

Abstract
A Glyoxalase inhibitor was synthesized and tested against Staphylococcus aureus for first time and showed MIC90 of 20 μg/ml. Henceforth, we synthesized unnatural azide derivative of the same inhibitor to improve the biological activity. In that order, an azide carboxylate was synthesized from dimethyl tartrate by tosylation and azide substitution. The synthesized, azide compound was coupled with glutathione derivative in high yield and tested against S. aureus and showed improved MIC90 of 5 μg/ml. In general, it can be also easily converted to unnatural β-amino acid in good yield. The shown methodology will be extended to study induced suicide in Burkholderia mallei, Francisella tularensis and Mycobacterium tuberculosis in future.
Keywords: Glyoxalase pathway, Metabolism, Staphylococcus aureus, Glutathione derivative, Inhibitor
Approximately 10 million people die every year due to bacterial infections and no new classes of antibacterial drugs have been produced since 1980’s1. The numbers of drug resistant pathogen strains are also increasing;2,3 therefore new active antibacterial drugs4 are necessary.
Recently, glyoxal pathway has been shown more interest and tested in E. coli,5 P. falciparum,6 and A. thaliana 7 and has led to identification of growth inhibitors.
In glycolytic metabolism, methylglyoxal (MG) is produced as a byproduct, which is a highly toxic α-oxoaldehyde and an unavoidable compound in microorganisms. MG modifies arginine, lysine and cysteine residues in proteins, forming glycation end products and impairing the biochemical functionality of those macromolecules. MG also reacts with guanyl nucleotide in nucleic acids forming adducts that can lead to DNA modification such as DNA adducts, mutations, chromosomal aberrations, DNA repair, sister chromatid exchanges and DNA single strand break. These processes ultimately lead to cell death. In the glyoxalase pathway, methylglyoxal is detoxified by reacting with glutathione to form hemithioacetal (HTA) (Fig. 1)8. HTA is then converted to S-D-lactoglutathione (SDLGSH) by GlxI. SDLGSH reacts in the presence of GlxII to form D-lactate and regenerates glutathione. The glutathione is then reused or recycled. The explained glyoxalase pathway is the major pathway for detoxification of MG.9–11 Eventhough GlxI and GlxII are well known in gram negative bacteria, they are not well explored in gram positive bacteria. Recently, GlxI enzyme is reported in gram positive bacteria - Clostridium acetobutylicum.12 Interestingly, the evidence for the functional protein has been well determined for the same microorganism. Most importantly, the same glyoxal pathway is not found in human cells. Therefore, it is considered to be a good drug target for the development of antimicrobial, antimalarial and herbicidal agents, a hypothesis not explored by many researchers. Here, we report the synthesis and inhibiton of glyoxalase pathway inhibitor against Staphylococcus aureus.
Fig. 1.

The glyoxalase pathway
N-phenylmethoxy carbonylglutathione13,7,14 a known A. thaliana GlxII inhibitor, 2 was synthesized from glutathione, 1 by reacting with benzyl chloroformate (Scheme 1). The synthesized compound, 2 was purified and characterized by 1H-NMR and MS and tested against Staphylococcus aureus, and Burkholderia thailandensis for the first time. The synthesized compound, 2 showed MIC90 20 μg/ml on testing against Staphylococcus aureus and 12. 5 μg/ml against Burkholderia thailandensis respectively (Table 1).15 This promising preliminary result displayed strong support for improving the glyoxalase pathway inhibitor using induced suicide methodology for Staphylococcus aureus and Burkholderia thailandensis.
Scheme 1.
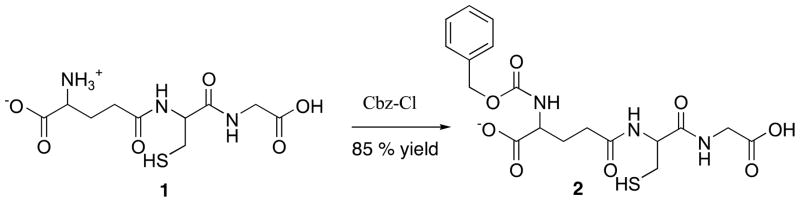
Synthesis of 2
Table 1.
MIC values against microorganisms
| Compound | Microorganism | MIC90 (μg/ml) |
|---|---|---|
| 2 | Burkholderia thailandensis | 12.5 |
| 2 | Staphylococcus aureus | 20.0 |
| 6 | Mycobacterium smegmatis | 6.0 |
| 6 | Staphylococcus aureus | 5.0 |
Earlier it was observed that the presence of β-amino acid at the terminal end enhances the biological activity of many compounds.16 Therefore, we decided to link azide carboxylate group, which is similar to the β-amino acid group, to the free terminal of the known inhibitor, 2 to increase the biological activity. The azide compound, 517 was synthesized from the available dimethyl tartrate (Scheme 2). Briefly, commercially available dimethyl L-tartrate, 3 was activated by monotosylating the single hydroxyl group with one equivalent of tosyl chloride in the presence of pyridine, triethylamine and catalytic amount of DMAP (dimethyl amino pyridine) in dicholoromathane. Concurrently, the azide group was introduced by nucleophilic substitution reaction using sodium azide. The azide ion displaced the tosyl group by SN2 reaction in DMF solvent. The synthesized unnatural azide derivative, 5 was successfully coupled with the known inhibitor, 2 to produce unnatural glutathione derivative, 6. Briefly, HATU ([Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate) was used to activate the terminal primary carboxylic acid group in 2 in presence of triethylamine. Fortunately, the secondary carboxylic acid group in glutathione derivative, 2 was not affected because of more steric hindrance. The newly synthesized 6, was characterized and confirmed by NMR and MS18 and tested against Staphylococcus aureus and showed MIC90 of 5 μg/ml (Table 1). The same compound was also tested against M. smegmatis and observed MIC90 of 6 μg/ml. Since the compound 6 showed better activity against S. aureus we were interested in evaluating the glyoxalase enzyme in S. aureus. Interestingly, through BLAST analysis we found that putative S. aureus glyoxalase had 47 % identity with E. coli glyoxalase and encoded a polypeptide of 229 amino acids with a molecular weight of 28.9 kDa. Designing and synthesis of novel compounds are in process to improve the biological activity against S. aureus.
Scheme 2.

Synthesis of 6, an unnatural glutathione derivative
The toxicity was tested by MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay in PBMC (peripheral blood mononuclear cell) cells. For determination of cytotoxicity of the inhibitors, MDM (monocyte derived macrophages) were cultured and treated with compounds at 0.05 mM for 8 hours at 37°C in 5% CO2. Following loading of each inhibitors, cells were washed with serum-free culture medium to remove excess compounds. The cytotoxicity was assessed over the subsequent 24 hours using the alamar Blue TM assay (AbD Serotec, Raleigh, NC).19 Interestingly, both the inhibitors, 2 and 6 are non-toxic and showed strong potential to be developed as drug candidate for future lead optimization and trials.
Hereby, we successfully synthesized unnatural azide carboxylate, 6 from commercially available dimethyl tartrate, 3. In addition 6 can be also easily converted to unnatural β-amino acid in good yield. We coupled 5 with the known glyoxalase pathway inihibitor, 2 to generate a novel compound, 6. The resulted azide glutathione derivative, 6 was tested against Staphylococcus aureus for the first time and observed moderate biological activity of MIC90 of 5 μg/ml as a glyoxalase pathway inhibitor. Presently, lead optimization is undergoing in our lab and in future it will be extended to study induced suicide in Burkholderia mallei, Francisella tularensis and Mycobacterium tuberculosis.
Acknowledgments
We are thankful to Dr. Dean Crick of CSU, Dr. Howard Gendelman, Dr. Bradley Britigan and Dr. Peter Iwen of UNMC for their kind suggestions and support. We would like to also thank NIH R01AI097550 for funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Organization, W. H. WHO. 2009. [Google Scholar]
- 2.Narayanasamy P, Eoh H, Brennan PJ, Crick DC. Chem Biol. 2010;17:117–22. doi: 10.1016/j.chembiol.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eoh H, Narayanasamy P, Brown AC, Parish T, Brennan PJ, Crick DC. Chem Biol. 2009;16:1230–9. doi: 10.1016/j.chembiol.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurosu M, Narayanasamy P, Biswas K, Dhiman R, Crick DC. J Med Chem. 2007;50:3973–5. doi: 10.1021/jm070638m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stokvis E, Clugston SL, Honek JF, Heck AJ. J Protein Chem. 2000;19:389–97. doi: 10.1023/a:1026439531005. [DOI] [PubMed] [Google Scholar]
- 6.Becker K, Rahlfs S, Nickel C, Schirmer RH. Biol Chem. 2003;384:551–66. doi: 10.1515/BC.2003.063. [DOI] [PubMed] [Google Scholar]
- 7.Zang TM, Hollman DA, Crawford PA, Crowder MW, Makaroff CA. J Biol Chem. 2001;276:4788–95. doi: 10.1074/jbc.M005090200. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson GP, Totemeyer S, MacLean MJ, Booth IR. Arch Microbiol. 1998;170:209–18. doi: 10.1007/s002030050635. [DOI] [PubMed] [Google Scholar]
- 9.Padmanabhan PK, Mukherjee A, Singh S, Chattopadhyaya S, Gowri VS, Myler PJ, Srinivasan N, Madhubala R. Biochem Biophys Res Commun. 2005;337:1237–48. doi: 10.1016/j.bbrc.2005.09.179. [DOI] [PubMed] [Google Scholar]
- 10.Mustafiz A, Sahoo KK, Singla-Pareek SL, Sopory SK. Methods Mol Biol. 2010;639:95–118. doi: 10.1007/978-1-60761-702-0_6. [DOI] [PubMed] [Google Scholar]
- 11.Greig N, Wyllie S, Vickers TJ, Fairlamb AH. Biochem J. 2006;400:217–23. doi: 10.1042/BJ20060882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suttisansanee U, Lau K, Lagishetty S, Rao KN, Swaminathan S, Sauder JM, Burley SK, Honek JF. J Biol Chem. 2011;286:38367–74. doi: 10.1074/jbc.M111.251603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang KW, Sobieski DN, Carenbauer AL, Crawford PA, Makaroff CA, Crowder MW. Arch Biochem Biophys. 2003;414:271–8. doi: 10.1016/s0003-9861(03)00193-0. [DOI] [PubMed] [Google Scholar]
- 14.Degani Y, Patchornik A, Maclaren JA. J Am Chem Soc. 1966;88:3460–1. doi: 10.1021/ja00966a069. [DOI] [PubMed] [Google Scholar]
- 15.96-well U-bottom assay plates were seeded with 1 × 106 CFU in a volume of 0.1 mL/well for Staphylococcus aureus assays. Testing compound was serially diluted in DMSO as 1:2 dilutions in a separate 96-well plate and then transferred to the assay plate. The final concentration of DMSO in the assay plates did not exceed 2% (S. aureus). Plates were incubated at 37°C for 24 hours (S. aureus). O.D.600 readings were taken once at the end of the 24-hour incubation for S. aureus. Sample wells exhibiting 20% or less of the average O.D.600 readings of the untreated control wells were considered to contain a drug concentration resulting in inhibition. Alamar Blue dye solution was added to the wells at the end of the incubation period to a final concentration of 10% (v/v) and assessed after 1 hour (S. aureus) to determine the point of activity. Each MIC assay included untreated controls, media blanks, and control compounds.
- 16.Sibi MP, Prabagaran N, Ghorpade SG, Jasperse CP. J Am Chem Soc. 2003;125:11796–7. doi: 10.1021/ja0372309. [DOI] [PubMed] [Google Scholar]
- 17.Saito SY, Ishikawa H, Niwa T, Moriwake N. Tet Lett. 1991;32:663–666. [Google Scholar]
- 18.Triethylamine (6 μL, 0.044 mmol, 1.0 equiv.) and HATU (16 mg, 0.044 mmol, 1.0 equiv.) were added to a solution of the tripeptide (20 mg, 0.044 mmol, 1.0 equiv.) in DMF (0.5 mL) at 0 °C under argon. A solution of the azide (9 mg, 0.044 mmol, 1.0 equiv.) and triethylamine (6 μL, 0.044 mmol, 1.0 equiv.) in DMF (0.5 mL) was then added dropwise. The mixture was stirred and gradually warmed to rt overnight and concentrated. The residue was diluted with EtOAc (10 mL) and washed with 1M HCl. The aqueous layer was extracted further with EtOAc (3 × 10 mL). The organic extracts were combined, dried over Na2SO4 and concentrated. The residue was purified by flash chromatography eluting with 9:1 CH2Cl2/CH3OH to give compound 6 (9 mg, 35 %). Rf 0.40 (9:1 CH2Cl2/MeOH). 1H NMR (500 MHz, CD3OD) δ 1.90–2.40 (m, 4H), 2.81 (s, 3H), 2.86 (s, 3H), 3.0–3.60 (m, 3H), 4.0–4.60 (m, 3H), 4.80–5.40 (m, 4H), 7.10–7.60 (m, 5H). 13C NMR (125 MHz, CD3OD) 25.2, 28.1, 33.1, 41.9, 53.2, 53.3, 57.5, 58.3, 63.9, 65.2, 83.1, 128.0, 128.3, 128.9, 129.4, 129.5, 135.9, 156.7, 171.8, 172.8, 175.4. δ MS (m/z) calcd for C24H30N6O12S (M + H)+: 626; obsd: 626.
- 19.Kadiu I, Narayanasamy P, Dash PK, Zhang W, Gendelman HE. J Immunol. 2012;189:744–54. doi: 10.4049/jimmunol.1102244. [DOI] [PMC free article] [PubMed] [Google Scholar]