNS to increase cardiac output |
[46–50] | [39, 49, 56–58] |
|
| ||||
| Leptin | (i) Levels correlate with adipose tissue mass (ii) Satiating factor decreases food intake (iii) Physiological regulation of feeding behavior trough hypothalamic receptors |
Vasocontractile effect related to SNS activation | [90, 92, 95, 97, 103, 104, 109, 110] | [90, 91, 99, 100, 102, 107, 111] |
|
| ||||
| NEFAs | Levels are increased in obesity and inversely correlated with insulin sensitivity | Induce a central activation of MNSA in lean subjects | [115–118] | [114, 119, 122] |
|
| ||||
| Adiponectin | (i) Levels are inversely related to obesity, DM, and insulin resistant states (ii) Ameliorates obesity-related hypertension |
[127, 128, 132, 133] | [129, 131] | |
|
| ||||
| Ghrelin | (i) Its infusion decreases blood pressure and HR (ii) Improves endothelial function (iii) Promotes weight gain and increases appetite |
Its infusion increases SNS activity | [136, 137, 141, 143] | [140, 142, 144] |
Figure 1.
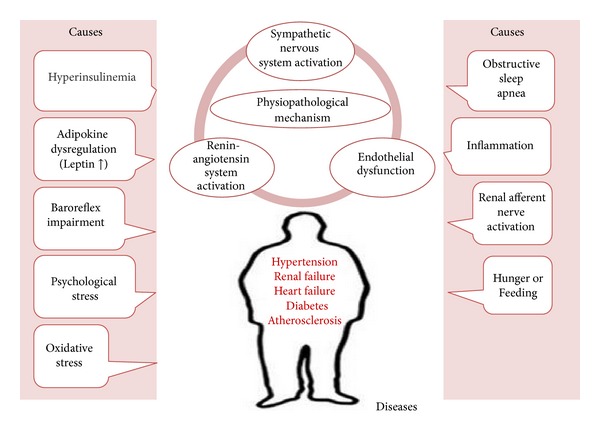
In obesity, infiltration of inflammatory cells in the white adipose tissue disturbs the secretion of adipokines and increases the activity of adipocyte renin-angiotensin system. Increased secretion of leptin and proinflammatory cytokines and decreased amounts of adiponectin contribute to the development of obesity-related hypertension.
5. Hyperinsulinemia and Sympathetic Overactivity
Hence, insulin plays a pivotal role in the development of DM, HT, and the MetS [38]. Insulin stimulates SNS to increase cardiac output and the delivery and enhances the utilization of glucose in the peripheral tissues [39]. I.R. is the inability of insulin to produce its numerous actions, in spite of its normal secretion from the pancreatic beta cells [15, 40]. Insulin elicits its various biological responses by binding to a specific receptor [41, 42]. The ability of insulin receptor to autophosphorylate and then phosphorylate intracellular substrates is crucial for the complex cellular responses to insulin [41–43]. The two major signaling pathways activated by insulin binding to its receptor, the phosphatidylinositol-3′-kinase (PI3K) pathway and the mitogenic-activated protein kinase (MAPK) pathway, among their effects, play a role in vasodilatation and in the decrease in nitric oxide (NO) production, respectively, [43, 44]. In the MetS I.R. mainly results from an impairment in the cellular events distal to the interaction insulin/insulin surface receptor [14]. Selective I.R., located primarily in the muscle and the adipose tissue, causes compensatory hyperinsulinemia which has an adverse impact on insulin-sensitive tissues [41, 42]. I.R. arises due to various genetic and acquired factors, including obesity [45]. The effects of insulin on BP are multifactorial, including sympathetic activation and direct antinatriuretic action [43]. In animal studies insulin increases sympathetic outflow via intracerebral administration [46, 47]. Recent investigations from Cassaglia and coworkers identify the arcuate nucleus, via the paraventricular nucleus of the hypothalamus, as the central site of action of insulin in the increase of SNS activity and in the sympathetic baroreflex gain [48]. Insulin receptors in the hypothalamus coactivate the SNS through a transport-mediated uptake of peripheral insulin across the blood-brain barrier [49]. Also, the presence of highly permeable capillaries in the arcuate nucleus (AN) allows insulin to activate receptors without a specific transport mechanism [50, 51]. In 1991, Anderson and coworkers showed that hyperinsulinemia causes sympathetic activation in humans [52]. Sympathetic overactivity occurring in the MetS is dependent on hyperinsulinemia and related I.R. [53]. Human studies suggest that hyperinsulinemia may contribute to the increased SNS activity observed in the obese MetS patients. Insulin secretion following a meal [54–56] or during a hyperinsulinemic euglycemic clamp [52, 57–60] determines an increase in MSNA and enhances the arterial baroreflex gain of SNA. Interestingly, chronic SNS overactivity contributes to a further decline of insulin sensitivity. Furthermore, SNS coactivation by the hypothalamic-pituitary-adrenal axis may also occur in the hyperinsulinemic state secondary to obesity [29]. In addition to the neural SNS overactivity at rest, obese subjects demonstrate baroreflex impairment and blunted responses to sympathoexcitatory manoeuvres. The exact cause of changes in baroreflex function in obese patients is not entirely clear. Changes in baroreceptor signalling may be a contributing factor to sympathetic overactivity as well as reduced baroreflex responsiveness [29]. Following hypocaloric diet, obese subjects showed a reduction in MSNA and whole body NE spillover rate [61]. However, during weight maintenance period following weight loss MSNA rebounded while NE spillover was preserved [29]. Recent findings from Straznicky et al. on obese MetS patients demonstrated that the progression to type II DM is associated with increased central sympathetic drive, blunted sympathetic responsiveness, and altered NE disposition [62]. Many studies showed that perturbed autonomic nervous system function in the MetS may be reversible. Recently, caloric restriction inhibited SNA via an antioxidant mechanism in the rostral ventrolateral medulla in obesity-induced hypertensive rats [63]. Also, data from Lambert et al. showed that in MetS patients dietary weight loss decreased sympathetic nerve firing and improved hemodynamic and metabolic parameters [64]. It is worth noting that in a very recent longitudinal investigation from Licht and coworkers, a dysregulation of autonomic nervous system in subjects under stress predicted 2-year development of the MetS [65]. However, other studies call into question the role of hyperinsulinemia in the determination of sympathetic overactivity. Obese subjects do not appear to retain their sensitivity to the stimulatory effects of insulin on SNS [29]. A greater increase in MSNA activity in response to euglycemic hyperinsulinemia was observed in lean subjects compared to obese by Vollenweider and coworkers [66]. Later on, similar results were obtained by Straznicky et al. in patients with I.R. and MetS, who exhibited a blunted sympathetic response to increased plasma insulin following a glucose load [67] consistent with central I.R. Certain adipokines expressed in central obesity may contribute to determining sympathetic overdrive and in case of overexpression may contribute to I.R., resulting in hyperinsulinemic state with greater sympathetic outflow [29].
Among the factors promoting the development of I.R. and progression of MetS, reactive oxygen species (ROS) play an important role. In obesity ROS are increased and ROS can be reduced with weight loss in humans [68]. In rats, obesity induced by a high-fat diet resulted in enhanced oxidative stress [69]. Ogihara and coworkers reported that ROS overproduction may cause insulin resistance in AngII-infused rats [70]. Blendea and coworkers confirmed these data and showed that insulin resistance was ameliorated by tempol in TG(mREN-2)27 (Ren-2) transgenic rats, which have a stimulated renin-angiotensin system [71]. Thus, ROS overproduction results in I.R. In mice, ROS overproduction in target organs of insulin, like adipose tissue and liver, preceded the onset of obesity and I.R. [72]. In a recent investigation, Yubero-Serrano and coworkers showed that the higher the number of MetS components, the greater the degree of oxidative stress, leading to increased plasma superoxide dismutase and glutathione peroxidase activities, plasma H2O2, lipid peroxidation products and sVCAM-1, and as well to decreased postischemic reactive hyperemia and total plasma nitrites. Also, oxidative stress increase was associated with higher body mass index, waist circumference, diastolic blood pressure, HOMAIR and triglycerides, glucose and insulin, and lower HDL-cholesterol and HOMAb and QUICKI indexes [73], confirming previous results from Fujita and coworkers [74].
6. Visceral Adiposity and MetS
Although adiposity is defined as an increase in total body mass, visceral fat expansion correlates with a cluster of metabolic abnormalities observed in the MetS [75]. Visceral fat represents a metabolically active organ and has been strongly related to insulin sensitivity [76, 77] and CVD both in humans and animals [77]. Subcutaneous fat, characterized by insulin-sensitive adipocytes, is mainly a fat depot. Instead, visceral fat adipocytes are insulin-resistant cells within a network of blood capillaries and infiltrating inflammatory cells [40]. Inflammatory cells within the visceral fat may play a role in adipocyte behaviour as a source of hormones and cytokines, called adipokines, with proinflammatory and proatherogenic action. Circulating cytokines including resistin and leptin are generally increased in obese subjects and in patients with DM [76, 78–80]. On the other hand, circulating adiponectin is decreased. Adiponectin is a tissue-specific circulating hormone with insulin-sensitizing and antiatherogenic properties. Also, adiponectin stimulates glucose and fatty acid oxidation in the muscle, enhances insulin sensitivity in the liver, increases free fatty acid oxidation, reduces hepatic glucose output, and inhibits monocyte adhesion and macrophage transformation to foam cells within the vascular wall [78–80].
I.R. and visceral obesity determine BP elevation by activating SNS and renin-angiotensin-aldosterone system (RAAS) [40], resulting in sodium retention and volume expansion as well as and endothelial and renal dysfunction [41, 44, 81]. Hyperinsulinemia activates RAAS in both heart and blood vessels, with production of angiotensin II which has proatherogenic effects. Angiotensin II inhibits vasodilator effects of insulin on blood vessels and glucose uptake into the skeletal muscle [41, 44, 82] resulting in decreased NO production, vasoconstriction, and GLUT 4 inhibition [40].
The presence of endothelial dysfunction in patients with obesity and I.R. was first reported by Steinberg and coworkers. Adipokines dysregulation and inflammatory state disrupt vascular homeostasis by causing an imbalance between the NO pathway and the endothelin 1 system, with impaired insulin-stimulated endothelium-dependent vasodilation [83].
It is noteworthy that in obesity vascular dysfunction also involves the other layers of the vessel wall. Obesity-induced changes in medial smooth muscle cells disrupt the physiological facilitatory action of insulin on the responsiveness to vasodilator stimuli, whereas the adventitia and perivascular fat appear to be a source of proinflammatory and vasoactive factors possibly contributing to the endothelial and smooth muscle cell dysfunction and to the pathogenesis of vascular disease as pointed out by Tesauro and coworkers [84, 85].
At the present time, body weight control has not yet proved to prevent metabolic and cardiovascular complications of obesity on a large scale. Recent data from the same investigators show that in the forearm circulation of hyperinsulinemic MetS patients, Rho-Kinase inhibition by fasudil improves both endothelium-dependent and independent vasodilator responsiveness, possibly by increased oxidative stress [86].
7. Effects of Adipokines on Sympathetic Overactivity
7.1. Adipokines
Adipose tissue produces bioactive substances, known as adipokines, and releases them into its direct surroundings and into the bloodstream [87]. Among the different actions of the adipokines is the regulation of arterial tone [80, 88]. Therefore, adipose tissue affects not only metabolism but also many functions of organs and tissues, such as brain, muscle, liver, and blood vessels. The presence of a normal amount of adipose tissue is essential. An imbalance can cause dysregulation in the release of adipokines that may result in vascular disturbances and inflammation [89]. Adipokines may alter SNS activity and impair insulin signaling. Adipokines mainly involved in obesity and MetS are leptin, nonesterified free fatty acids (NEFAs), reactive oxygen species (ROS), adipocytic angiotensinogen and resistin. A reduction of adiponectin may be involved as well. An unbalanced interplay between these adipokines may lead to an impaired insulin signaling as well as to a state of inflammation and/or alter sympathetic regulation [29]. The aim of this section is to focus on the main SNS-activating adipokine, leptin, and on the main SNS-inhibiting one, adiponectin.
7.2. Leptin and Leptin Resistance
Leptin is a 167 aminoacid-16 kDa protein. It is secreted from adipocytes proportionally to the adipose tissue mass [90–92]. Physiologically, leptin represents the inhibitory signal from fat that informs the brain about the body's stocks of stored energy [93]. Its ability to produce anorexigenic effects has been extensively studied and is beyond the scope of this review. Leptin decreases food intake, and this is particularly dependent on the depolarization and hyperpolarization of neurons in the AN of the hypothalamus [94, 95]. Leptin resistance develops in obesity because the ARH neurons expressing leptin receptors do not become further activated from baseline in response to exogenous leptin; consequently, increased leptin levels do not increase energy expenditure or decrease food intake [96, 97]. How leptin resistance develops and how it could be treated in obesity is now under investigation.
Whether SNA increase in obesity [31, 98, 99] leads to innervation of all organs or it is organ-specific is still matter of debate. An interesting finding, as it is now readily hypothesized, is that a chronic increase of SNA to the kidney contributes to the development of HT. HT is part of the MetS and is well known to contribute significantly to the development of CVD. Leptin acutely increases SNA [100, 101], although, at the present time, no conclusive data demonstrate that leptin increases SNA chronically, leading to HT. The increase of SNA observed in obesity also appears to cause organ damage, which exacerbates the risk of MetS and CVD [102]. When acutely injected into the dorsomedial hypothalamus of anesthetized rats leptin determines an increase in HR and BP [103]. Very recent animal studies show that leptin exerts its action at the level of the nucleus of the solitary tract where it alters the activity of neurons that mediate the cardiovascular responses to the activation of the aortic baroreceptor reflex [104] and in the forebrain to influence the baroreflex control of lumbar, renal, and splanchnic SNA and finally the HR [105]. Also, leptin activates brain centers that regulate SNS activity through a melanocortin-system-dependent pathway [106].
The interactions between the brain melanocortin system and leptin represent an important area of research to further understand the mechanisms leading to SNS activation in obesity.
Determining whether hyperleptinemia may be the cause of chronically elevated SNA in obesity, via activation of leptin receptors in higher brain regions, will hopefully lead to new treatment options for obesity. In a recent work, Curry and coworkers showed that a low MSNA and a lack of SNS-mediated support of resting energy expenditure 3 years after gastric by-pass should possibly be multifactorial in origin and involve changes in insulin sensitivity, body composition, and leptin [107].
Physiologically, leptin contributes to BP by its vasorelaxing and vasocontractile effects [108, 109]. While the contractile effect of leptin is attributed to SNS activation [110], various mechanisms seem to be responsible for leptin-induced vasorelaxation. This latter effect can be endothelium-dependent, either through the release of NO [110] or by other mechanisms [109, 111]. Recent findings from Schinzari and coworkers suggest that, under physiologic conditions, leptin stimulates both endothelin-1 and NO activity in the human circulation. This effect is absent in hyperleptinemic patients with MetS who are unresponsive to additional leptin [112].
7.3. Nonesterified Fatty Acids
Nonesterified fatty acids (NEFAs) act on all the aspects of glucose homeostasis, from uptake at peripheral tissue to hepatic production and disposal. NEFAs are increased in obesity and are inversely correlated with insulin sensitivity [113, 114]. Increased NEFAs may inhibit glucose uptake into peripheral tissues by impairing PI3-kinase activation [115]. PI3-kinase activation is lost indeed as a result of a high fat diet in mice [116]. Impaired PI3-kinase activation may also be due to excess of protein kinase C [117]. NEFAs can cross the blood-brain barrier, cause a central activation of MSNA in lean subjects [118, 119], and reduce cardiovagal baroreflex in lean and obese subjects [120]. NEFAs also stimulate plasminogen activator inhibitor-1, which may contribute to the association between increased plasma NEFA in obesity and the augmentation of MSNA [121]. However, other investigations demonstrated no significant effect of NEFAs on MSNA or sympathetic baroreflex sensitivity [122]. Furthermore, whole body and renal NE spillover did not change during infusion of NEFAs [123].
7.4. Adiponectin
Adiponectin is an anti-inflammatory, insulin-sensitizing, and antiatherogenic protein exclusively secreted by adipocytes. Adiponectin is an adipose tissue hormone, also known as gelatin-binding protein-28 (GBP28), AdipoQ, adipocyte complement-related protein (ACRP30), or apM1. Adiponectin circulates in trimeric, hexameric, and high-molecular-mass species. Different forms of adiponectin may play distinct roles in the balance of energy homoeostasis. Adiponectin is an insulin sensitizing hormone that exerts its action through its receptors Adipo R1, Adipo R2, and T-cadherin. Adipo R1 is expressed mainly in muscle, whereas Adipo R2 is predominantly expressed in the liver.
Adiponectin is inversely related to obesity, DM, and other I.R. states that cause metabolic dysfunction; adiponectin deficiency may also contribute to coronary heart disease, steatohepatitis, I.R., nonalcoholic fatty liver disease, and a wide array of cancers.
The human adiponectin gene was cloned through systematic sequencing of an adipose-tissue library [124, 125]. The apM1 gene encodes a 244 amino acid open reading frame containing a putative signal sequence repeat (66 amino acids) followed by a cluster of aromatic residues near the C terminus that shows a high local resemblance to collagens X and VIII and complement factor C1q [126].
The regulation of adiponectin receptors Adipo R1 and Adipo R2 is important to facilitate essential physiological functions. Adipo R1 is expressed ubiquitously and exhibits high affinity to the ligand, whereas Adipo R2 exhibits intermediate affinity. The expression of adiponectin and its receptors has been investigated in streptozotocin (STZ)-induced diabetic rat heart and in mouse skeletal muscle. STZ-induced DM upregulates adiponectin receptors in the heart [127].
Some evidence suggests that T-cadherin can bind to the hexameric and HMW forms of adiponectin but not to monomer globular and trimeric forms. T-cadherin is expressed ubiquitously, with the highest expression found in the heart and the aortic, carotid, iliac, and kidney arteries [128]. T-cadherin is bound to adiponectin and is critical for the association of adiponectin protection with cardiac stress in mice.
The pleiotropic roles of adiponectin have been studied in multiple in vitro and in vivo models. The multiple molecular targets of adiponectin mediate multiple pharmacological actions.
A steep rise in the prevalence of obesity has occurred over the past few decades. Obesity is inversely related to adiponectin, making adiponectin a negative marker of MetS. Furthermore, the expression of the receptors Adipo R1 and Adipo R2 declines by 30% in the subcutaneous fat of obese individuals, while they normalize following weight loss [129]. Adiponectin may play an important role in type II DM, HT, multiple sclerosis, and the dyslipidemias. The most significant role played by adiponectin is that of its insulin-sensitizing effect. Adiponectin in the diabetic's blood is lower than normal, whereas higher adiponectin in plasma minimizes the risk of type II DM [130]. Adiponectin relates negatively to blood glucose and insulin. Total adiponectin, HMW adiponectin, and the total/HMW ratio are all inversely related to homeostasis model assessment I.R. index. The total/HMW ratio is considered a better indicator of I.R. than total plasma adiponectin [131]. The role of adiponectin in I.R. was determined using knockout mice. These mice had normal plasma insulin, but its capability of lowering blood glucose was severely impaired, this clearly pointing to the role of adiponectin in glucose tolerance [132]. The absence of serum adiponectin in lipoatrophic mice causes hyperglycemia and hyperinsulinemia, which can be normalized by adiponectin injections. All studies on the putative role of adiponectin in IR and type II DM suggest that low plasma adiponectin causes susceptibility to these disorders.
In conclusion, adiponectin exerts an insulin-sensitizing action with profound effects on fatty acid oxidation and inflammation. Drugs affecting serum adiponectin may have a role in the treatment of type II DM and possibly the Mets. In obese adiponectin-knockout mice with HT adiponectin replenishment lowers elevated BP [133]. Existing drugs such as peroxisome proliferator-activated receptor agonists (thiazolidinediones), some angiotensin Adipo R1 receptor blockers (telmisartan), angiotensin-converting enzyme inhibitors, and cannabinoid Adipo R1 receptor blockers (rimonabant and taranabant) may increase circulating adiponectin [134]. However, future strategies should focus on upregulation of adiponectin/adiponectin receptors expression or on targeting adiponectin receptors with specific agonists [132]. Modulation of adiponectin actions through the expression of adiponectin receptors may thus be a novel and promising therapeutic option.
7.5. Ghrelin
Ghrelin is a 28-amino-acid growth-hormone-releasing peptide secreted by the stomach [135]. It causes increased food intake and weight gain in rodents [136, 137]. But in obese subjects circulating levels are decreased. This observation is against a central role of ghrelin in the determination of common obesity [138, 139]. It is noteworthy that ghrelin infusion decreases BP by 5–10 mmHg, although it also increases SNS activity, perhaps through compensatory baroreflex activation [140]. The long-term central nervous system mediated cardiovascular actions of ghrelin are still unknown. A recent work of Freeman et al. demonstrated that chronic central ghrelin infusion reduces BP and HR despite increasing appetite and promoting weight gain in normotensive and hypertensive rats [141].
Ghrelin may also improve endothelial function mimicking phosphoinositol 3-kinase-dependent actions of insulin to stimulate production of NO by endothelial cells and restoring the endothelin 1/nitric oxide balance in patients with obesity-related MetS, as observed by Tesauro and coworkers [142–144].
7.6. Further Perspectives
The prevalence of the MetS is escalating worldwide. Sympathetic overdrive may be the common thread of visceral adiposity, HT, dyslipidemia, and glucose intolerance in the clinical diagnosis of the MetS [29]. MetS is thus characterized by sympathetic overdrive with outflow activation to both kidneys and adipose tissue among others. Although the mechanisms responsible for the initial activation of SNS are still to be fully elucidated, hyperinsulinemia, derangement of circulating adipokines, and beta receptor polimorphisms are all implicated and may cause the development of HT, I.R., diastolic dysfunction, and finally renal disease. Although lifestyle correction and hypotensive medications are the first-line therapy for obesity and HT in the MetS, interventions that target the SNS directly may be of further benefit. Such benefits may even be weight-unrelated and associated with a significant reduction in end-organ damage [37]. Afferent adrenergic signaling from the kidneys was recently identified as an important contributor to central SNS overdrive and SNS outflow to the kidneys is involved in cardiovascular, renal, and metabolic control [145, 146]. In recent clinical investigations, functional renal denervation obtained by means of catheter-based radiofrequency or ultrasound technologies achieved BP control in patients with resistant HT [147, 148] and polycystic ovary syndrome [149], further emphasizing the link between SNS overdrive and I.R., HT, and other comorbidities in the MetS. Finally, catheter-based renal denervation was investigated in end-stage renal disease (ESRD) patients with resistant intradialytic HT, who are supposed to compel a significant SNS overdrive [150, 151]. In preliminary investigations, a reduction of SNS overdrive with good control of blood pressure was obtained in small series of ESRD disease patients on maintenance hemodialysis by Di Daniele and coworkers [152], Ott et al. [153], and Schlaich and Coworkers [154].
Further investigations addressing the still open questions in the treatment of resistant HT and evaluating potential new indications such as the MetS or heart failure are still necessary to prove the safety and effectiveness of renal denervation in these patients. By modulating sympathetic activity, renal denervation may have the potential to provide significant benefits in a variety of diseases.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Abbreviations
- MetS:
Metabolic syndrome
- CVD:
Cardiovascular disease
- BP:
Blood pressure
- HT:
Hypertension
- CKD:
Chronic kidney disease
- SNS:
Sympathetic nervous system
- RAAS:
Renin-angiotensin system
- I.R.:
Insulin resistance
- SNA:
Sympathetic nerve activity
- MSNA:
Muscle sympathetic nerve activity
- NE:
Norepinephrine
- AN:
Arcuate nucleus
- DM:
Diabetes
- HR:
Heart rate
- NO:
Nitric oxide
- PI3K:
Phosphatidylinositol-3′-kinase
- MAPK:
Mitogenic-activated protein-kinase
- NEFAs:
Nonesterified fatty acids
- STZ:
Streptozotocin
- HMW:
High molecular weight
- ESRD:
End-stage renal disease
- ROS:
Reactive oxygen species.
References
- 1.Grundy SM, Brewer HB, Jr., Cleeman JI, Smith SC, Jr., Lenfant C. Definition of Metabolic Syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation. 2004;109(3):433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 2.Third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation and treatment of high blood cholesterol in adults (Adult Treatment Panel III). Final report. Circulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- 3.Alberti KGMM, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the international diabetes federation task force on epidemiology and prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 4.Grundy SM. Metabolic syndrome pandemic. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(4):629–636. doi: 10.1161/ATVBAHA.107.151092. [DOI] [PubMed] [Google Scholar]
- 5.Alexander CM, Landsman PB, Teutsch SM, Haffner SM. NCEP-defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes. 2003;52(5):1210–1214. doi: 10.2337/diabetes.52.5.1210. [DOI] [PubMed] [Google Scholar]
- 6.Cameron AJ, Shaw JE, Zimmet PZ. The metabolic syndrome: prevalence in worldwide populations. Endocrinology and Metabolism Clinics of North America. 2004;33(2):351–375. doi: 10.1016/j.ecl.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Ford ES, Giles WH, Mokdad AH. Increasing prevalence of the metabolic syndrome among U.S. adults. Diabetes Care. 2004;27(10):2444–2449. doi: 10.2337/diacare.27.10.2444. [DOI] [PubMed] [Google Scholar]
- 8.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. The Lancet. 2005;365(9468):1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 9.Isomaa B, Almgren P, Tuomi T, et al. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24(4):683–689. doi: 10.2337/diacare.24.4.683. [DOI] [PubMed] [Google Scholar]
- 10.Lakka H-M, Laaksonen DE, Lakka TA, et al. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. The Journal of the American Medical Association. 2002;288(21):2709–2716. doi: 10.1001/jama.288.21.2709. [DOI] [PubMed] [Google Scholar]
- 11.Poirier P, Giles TD, Bray GA, et al. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on obesity and heart disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2006;113(6):898–918. doi: 10.1161/CIRCULATIONAHA.106.171016. [DOI] [PubMed] [Google Scholar]
- 12.Tesauro M, Canale MP, Rodia G, et al. Metabolic syndrome, chronic kidney, and cardiovascular diseases: role of adipokines. Cardiology Research and Practice. 2011;2011:11 pages. doi: 10.4061/2011/653182.653182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirk EP, Klein S. Pathogenesis and pathophysiology of the cardiometabolic syndrome. Journal of Clinical Hypertension. 2009;11(12):761–765. doi: 10.1111/j.1559-4572.2009.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 15.Hall JE, da Silva AA, do Carmo JM, et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. The Journal of Biological Chemistry. 2010;285(23):17271–17276. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaudhary K, Buddineni JP, Nistala R, Whaley-Connell A. Resistant hypertension in the high-risk metabolic patient. Current Diabetes Reports. 2011;11(1):41–46. doi: 10.1007/s11892-010-0155-x. [DOI] [PubMed] [Google Scholar]
- 17.Mancia G, Bousquet P, Elghozi JL, et al. The sympathetic nervous system and the metabolic syndrome. Journal of Hypertension. 2007;25(5):909–920. doi: 10.1097/HJH.0b013e328048d004. [DOI] [PubMed] [Google Scholar]
- 18.Reaven GM. Pathophysiology of insulin resistance in human disease. Physiological Reviews. 1995;75(3):473–486. doi: 10.1152/physrev.1995.75.3.473. [DOI] [PubMed] [Google Scholar]
- 19.Shirai K. Obesity as the core of the metabolic syndrome and the management of coronary heart disease. Current Medical Research and Opinion. 2004;20(3):295–304. doi: 10.1185/030079903125003008. [DOI] [PubMed] [Google Scholar]
- 20.Després J-P, Lemieux I, Bergeron J, et al. Abdominal Obesity and the Metabolic Syndrome: contribution to global cardiometabolic risk. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(6):1039–1049. doi: 10.1161/ATVBAHA.107.159228. [DOI] [PubMed] [Google Scholar]
- 21.Phillips LK, Prins JB. The link between abdominal obesity and the metabolic syndrome. Current Hypertension Reports. 2008;10(2):156–164. doi: 10.1007/s11906-008-0029-7. [DOI] [PubMed] [Google Scholar]
- 22.Sowers JR, Frohlich ED. Insulin and insulin resistance: impact on blood pressure and cardiovascular disease. Medical Clinics of North America. 2004;88(1):63–82. doi: 10.1016/s0025-7125(03)00128-7. [DOI] [PubMed] [Google Scholar]
- 23.Bays H, Abate N, Chandalia M. Adiposopathy: sick fat causes high blood sugar, high blood pressure and dyslipidaemia. Future Cardiology. 2005;1:39–59. doi: 10.1517/14796678.1.1.39. [DOI] [PubMed] [Google Scholar]
- 24.Alberti KG, Zimmet P, Shaw J, IDF Epidemiology Task Force Consensus Group The metabolic syndrome—a new worldwide definition. The Lancet. 2005;366(9491):1059–1062. doi: 10.1016/S0140-6736(05)67402-8. [DOI] [PubMed] [Google Scholar]
- 25.Grassi G. Role of the sympathetic nervous system in human hypertension. Journal of Hypertension. 1998;16(12):1979–1987. doi: 10.1097/00004872-199816121-00019. [DOI] [PubMed] [Google Scholar]
- 26.Amerena J, Julius S. The role of the autonomic nervous system in hypertension. Hypertension Research. 1995;18(2):99–110. doi: 10.1291/hypres.18.99. [DOI] [PubMed] [Google Scholar]
- 27.Grassi G. Sympathetic overdrive and cardiovascular risk in the metabolic syndrome. Hypertension Research. 2006;29(11):839–847. doi: 10.1291/hypres.29.839. [DOI] [PubMed] [Google Scholar]
- 28.Alvarez GE, Ballard TP, Beske SD, Davy KP. Subcutaneous obesity is not associated with sympathetic neural activation. American Journal of Physiology—Heart and Circulatory Physiology. 2004;287(1):H414–H418. doi: 10.1152/ajpheart.01046.2003. [DOI] [PubMed] [Google Scholar]
- 29.Smith MM, Minson CT. Obesity and adipokines: effects on sympathetic overactivity. Journal of Physiology. 2012;590(8):1787–1801. doi: 10.1113/jphysiol.2011.221036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalil GZ, Haynes WG. Sympathetic nervous system in obesity-related hypertension: mechanisms and clinical implications. Hypertension Research. 2012;35(1):4–16. doi: 10.1038/hr.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaz M, Jennings G, Turner A, Cox H, Lambert G, Esler M. Regional sympathetic nervous activity and oxygen consumption in obese normotensive human subjects. Circulation. 1997;96(10):3423–3429. doi: 10.1161/01.cir.96.10.3423. [DOI] [PubMed] [Google Scholar]
- 32.Grassi G, Dell’Oro R, Facchini A, Trevano FQ, Bolla GB, Mancia G. Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. Journal of Hypertension. 2004;22(12):2363–2369. doi: 10.1097/00004872-200412000-00019. [DOI] [PubMed] [Google Scholar]
- 33.Straznicky NE, Lambert EA, Lambert GW, Masuo K, Esler MD, Nestel PJ. Effects of dietary weight loss on sympathetic activity and cardiac risk factors associated with the metabolic syndrome. The Journal of Clinical Endocrinology & Metabolism. 2005;90(11):5998–6005. doi: 10.1210/jc.2005-0961. [DOI] [PubMed] [Google Scholar]
- 34.Landsberg L. Diet, obesity and hypertension: an hypothesis involving insulin, the sympathetic nervous system, and adaptive thermogenesis. Quarterly Journal of Medicine. 1986;61(236):1081–1090. [PubMed] [Google Scholar]
- 35.Julius S, Valentini M, Palatini P. Overweight and hypertension: a 2-way street? Hypertension. 2000;35(3):807–813. doi: 10.1161/01.hyp.35.3.807. [DOI] [PubMed] [Google Scholar]
- 36.Grundy SM. What is the contribution of obesity to the metabolic syndrome? Endocrinology and Metabolism Clinics of North America. 2004;33:267–282. doi: 10.1016/j.ecl.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Lambert GW, Straznicky NE, Lambert EA, Dixon JB, Schlaich MP. Sympathetic nervous activation in obesity and the metabolic syndrome-Causes, consequences and therapeutic implications. Pharmacology and Therapeutics. 2010;126(2):159–172. doi: 10.1016/j.pharmthera.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 38.Cheung BMY, Li C. Diabetes and hypertension: is there a common metabolic pathway? Current Atherosclerosis Reports. 2012;14:160–166. doi: 10.1007/s11883-012-0227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deedwania P. Hypertension, dyslipidemia, and insulin resistance in patients with diabetes mellitus or the cardiometabolic syndrome: benefits of vasodilating beta-blockers. Journal of Clinical Hypertension. 2011;13(1):52–59. doi: 10.1111/j.1751-7176.2010.00386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duvnjak L, Duvnjak M. The metabolic syndrome—an ongoing story. Journal of Physiology and Pharmacology. 2009;60:19–24. [PubMed] [Google Scholar]
- 41.Reaven GM. Insulin resistance, compensatory hyperinsulinemia, and coronary heart disease: syndrome X revisited. In: Jefferson LS, Cherrington AD, editors. Handbook of Physiology. section 7. Vol. 2. New York, NY, USA: Oxford University Press; 2001. pp. 1169–1197. (The Endocrine Pancreas and Regulation of Metabolism). [Google Scholar]
- 42.Reaven GM. Insulin resistance and its consequences. In: LeRoith D, Taylor SI, Olefasky JM, editors. Diabetes Mellitus: A Fundamental and Clinical Text. Philadelphia, Pa, USA: Lippincott Williams & Wilkins; 2004. pp. 899–915. [Google Scholar]
- 43.Flakoll PJ, Jensen MD, Cherrington AD. Physiological action of insulin. In: LeRoith D, Taylor SI, Olefasky JM, editors. Diabetes Mellitus: A Fundamental and Clinical Text. Philadelphia, Pa, USA: Lippincott Williams & Wilkins; 2004. pp. 165–181. [Google Scholar]
- 44.Wang CCL, Goalstone ML, Draznin B. Molecular mechanisms of insulin resistance that impact cardiovascular biology. Diabetes. 2004;53(11):2735–2740. doi: 10.2337/diabetes.53.11.2735. [DOI] [PubMed] [Google Scholar]
- 45.Stump CS, Clark SE, Sowers JR. Oxidative stress in insulin-resistant conditions: cardiovascular implications. Treatments in Endocrinology. 2005;4(6):343–351. doi: 10.2165/00024677-200504060-00003. [DOI] [PubMed] [Google Scholar]
- 46.Muntzel MS, Morgan DA, Mark AL, Johnson AK. Intracerebroventricular insulin produces nonuniform regional increases in sympathetic nerve activity. American Journal of Physiology, Regulatory—Integrative and Comparative Physiology. 1994;267(5):R1350–R1355. doi: 10.1152/ajpregu.1994.267.5.R1350. [DOI] [PubMed] [Google Scholar]
- 47.Rahmouni K, Morgan DA, Morgan GM, et al. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. The Journal of Clinical Investigation. 2004;114(5):652–658. doi: 10.1172/JCI21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cassaglia PA, Hermes SM, Aicher SA, Brooks VL. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. Journal of Physiology. 2011;589(7):1643–1662. doi: 10.1113/jphysiol.2011.205575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hopkins DF, Williams G. Insulin receptors are widely distributed in human brain and bind human and porcine insulin with equal affinity. Diabetic Medicine. 1997;14:1044–1050. doi: 10.1002/(SICI)1096-9136(199712)14:12<1044::AID-DIA508>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 50.Ciofi P. The arcuate nucleus as a circumventricular organ in the mouse. Neuroscience Letters. 2011;487(2):187–190. doi: 10.1016/j.neulet.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 51.Dampney RAL. Arcuate nucleus—a gateway for insulin’s action on sympathetic activity. Journal of Physiology. 2011;589(9):2109–2110. doi: 10.1113/jphysiol.2011.208579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson EA, Hoffman RP, Balon TW, Sinkey CA, Mark AL. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. The Journal of Clinical Investigation. 1991;87(6):2246–2252. doi: 10.1172/JCI115260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Landsberg L. Obesity and the insulin resistance syndrome. Hypertension Research. 1996;19(1):S51–S55. doi: 10.1291/hypres.19.supplementi_s51. [DOI] [PubMed] [Google Scholar]
- 54.Fagius J, Berne C. Increase in muscle nerve sympathetic activity in humans after food intake. Clinical Science. 1994;86(2):159–167. doi: 10.1042/cs0860159. [DOI] [PubMed] [Google Scholar]
- 55.Cox HS, Kaye DM, Thompson JM, et al. Regional sympathetic nervous activation after a large meal in humans. Clinical Science. 1995;89(2):145–154. doi: 10.1042/cs0890145. [DOI] [PubMed] [Google Scholar]
- 56.Young CN, Deo SH, Chaudhary K, Thyfault JP, Fadel PJ. Insulin enhances the gain of arterial baroreflex control of muscle sympathetic nerve activity in humans. Journal of Physiology. 2010;588(18):3593–3603. doi: 10.1113/jphysiol.2010.191866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berne C, Fagius J, Pollare T, Hjemdahl P. The sympathetic response to euglycaemic hyperinsulinaemia. Evidence from microelectrode nerve recordings in healthy subjects. Diabetologia. 1992;35(9):873–879. doi: 10.1007/BF00399935. [DOI] [PubMed] [Google Scholar]
- 58.Vollenweider P, Tappy L, Randin D, et al. Differential effects of hyperinsulinemia and carbohydrate metabolism on sympathetic nerve activity and muscle blood flow in humans. The Journal of Clinical Investigation. 1993;92(1):147–154. doi: 10.1172/JCI116542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hausberg M, Mark AL, Hoffman RP, Sinkey CA, Anderson EA. Dissociation of sympathoexcitatory and vasodilator actions of modestly elevated plasma insulin levels. Journal of Hypertension. 1995;13(9):1015–1021. doi: 10.1097/00004872-199509000-00012. [DOI] [PubMed] [Google Scholar]
- 60.Van De Borne P, Hausberg M, Hoffman RP, Mark AL, Anderson EA. Hyperinsulinemia produces cardiac vagal withdrawal and nonuniform sympathetic activation in normal subjects. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 1999;276(1):R178–R183. doi: 10.1152/ajpregu.1999.276.1.R178. [DOI] [PubMed] [Google Scholar]
- 61.Straznicky NE, Grima MT, Eikelis N, et al. The effects of weight loss versus weight loss maintenance on sympathetic nervous system activity and metabolic syndrome components. The Journal of Clinical Endocrinology & Metabolism. 2011;96(3):E503–E508. doi: 10.1210/jc.2010-2204. [DOI] [PubMed] [Google Scholar]
- 62.Straznicky NE, Grima MT, Sari CI, et al. Neuroadrenergic dysfunction along the diabetes continuum: a comparative study in obese metabolic syndrome subjects. Diabetes. 2012;61(10):2506–2516. doi: 10.2337/db12-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kishi T, Hirooka Y, Ogawa K, Konno S, Sunagawa K. Calorie restriction inhibits sympathetic nerve activity via anti-oxidant effect in the rostral ventrolateral medulla of obesity-induced hypertensive rats. Clinical and Experimental Hypertension. 2011;33(4):240–245. doi: 10.3109/10641963.2011.583969. [DOI] [PubMed] [Google Scholar]
- 64.Lambert E, Straznicky NE, Dawood T, et al. Change in sympathetic nerve firing pattern associated with dietary weight loss in the metabolic syndrome. Frontiers in Physiology. 2011;2, article 52 doi: 10.3389/fphys.2011.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Licht CM, de Geus EJ, Penninx BW. Dysregulation of the autonomic nervous system predicts the developement of the metabolic syndrome. The Journal of Clinical Endocrinology & Metabolism. 2013;98(6):2484–2493. doi: 10.1210/jc.2012-3104. [DOI] [PubMed] [Google Scholar]
- 66.Vollenweider P, Randin D, Tappy L, Jéquier E, Nicod P, Scherrer U. Impaired insulin-induced sympathetic neural activation and vasodilation in skeletal muscle in obese humans. The Journal of Clinical Investigation. 1994;93(6):2365–2371. doi: 10.1172/JCI117242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Straznicky NE, Lambert GW, Masuo K, et al. Blunted sympathetic neural response to oral glucose in obese subjects with the insulin-resistant metabolic syndrome. American Journal of Clinical Nutrition. 2009;89(1):27–36. doi: 10.3945/ajcn.2008.26299. [DOI] [PubMed] [Google Scholar]
- 68.Vincent HK, Taylor AG. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. International Journal of Obesity. 2006;30(3):400–418. doi: 10.1038/sj.ijo.0803177. [DOI] [PubMed] [Google Scholar]
- 69.Dobrian AD, Davies MJ, Schriver SD, Lauterio TJ, Prewitt RL. Oxidative stress in a rat model of obesity-induced hypertension. Hypertension. 2001;37(2):554–560. doi: 10.1161/01.hyp.37.2.554. [DOI] [PubMed] [Google Scholar]
- 70.Ogihara T, Asano T, Ando K, et al. Angiotensin II-induced insulin resistance is associated with enhanced insulin signaling. Hypertension. 2002;40(6):872–879. doi: 10.1161/01.hyp.0000040262.48405.a8. [DOI] [PubMed] [Google Scholar]
- 71.Blendea MC, Jacobs D, Stump CS, et al. Abrogation of oxidative stress improves insulin sensitivity in the Ren-2 rat model of tissue angiotensin II overexpression. American Journal of Physiology—Endocrinology and Metabolism. 2005;288(2):E353–E359. doi: 10.1152/ajpendo.00402.2004. [DOI] [PubMed] [Google Scholar]
- 72.Matsuzawa-Nagata N, Takamura T, Ando H, et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism. 2008;57(8):1071–1077. doi: 10.1016/j.metabol.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 73.Yubero-Serrano EM, Delgado-Lista J, Peña-Orihuela P, et al. Oxidative stress is associated with the number of components of metabolic syndrome: LIPGENE study. Experimental & Molecular Medicine. 2013;45, article e28 doi: 10.1038/emm.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fujita K, Nishizawa H, Funahashi T, Shimomura I, Shimabukuro M. Systemic oxidative stress is associated with visceral fat accumulation and the metabolic syndrome. Circulation Journal. 2006;70(11):1437–1442. doi: 10.1253/circj.70.1437. [DOI] [PubMed] [Google Scholar]
- 75.Despres JP. Health consequences of visceral adiposity. Annals of Internal Medicine. 2001;33:534–541. doi: 10.3109/07853890108995963. [DOI] [PubMed] [Google Scholar]
- 76.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocrine Reviews. 2000;21(6):697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- 77.Haffner SM. Abdominal adiposity and cardiometabolic risk: do we have all the answer? American Journal of Medicine. 2007;120(9, supplement 1):S10–S16. doi: 10.1016/j.amjmed.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 78.Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. The Journal of Clinical Investigation. 2003;112(12):1785–1788. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neels JG, Olefsky JM. Inflamed fat: what starts the fire? The Journal of Clinical Investigation. 2006;116(1):33–35. doi: 10.1172/JCI27280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. British Journal of Nutrition. 2004;92(3):347–355. doi: 10.1079/bjn20041213. [DOI] [PubMed] [Google Scholar]
- 81.Sowers JR, Frohlich ED. Insulin and insulin resistance: impact on blood pressure and cardiovascular disease. Medical Clinics of North America. 2004;88(1):63–82. doi: 10.1016/s0025-7125(03)00128-7. [DOI] [PubMed] [Google Scholar]
- 82.Rahmouni K, Correia MLG, Haynes WG, Mark AL. Obesity-associated hypertension: new insights into mechanisms. Hypertension. 2005;45(1):9–14. doi: 10.1161/01.HYP.0000151325.83008.b4. [DOI] [PubMed] [Google Scholar]
- 83.Schinzari F, Tesauro M, Rovella V, et al. Generalized impairment of vasodilator reactivity during hyperinsulinemia in patients with obesity-related metabolic syndrome. American Journal of Physiology—Endocrinology and Metabolism. 2010;299(6):E947–E952. doi: 10.1152/ajpendo.00426.2010. [DOI] [PubMed] [Google Scholar]
- 84.Tesauro M, Cardillo C. Obesity, blood vessels and metabolic syndrome. Acta Physiologica. 2011;203(1):279–286. doi: 10.1111/j.1748-1716.2011.02290.x. [DOI] [PubMed] [Google Scholar]
- 85.Campia U, Tesauro M, Cardillo C. Human obesity and endothelium-dependent responsiveness. British Journal of Pharmacology. 2012;165(3):561–573. doi: 10.1111/j.1476-5381.2011.01661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schinzari F, Tesauro M, Rovella V, et al. Rho-kinase inhibition improves vasodilator responsiveness during hyperinsulinemia in the metabolic syndrome. American Journal of Physiology—Endocrinology and Metabolism. 2012;303(6):806–811. doi: 10.1152/ajpendo.00206.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mohamed-Ali V, Pinkney JH, Coppack SW. Adipose tissue as an endocrine and paracrine organ. International Journal of Obesity. 1998;22(12):1145–1158. doi: 10.1038/sj.ijo.0800770. [DOI] [PubMed] [Google Scholar]
- 88.Hajer GR, van Haeften TW, Visseren FLJ. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. European Heart Journal. 2008;29(24):2959–2971. doi: 10.1093/eurheartj/ehn387. [DOI] [PubMed] [Google Scholar]
- 89.Maenhaut N, Van de Voorde J. Regulation of vascular tone by adipocytes. BMC Medicine. 2011;9, article 25 doi: 10.1186/1741-7015-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mapfei M, Halaas J, Ravussin E, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nature Medicine. 1995;1(11):1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 91.Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. The New England Journal of Medicine. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 93.Simonds SE, Cowley MA, Enriori PJ. Leptin increasing sympathetic nerve outflow in obesity. A cure for obesity or a potential contributor to metabolic syndrome? Adipocyte. 2012;1(3):177–181. doi: 10.4161/adip.20690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford MLJ. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature. 1997;390(6659):521–525. doi: 10.1038/37379. [DOI] [PubMed] [Google Scholar]
- 95.Cowley MA, Smart JL, Rubinstein M, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411(6836):480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 96.Enriori PJ, Evans AE, Sinnayah P, et al. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metabolism. 2007;5(3):181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 97.Münzberg H, Flier JS, Bjørbæk C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145(11):4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 98.Landsberg L, Young JB. Fasting, feeding and regulation of the sympathetic nervous system. The New England Journal of Medicine. 1978;298(23):1295–1301. doi: 10.1056/NEJM197806082982306. [DOI] [PubMed] [Google Scholar]
- 99.Scherrer U, Randin D, Tappy L, Vollenweider P, Jéquier E, Nicod P. Body fat and sympathetic nerve activity in healthy subjects. Circulation. 1994;89(6):2634–2640. doi: 10.1161/01.cir.89.6.2634. [DOI] [PubMed] [Google Scholar]
- 100.Galletti F, D’Elia L, Barba G, et al. High-circulating leptin levels are associated with greater risk of hypertension in men independently of body mass and insulin resistance: results of an eight-year follow-up study. The Journal of Clinical Endocrinology & Metabolism. 2008;93(10):3922–3926. doi: 10.1210/jc.2008-1280. [DOI] [PubMed] [Google Scholar]
- 101.Eikelis N, Schlaich M, Aggarwal A, Kaye D, Esler M. Interactions between leptin and the human sympathetic nervous system. Hypertension. 2003;41(5):1072–1079. doi: 10.1161/01.HYP.0000066289.17754.49. [DOI] [PubMed] [Google Scholar]
- 102.Lambert E, Sari CI, Dawood T, et al. Sympathetic nervous system activity is associated with obesity-induced subclinical organ damage in young adults. Hypertension. 2010;56(3):351–358. doi: 10.1161/HYPERTENSIONAHA.110.155663. [DOI] [PubMed] [Google Scholar]
- 103.Marsh AJ, Fontes MAP, Killinger S, Pawlak DB, Polson JW, Dampney RAL. Cardiovascular responses evoked by leptin acting on neurons in the ventromedial and dorsomedial hypothalamus. Hypertension. 2003;42(4):488–493. doi: 10.1161/01.HYP.0000090097.22678.0A. [DOI] [PubMed] [Google Scholar]
- 104.Ciriello J. Leptin in nucleus of the solitary tract alters the cardiovascular responses to aortic baroreceptor activation. Peptides. 2013;44:1–7. doi: 10.1016/j.peptides.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 105.Li B, Shi Z, Cassaglia PA, Brooks VL. Leptin acts in the forebrain to differentially influence baroreflex control of lumbar, renal, and splanchnic sympathetic nerve activity and heart rate. Hypertension. 2013;61(4):812–819. doi: 10.1161/HYPERTENSIONAHA.111.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.da Silva AA, do Carmo JM, Hall JE. Role of leptin and central nervous system melanocortins in obesity hypertension. Current Opinion in Nephrology and Hypertension. 2013;22(2):135–140. doi: 10.1097/MNH.0b013e32835d0c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Curry TB, Somaraju M, Hines CN, et al. Sympathetic support of energy expenditure and sympathetic nervous system activity after gastric bypass surgery. Obesity. 2013;21(3):480–485. doi: 10.1002/oby.20106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Frühbeck G. Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes. 1999;48(4):903–908. doi: 10.2337/diabetes.48.4.903. [DOI] [PubMed] [Google Scholar]
- 109.Lembo G, Vecchione C, Fratta L, et al. Leptin induces direct vasodilation through distinct endothelial mechanisms. Diabetes. 2000;49(2):293–297. doi: 10.2337/diabetes.49.2.293. [DOI] [PubMed] [Google Scholar]
- 110.Vecchione C, Maffei A, Colella S, et al. Leptin effect on endothelial nitric oxide is mediated through Akt-endothelial nitric oxide synthase phosphorylation pathway. Diabetes. 2002;51(1):168–173. doi: 10.2337/diabetes.51.1.168. [DOI] [PubMed] [Google Scholar]
- 111.Matsuda K, Teragawa H, Fukuda Y, Nakagawa K, Higashi Y, Chayama K. Leptin causes nitric-oxide independent coronary artery vasolidation in humans. Hypertension Research. 2003;26(2):147–152. doi: 10.1291/hypres.26.147. [DOI] [PubMed] [Google Scholar]
- 112.Schinzari F, Tesauro M, Rovella V, et al. Leptin stimulates both endothelin-1 and nitric oxide activity in lean subjects but not in patients with obesity-related metabolic syndrome. The Journal of Clinical Endocrinology & Metabolism. 2013;98(3):1235–1241. doi: 10.1210/jc.2012-3424. [DOI] [PubMed] [Google Scholar]
- 113.Bruce R, Godsland I, Walton C, Crook D, Wynn V. Associations between insulin sensitivity, and free fatty acid and triglyceride metabolism independent of uncomplicated obesity. Metabolism. 1994;43(10):1275–1281. doi: 10.1016/0026-0495(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 114.Perseghin G, Ghosh S, Gerow K, Shulman GI. Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes. 1997;46(6):1001–1009. doi: 10.2337/diab.46.6.1001. [DOI] [PubMed] [Google Scholar]
- 115.Kruszynska YT, Worrall DS, Ofrecio J, Frias JP, Macaraeg G, Olefsky JM. Fatty acid-induced insulin resistance: decreased muscle PI3K activation but unchanged Akt phosphorylation. The Journal of Clinical Endocrinology & Metabolism. 2002;87(1):226–234. doi: 10.1210/jcem.87.1.8187. [DOI] [PubMed] [Google Scholar]
- 116.Zierath JR, Houseknecht KL, Gnudi L, Kahn BB. High-fat feeding impairs insulin-stimulated GLUT4 recruitment via an early insulin-signaling defect. Diabetes. 1997;46(2):215–223. doi: 10.2337/diab.46.2.215. [DOI] [PubMed] [Google Scholar]
- 117.Griffin ME, Marcucci MJ, Cline GW, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C θ and alterations in the insulin signaling cascade. Diabetes. 1999;48(6):1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 118.Lam TKT, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nature Neuroscience. 2005;8(5):579–584. doi: 10.1038/nn1456. [DOI] [PubMed] [Google Scholar]
- 119.Florian JP, Pawelczyk JA. Non-esterified fatty acids increase arterial pressure via central sympathetic activation in humans. Clinical Science. 2010;118(1):61–69. doi: 10.1042/CS20090063. [DOI] [PubMed] [Google Scholar]
- 120.Gadegbeku CA, Dhandayuthapani A, Sadler ZE, Egan BM. Raising lipids acutely reduces baroreflex sensitivity. American Journal of Hypertension. 2002;15(6):479–485. doi: 10.1016/s0895-7061(02)02275-6. [DOI] [PubMed] [Google Scholar]
- 121.Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. The New England Journal of Medicine. 2000;342(24):1792–1801. doi: 10.1056/NEJM200006153422406. [DOI] [PubMed] [Google Scholar]
- 122.Monahan KD, Dyckman DJ, Ray CA. Effect of acute hyperlipidemia on autonomic and cardiovascular control in humans. Journal of Applied Physiology. 2007;103(1):162–169. doi: 10.1152/japplphysiol.00167.2007. [DOI] [PubMed] [Google Scholar]
- 123.Grekin RJ, Ngarmukos C-O, Williams DM, Supiano MA. Renal norepinephrine spillover during infusion of nonesterified fatty acids. American Journal of Hypertension. 2005;18(3):422–426. doi: 10.1016/j.amjhyper.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 124.Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. The Journal of Biological Chemistry. 1996;271(18):10697–10703. doi: 10.1074/jbc.271.18.10697. [DOI] [PubMed] [Google Scholar]
- 125.Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (adipose most abundant gene transcript 1) Biochemical and Biophysical Research Communications. 1996;221(2):286–289. doi: 10.1006/bbrc.1996.0587. [DOI] [PubMed] [Google Scholar]
- 126.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. The Journal of Biological Chemistry. 1995;270(45):26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 127.Guo Z, Xia Z, Yuen VG, McNeill JH. Cardiac expression of adiponectin and its receptors in streptozotocin-induced diabetic rats. Metabolism. 2007;56(10):1363–1371. doi: 10.1016/j.metabol.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 128.Hug C, Wang J, Ahmad NS, Bogan JS, Tsao T-S, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(28):10308–10313. doi: 10.1073/pnas.0403382101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cohen SS, Gammon MD, North KE, et al. ADIPOQ, ADIPOR1, and ADIPOR2 polymorphisms in relation to serum adiponectin levels and BMI in black and white women. Obesity. 2011;19(10):2053–2062. doi: 10.1038/oby.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Haluzik M, Parizkova J, Haluzik MM. Adiponectin and its role in the obesity-induced insulin resistance and related complications. Physiological Research. 2004;53:123–129. [PubMed] [Google Scholar]
- 131.Salani B, Briatore L, Andraghetti G, Adami GF, Maggi D, Cordera R. High-molecular weight adiponectin isoforms increase after biliopancreatic diversion in obese subjects. Obesity. 2006;14(9):1511–1514. doi: 10.1038/oby.2006.174. [DOI] [PubMed] [Google Scholar]
- 132.Trevaskis JL, Gawronska-Kozak B, Sutton GM, et al. Role of adiponectin and inflammation in insulin resistance of Mc3r and Mc4r knockout mice. Obesity. 2007;15(11):2664–2672. doi: 10.1038/oby.2007.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ohashi K, Kihara S, Ouchi N, et al. Adiponectin replenishment ameliorates obesity-related hypertension. Hypertension. 2006;47(6):1108–1116. doi: 10.1161/01.HYP.0000222368.43759.a1. [DOI] [PubMed] [Google Scholar]
- 134.Antoniades C, Antonopoulos AS, Tousoulis D, Stefanadis C. Adiponectin: from obesity to cardiovascular disease: etiology and Pathophysiology. Obesity Reviews. 2009;10(3):269–279. doi: 10.1111/j.1467-789X.2009.00571.x. [DOI] [PubMed] [Google Scholar]
- 135.Tesauro M, Schinzari F, Caramanti M, Lauro R, Cardillo C. Metabolic and cardiovascular effects of ghrelin. International Journal of Peptides. 2010;2010 doi: 10.1155/2010/864342.864342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pulkkinen L, Ukkola O, Kolehmainen M, Uusitupa M. Ghrelin in diabetes and metabolic syndrome. International Journal of Peptides. 2010;2010:11 pages. doi: 10.1155/2010/248948.248948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407(6806):908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- 138.Tschöp M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating ghrelin levels are decreased in human obesity. Diabetes. 2001;50(4):707–709. doi: 10.2337/diabetes.50.4.707. [DOI] [PubMed] [Google Scholar]
- 139.Langenberg C, Bergstrom J, Laughlin GA, Barrett-Connor E. Ghrelin and the metabolic syndrome in older adults. The Journal of Clinical Endocrinology & Metabolism. 2005;90(12):6448–6453. doi: 10.1210/jc.2005-1358. [DOI] [PubMed] [Google Scholar]
- 140.Lambert E, Lambert G, Ika-Sari C, et al. Ghrelin modulates sympathetic nervous system activity and stress response in lean and overweight men. Hypertension. 2011;58(1):43–50. doi: 10.1161/HYPERTENSIONAHA.111.171025. [DOI] [PubMed] [Google Scholar]
- 141.Freeman JN, do Carmo JM, Adi AH, da Silva AA. Chronic central ghrelin infusion reduces blood pressure and heart rate despite increasing appetite and promoting weight gain in normotensive and hypertensive rats. Peptides. 2013;42:35–42. doi: 10.1016/j.peptides.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tesauro M, Schinzari F, Iantorno M, et al. Ghrelin improves endothelial function in patients with metabolic syndrome. Circulation. 2005;112(19):2986–2992. doi: 10.1161/CIRCULATIONAHA.105.553883. [DOI] [PubMed] [Google Scholar]
- 143.Iantorno M, Chen H, Kim J-A, et al. Ghrelin has novel vascular actions that mimic PI 3-kinase-dependent actions of insulin to stimulate production of NO from endothelial cells. American Journal of Physiology—Endocrinology and Metabolism. 2007;292(3):E756–E764. doi: 10.1152/ajpendo.00570.2006. [DOI] [PubMed] [Google Scholar]
- 144.Tesauro M, Schinzari F, Rovella V, et al. Ghrelin restores the endothelin 1/nitric oxide balance in patients with obesity-related metabolic syndrome. Hypertension. 2009;54(5):995–1000. doi: 10.1161/HYPERTENSIONAHA.109.137729. [DOI] [PubMed] [Google Scholar]
- 145.Dibona GF, Kopp UC. Neural control of renal function. Physiological Reviews. 1997;77(1):75–197. doi: 10.1152/physrev.1997.77.1.75. [DOI] [PubMed] [Google Scholar]
- 146.Sobotka PA, Mahfoud F, Schlaich MP, Hoppe UC, Böhm M, Krum H. Sympatho-renal axis in chronic disease. Clinical Research in Cardiology. 2011;100(12):1049–1057. doi: 10.1007/s00392-011-0335-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schlaich MP, Hering D, Sobotka P, et al. Effects of renal denervation on sympathetic activation, blood pressure, and glucose metabolism in patients with resistant hypertension. Frontiers in Physiology. 2012;3, article 10 doi: 10.3389/fphys.2012.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mabin T, Sapoval M, Cabane V, Stemmett J, Iyer M. First experience with endovascular ultrasound renal denervation for the treatment of resistant hypertension. EuroIntervention. 2012;8:57–61. doi: 10.4244/EIJV8I1A10. [DOI] [PubMed] [Google Scholar]
- 149.Schlaich MP, Straznicky N, Grima M, et al. Renal denervation: a potential new treatment modality for polycystic ovary syndrome? Journal of Hypertension. 2011;29(5):991–996. doi: 10.1097/HJH.0b013e328344db3a. [DOI] [PubMed] [Google Scholar]
- 150.Horl MP, Horl WH. Hemodialysis-associated hypertension: pathophysiology and therapy. American Journal of Kidney Diseases. 2002;39(2):227–244. doi: 10.1053/ajkd.2002.30542. [DOI] [PubMed] [Google Scholar]
- 151.Rubinger D, Backenroth R, Sapoznikov D. Sympathetic nervous system function and dysfunction in chronic hemodialysis patients. Seminars in Dialysis. 2013;26:333–343. doi: 10.1111/sdi.12093. [DOI] [PubMed] [Google Scholar]
- 152.Di Daniele N, De Francesco M, Violo L, Spinelli A, Simonetti G. Renal sympathetic nerve ablation for the treatment of difficult-to-control or refractory hypertension in a haemodialysis patient. Nephrology Dialysis Transplantation. 2012;27(4):1689–1690. doi: 10.1093/ndt/gfs044. [DOI] [PubMed] [Google Scholar]
- 153.Ott C, Schmid A, Ditting T, et al. Renal denervation in a hypertensive patient with end-stage renal disease and small arteries: a direction for future research. Journal of Clinical Hypertension. 2012;14:799–801. doi: 10.1111/jch.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schlaich MP, Bart B, Hering D, et al. Feasibility of catheter-based renal nerve ablation and effects on sympathetic nerve activity and blood pressure in patients with end-stage renal disease. International Journal of Cardiology. 2013 doi: 10.1016/j.ijcard.2013.01.218. [DOI] [PubMed] [Google Scholar]